

Abstract
Background
Sepsis, iron loading and aging cause independent increases in gut epithelial and splenic apoptosis. It is unknown how their combination will affect apoptosis and systemic cytokine levels.
Methods
Hfe−/− mice (a murine homolog of hemochromatosis) abnormally accumulate iron in their tissues. Aged (24–26 months) or mature (16–18 months) Hfe−/− mice and wild type (WT) littermates were subjected to cecal ligation and puncture (CLP) or sham laparotomy. Intestine, spleen, and blood were harvested 24 hours later and assessed for apoptosis and cytokine levels.
Results
Gut epithelial and splenic apoptosis were low in both aged septic and sham Hfe−/− mice, regardless of the amount of iron in their diet. Mature septic WT mice had increased apoptosis compared to age-matched sham WT mice. Mature septic Hfe−/− mice had similar levels of intestinal cell death to age-matched septic WT mice but higher levels of splenic apoptosis. Apoptosis was significantly lower in septic aged Hfe−/− mice than septic mature Hfe−/− animals. Interleukin-6 was elevated in septic aged Hfe−/− mice compared to sham mice.
Conclusions
Although sepsis, chronic iron dysregulation, and aging each increase gut and splenic apoptosis, their combination yields cell death levels similar to sham animals despite the fact that aged Hfe−/− mice are able to mount an inflammatory response following CLP and mature Hfe−/− mice have elevated sepsis-induced apoptosis. Combining sepsis with two risk factors that ordinarily increase cell death and increase mortality in CLP yields an apoptotic response that could not have been predicted based upon each element in isolation.
Keywords: Apoptosis, gut, spleen, sepsis, iron, aging, intestine, cytokines, interleukin 6, hemochromatosis
INTRODUCTION
Sepsis causes a disproportionate increase in apoptosis in the intestinal epithelium and lymphocytes in both young mice [1–4] and critically ill patients [5]. Elevated gut cell death appears to be detrimental in sepsis in young animals since tissue specific overexpression of the anti-apoptotic protein Bcl-2 prevents intestinal epithelial apoptosis and improves survival in mice subjected to cecal ligation and puncture (CLP) or Pseudomonas aeruginosa pneumonia [6, 7]. Similarly prevention of splenic apoptosis by overexpression of Bcl-2 in lymphocytes of transgenic mice or administration of caspase inhibitors decreases lymphocyte apoptosis and improves survival in young septic mice [8–10].
Critical illness also causes dysregulated iron metabolism with functional iron deficiency [11, 12]. Patients in the intensive care unit frequently have anemia of critical illness, and because biologically available iron is necessary for hematopoiesis, iron supplementation is often given to septic patients [13, 14]. However, acute high-dose iron supplementation after the onset of sepsis increases mortality in CLP in young mice, and this is associated with increased gut epithelial and splenic apoptosis [15].
Iron may also be detrimental in the setting of chronic heritable dysregulations in iron metabolism such as occurs in hemochromatosis, a disease that affects between 1 in 100 and 1 in 400 adults [16, 17]. This autosomal recessive disorder is caused by a mutation in the Hfe gene, and patients who are homozygous for the abnormality have elevated transferrin saturation. The disease normally does not cause symptoms in those who are affected until the fourth to sixth decade of life, and the number of patients with the genetic abnormality who progress to organ damage is unclear [17, 18]. While the effect of chronic iron dysregulation in acutely ill middle aged or elderly patients is unknown, iron overload given prior to CLP causes increased mortality in young Hfe−/− mice [19], suggesting that both acute and chronic abnormalities in iron metabolism affect outcome in critical illness.
Sepsis is primarily a disease of the aged. While 660,000 to 750,000 people develop sepsis annually in the United States, nearly 60% of all cases occur in patients greater than 65 years of age, with 80% of deaths occurring in this group [20, 21]. Despite the fact that sepsis primarily affects the elderly, the majority of animal studies modeling the disease use 6 to 16 week old rodents, corresponding to a human age of 10–17 years [22]. Recent studies have demonstrated that similar to results in humans, age plays an important role in the murine response sepsis, with aged mice having a marked increase in mortality, decreased sensitivity to antibiotics, and altered inflammatory profiles compared to young animals [22, 23].
Aging has also been reported to cause increased apoptosis in gut epithelial and splenic apoptosis. Lymphocytes from elderly humans are more prone to undergo apoptosis, and aged mice and rats have increased basal and activation-induced lymphocyte cell death [24–27]. Intestinal cell death is also elevated in elderly humans and in aged mice subjected to ionizing radiation or calorie restriction (although not under basal conditions) [28–30]. Importantly the combination of aging and sepsis disproportionately increases both gut epithelial and splenic apoptosis, since septic aged mice have substantially more intestinal and lymphocyte death than could be predicted by examining either variable in isolation [31].
It is therefore known that a) sepsis, iron loading and aging each independently increase apoptosis, b) sepsis, combined with either acute iron loading or aging, causes more cell death than would be seen with each variable in isolation, c) mortality is increased in aged septic mice as well as in young septic mice with either chronic iron dysregulation or acute iron overload. It is not known how the combination of sepsis, chronic iron dysregulation and aging interact. To address this question, we studied gut and splenic apoptosis in septic aged and mature Hfe−/− mice with or without iron loading.
MATERIALS AND METHODS:
Animals:
Breeding pairs of 129/SvJ × C57Bl/6 progeny mice, deleted in the Hfe gene (a generous gift of Dr. William Sly, St. Louis University [32] created a colony of animals that were aged for 16 to 26 months. Previously published survival curves demonstrate that a mouse age of 16–18 months correlates to a human age of 50–60 years, while a murine age of 24–26 months correlates to a human age of 80–90 years [22]. 129/SvJ and wild type (WT) C57Bl/6 mice were also mated and aged for a similar length of time. Mice were maintained on strict 12 hour light-dark cycle with free access to food and water at all times. Animals received Purina Lab Diet #5053 (Richmond, IN) which contains 168.1 ppm iron except as described below. All experiments were conducted in accordance with the National Institutes of Health guidelines for the use of laboratory animals and were approved by Washington University Animal Studies Committee.
Sepsis Model:
CLP was performed on male and female mice by the methods of Baker et al and as described previously [15, 19, 33]. Briefly, a midline laparotomy was performed, the cecum was externalized and ligated distal to the ileocecal valve, taking care to prevent intestinal obstruction. The cecum was punctured once with a 25-gauge needle and gently squeezed to extrude some stool, then replaced into the abdomen, which was closed in layers. Published data demonstrates that double-puncture with a 25-gauge needle results in 30–60% 7-day survival in young Hfe−/− or WT mice given high or iron deficient diet two weeks prior to the onset of sepsis [19]. The reason a smaller injury was used is this study is based upon the fact that aged (24 month) C57Bl/6 WT mice have a 3-fold increase in mortality compared to young (4 months) WT mice subjected to the same insult [22]. Sham operated animals had a similar procedure except the cecum was neither ligated nor punctured. Anesthesia was induced with 5% halothane and maintained with 2.5% halothane.
Iron loading:
Two weeks before the first set of aged animals were subjected to CLP, animals were randomly assigned to receive either iron deficient (0.0 ppm) or high iron (24,310 ppm, 2.5% wt/wt) iron carbonyl ad libitum. During this time, there were no differences in chow consumption or the general health of the animals. Following the first set of experiments demonstrating no difference in apoptosis between mice that received high iron or iron deficient diets, all subsequent animals (including all mature animals) received iron deficient diets for two weeks prior to CLP.
Quantification of apoptosis:
Mice were euthanized 1 day following CLP, and had their small intestine, spleen, kidney, heart, and lung fixed in 10% buffered formalin for 24 hours before being placed in 70% ethanol for sectioning. Prior to fixing, intestines were opened from proximal to distal and washed with 0.9% NaCl, to remove luminal contents. After formalin fixation, intestines were rolled with the lumen facing outward.
Apoptosis was identified using two independent techniques: hematoxylin and eosin (H&E) staining and active caspase 3 staining. Apoptotic intestinal epithelial cells were quantified in 100 contiguous crypts, in well-orientated crypt-villus units. “Well-oriented” sections had a crypt parallel to a crypt -villus axis with Paneth cells at the base and an unbroken epithelial column extending to the villus tip. Apoptotic splenic cells were quantified in 5 random high power (40X) fields. Quantitation was performed in both tissues by an investigator (PJ) blinded to sample identity. Low or no apoptosis was identified in kidney, heart, or lung in 5 high powered fields in all animals, and no differences were identified between groups (data not shown).
Apoptosis was identified in H&E stained sections by characteristic morphology of nuclear fragmentation (karyorrhexis) and cell shrinkage with condensed nuclei (pyknosis). All data presented was quantified using H&E staining. Qualitatively similar results were obtained using active caspase 3 staining (data not shown). Previous publications from our laboratory have shown that these methods of detecting apoptosis correlate closely when quantifying levels of cell death in sepsis, aging or iron dysregulation [6, 7, 15, 31, 34]. Active caspase 3 staining was performed as previously described [6, 7, 15].
Cytokines:
All aged animals examined for apoptosis also had 400 μl of blood obtained from the inferior vena cava (IVC) using sterile techniques under halothane anesthesia (following blood harvest the animals were immediately euthanized by cervical dislocation while still anesthetized). Blood was transferred to a 1.5 ml micro centrifuge tube, and centrifuged at 3300g for five minutes to separate plasma. IL-6, IL-10 and TNFα were measured by enzyme-linked immunosorbent assay (ELISA) using commercially available kits (R&D Systems, Minneapolis, MN) according to manufacturer specifications. All cytokine levels represent the average of duplicates of each sample.
Statistics:
Pairwise comparisons were performed using unpaired t-tests. Multigroup comparisons of apoptosis or cytokines were analyzed using one way analysis of variance followed by the Newman-Keuls multiple comparison test using the statistical software Prism 3.0 (GraphPad Software, San Diego, CA). Data are presented as mean ± SEM. P values <0.05 were considered to be statistically significant.
RESULTS:
Gut epithelial apoptosis
Aged (24–26 months) Hfe−/− animals given either a high iron diet (n=8) or an iron deficient diet (n=6) had low levels of gut epithelial apoptosis following sham laparotomy (Fig. 1, 2). Addition of a septic insult failed to increase gut epithelial apoptosis in aged Hfe−/− animals given either a high iron diet (n=10) or an iron deficient diet (n=4), with similar levels noted in animals subjected to either CLP or sham laparotomy (p>0.05, Fig. 1, 2).
FIG. 1.
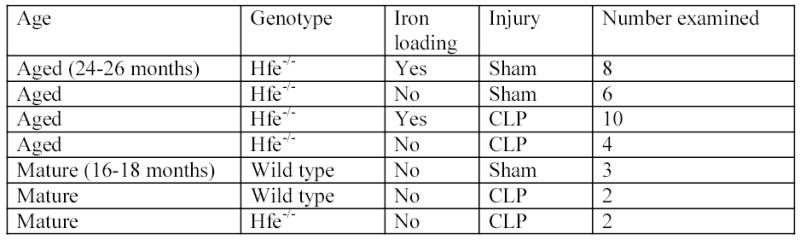
Graph demonstrating all variables examined in overall study design including age, genotype, iron loading status, injury and number of animals examined.
FIG. 2.
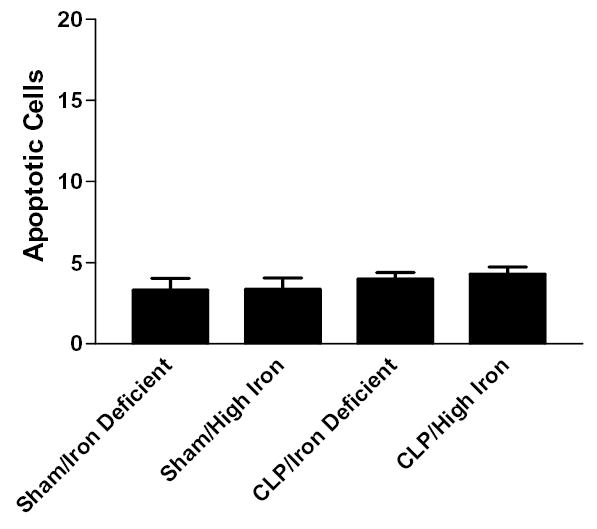
Number of gut epithelial apoptotic cells in 100 crypts in aged Hfe−/− mice subjected to either sham laparotomy or CLP, fed either an iron deficient or high iron diet 2 weeks prior to surgical manipulation. Apoptosis is low in all groups.
Since sepsis [1, 5–7], iron [15], and aging [28, 29] have all been demonstrated to increase gut epithelial apoptosis, we sought to isolate each of these variables to determine why aged septic Hfe−/− animals did not have increased intestinal cell death. As acute iron loading did not affect intestinal cell death, all subsequent experiments were performed in mice that received an iron deficient diet 2 weeks prior to CLP.
The first variable examined was strain. Since a mouse’s genetic background alters its pathobiologic response to sepsis [35, 36] and gut epithelial apoptosis has not been reported in 129/SvJ x C57Bl/6 mice, we first examined how their guts responded to CLP. Mature (16–18 months) WT mice subjected to either sham laparotomy (n=3) or CLP (n=2) showed the expected statistically significant increase in crypt cell death following sepsis (p<0.05, Fig. 1, 3A, 4A, 4B) demonstrating that animals with this genetic background have the capacity to undergo sepsis-induced gut epithelial apoptosis.
FIG. 3.

Comparison of gut epithelial apoptosis between (A) mature WT mice subjected to sham laparotomy or CLP, (B) mature WT and Hfe−/− mice subjected to CLP, and (C) mature and aged Hfe−/− mice subjected to CLP. Asterisks represent p values <0.05.
FIG. 4.

Gut epithelial apoptosis in (A) mature WT mice subjected to sham laparotomy, (B) mature WT mice subjected to CLP, (C) mature Hfe−/− mice subjected to CLP, and (D) aged Hfe−/−mice subjected to CLP. Apoptotic cells are identified by arrows.
The next variable examined was iron. To determine whether chronic abnormalities in iron handling in Hfe−/− animals caused the same alterations in sepsis-induced intestinal cell death seen in age-matched WT mice, mature Hfe−/− animals and WT mice with the same genetic background were examined. Each had had similar levels of gut epithelial apoptosis 24 hours following CLP (n=2, p>0.05, Fig. 1, 3B, 4C), demonstrating that Hfe−/− mice can respond to infection by increasing intestinal cell death, although the effects of iron loading and sepsis are not additive in this model.
The final variable examined was age. A post-hoc analysis of the above experiments was performed comparing 16–18 month old Hfe−/− animals to 24–26 month old Hfe−/− animals subjected to CLP after receiving an identical diet prior to injury. A greater than 3-fold difference in gut epithelial apoptosis was noted with elevated levels in middle aged mice that disappeared in elderly mice (p<0.0001, Fig. 1, 3C, 4C, 4D).
Splenic apoptosis
Similar analysis was performed on the same mice for splenic apoptosis. Aged Hfe−/−animals given either an iron deficient diet or high iron diet had low levels of splenic apoptosis following sham laparotomy (Fig. 1, 5). Addition of a septic insult failed to increase splenic apoptosis in aged Hfe−/− animals given either a high iron diet or an iron deficient diet with similar levels noted in animals subjected to either CLP or sham laparotomy (p>0.05, Fig. 1, 5).
FIG. 5.
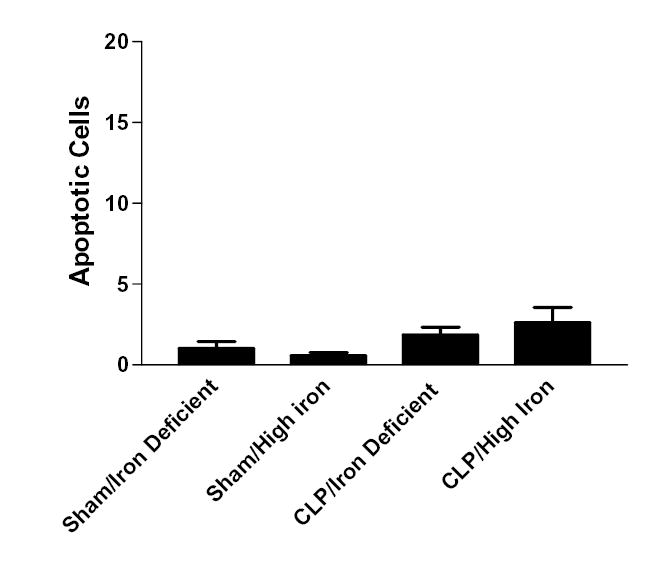
Splenic apoptosis in 5 random high powered fields in aged Hfe−/− mice subjected to either sham laparotomy or CLP, fed either an iron deficient or high iron diet 2 weeks prior to surgical manipulation. Apoptosis is low in all groups.
Similar to results seen in the intestine, mature WT mice subjected to CLP had a significant increase in splenic death compared to WT mice that underwent sham laparotomy (p<0.05, Fig. 1, 6A). However, unlike in the gut, mature Hfe−/− animals had a further increase in sepsis-induced splenic apoptosis compared to WT mice of same genetic background 24 hours following CLP (p<0.05, Fig. 1, 6B). In addition, a post-hoc analysis of the above experiments comparing 16–18 month old Hfe−/− animals to 24–26 month old Hfe−/− animals subjected to CLP after receiving an identical diet prior to injury showed a nearly 20-fold difference in splenic apoptosis (p=0.0005, Fig. 1, 6C).
FIG. 6.

Comparison of splenic apoptosis between (A) mature WT mice subjected to sham laparotomy or CLP, (B) mature WT and Hfe−/− mice subjected to CLP, and (C) mature and aged Hfe−/− mice subjected to CLP. All groups are statistically different.
Cytokines
Since aged Hfe−/− animals had no increase in either gut epithelial or splenic apoptosis following CLP regardless of whether they received a high iron or iron deficient diet, we examined cytokine levels 24 hours post-operatively as a surrogate of their systemic inflammatory response. Levels of the pro-inflammatory cytokine interleukin (IL)-6 were elevated in Hfe−/− mice subjected to CLP regardless of diet compared to sham animals (p<0.05, Fig. 1, 7A). Levels of tumor necrosis factor-α (TNFα) were elevated in septic animals given a high iron diet prior to CLP (p<0.05) but not animals given an iron deficient diet (Fig. 1, 7B). Although absolute values of the anti-inflammatory mediator IL-10 were higher in septic mice, no statistically significant differences were seen between any groups (Fig. 1, 7C).
FIG. 7.
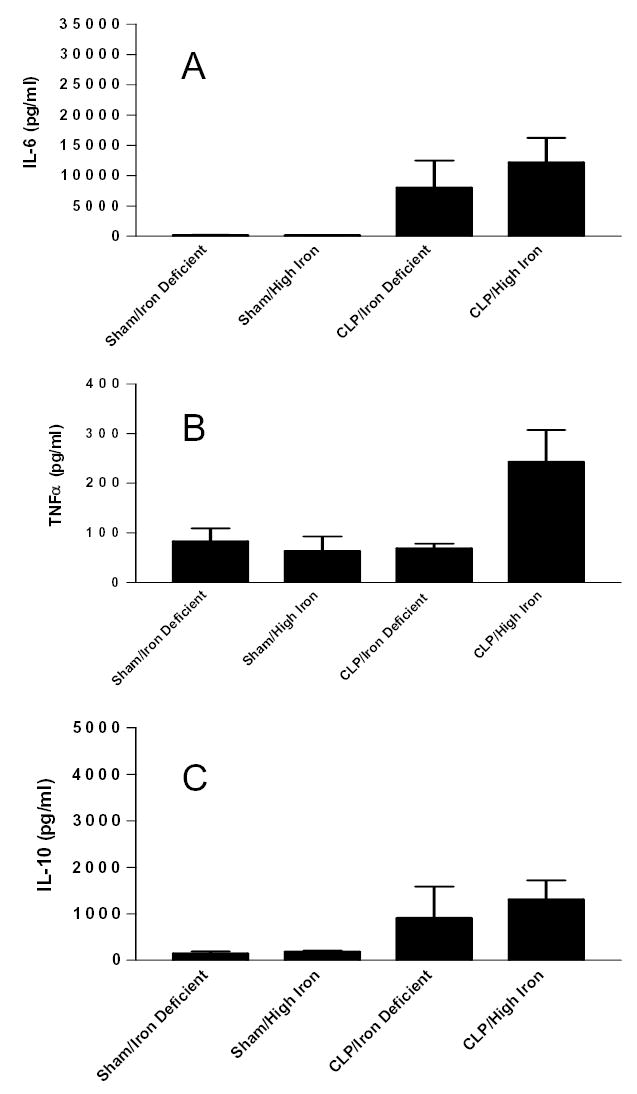
Systemic levels of inflammatory cytokines (A) IL-6, (B) TNFα, and (C) IL-10 in aged Hfe−/− mice subjected to either sham laparotomy or CLP, fed either an iron deficient or high iron diet 2 weeks prior to surgical manipulation. Absolute levels of all mediators are generally higher in septic animals demonstrating CLP changes the inflammatory profile without altering gut and splenic apoptosis (compare to fig. 1, 4).
DISCUSSION
These experiments demonstrate that although sepsis, dysregulated iron handling, and aging all independently increase gut epithelial and splenic apoptosis, their combination leads to a complete abrogation of this response, resulting in cell death levels that are similar to animals that undergo sham laparotomy. An animal’s age appears to be the critical determinant of its apoptotic response based upon our findings that a) middle aged WT animals subjected to CLP have elevated apoptosis compared to sham (demonstrating that animals of this genetic background have the capacity to undergo cell death), b) mature septic Hfe−/− mice have similar or elevated levels of apoptosis to septic age-matched WT mice (demonstrating that Hfe−/− animals also have the capacity to undergo cell death), but c) aged septic Hfe−/− mice have significantly less apoptosis than genetically-matched, diet-matched, middle-aged Hfe−/− mice that undergo the same CLP insult.
These results are surprising given that both sepsis and aging as well as sepsis and acute iron loading result in higher levels of apoptosis than would be expected based upon each variable in isolation. This is important because our result models the clinically relevant situation where an acute insult (CLP) is superimposed upon chronic, pre-existing conditions. This demonstrates a potential limitation of commonly used reductionist animal models of critical illness such as performing CLP on healthy 6-week old mice. While valuable insights may obviously be gained by studying sepsis in this group, it is unclear if these are appropriate surrogates for aged patients, which make up the majority of ICU admissions and deaths. Further, even studying sepsis in aged mice – with the genetic and immunological changes they accumulate throughout life – may model previously healthy elderly patients but not those with multiple medical problems. This is demonstrated comparing sepsis-induced gut epithelial or splenic apoptosis in the following groups: young WT, mature WT, aged WT, mature Hfe−/−, aged Hfe−/−. Although each group differs in a only a single variable from the previous one, cell death ranges from increased compared to sham (young WT, mature WT, mature Hfe−/−), markedly increased compared to sham (aged WT) [31] to unchanged compared to sham (aged Hfe−/−) with age or chronically abnormal iron handling having varying results based upon the host’s underlying condition. It also demonstrates that the body does not respond in a uniform fashion to sepsis, since IL-6 and TNFα were elevated compared to sham 24 hours following CLP in aged Hfe−/− mice while apoptosis was unchanged.
This is complimentary to data demonstrating that the pathobiological response to two separate “hits” is dependent upon an animal’s genetic background and both the timing between and specific insults used [35, 37–40]. In “two hit” models of critical illness, the body’s response frequently is markedly different from what would have been predicted based upon the reaction to a single insult. Together, this demonstrates that a host subjected to multiple acute insults or an acute insult superimposed on chronic underlying conditions can have an unexpected response to sepsis, which may impact the response to therapy.
While our results are best globally interpreted in the context of combining acute (CLP) and chronic (aging and hemochromatosis) conditions, insights can also be gained by examining the effect of iron loading in isolation. First, iron loading prior to CLP had no impact on any variable examined in Hfe−/− mice except for TNFα levels at 24 hours. This was surprising given that a high iron diet given two weeks prior to CLP in young animals with this genetic abnormality causes increased mortality over those given a low iron diet [19]. We do not have a good explanation for this unexpected result. Since we have not examined apoptosis or cytokines in young mice or mortality in aged mice, it is not clear if cell death or systemic cytokine levels correlate to survival or are unrelated. However the fact that apoptosis and most cytokines were similar between mice that were iron loaded or received an iron deficient diet two weeks prior to CLP suggests that the chronic state of iron dysregulation is more important than acute supplementation of iron although it does not delineate the impact of iron given after the septic insult. How this is related to the mechanism of overload in Hfe−/− animals (wherein inappropriately low crypt cell iron results in a stabilization of the divalent metal transporter, ultimately leading to increased absorption of dietary iron) is unclear [32].
It was also surprising that aged sham Hfe−/− mice had low basal levels of apoptosis, regardless of iron loading. Although these were control animals, aging, by itself, has been shown to cause increased gut epithelial and splenic apoptosis, and we would have expected these mice would have at least mildly elevated levels of cell death compared to genotypically identical younger animals. This did not appear to be the case, although we cannot definitively state this since we did not examine young or mature sham Hfe−/− mice and therefore could not compare apoptosis between the sham mice of different ages. We believe it is likely however that sham Hfe−/− mice have similar levels of basal cell death regardless of their age, suggesting that the body’s response to a lifelong deficiency in the Hfe gene impacts gut and splenic apoptosis via an unknown mechanism.
In young animals, acute iron loading increases mortality whether given before sepsis in Hfe−/− mice or following CLP in C57Bl/6 mice. While this is clearly an important consideration in evaluating the role of iron in critical illness, those experiments were performed on animals that chronologically mimic human teenagers. In contrast, hemochromatosis affects older patients and iron supplementation in sepsis is generally given to anemic elderly patients. Since apoptosis is elevated in the liver of patients with hemochromatosis [41] who are unlikely to be on long-term high iron diets, this suggests that chronic dysregulation of iron metabolism can affect cell death independent of the composition of oral intake. The fact that apoptosis was effectively decreased (i.e. no sepsis-induced increase) in our results also demonstrates that the response to iron loading must be interpreted in the context of co-existing factors.
Although this study provides new insights into the interplay between sepsis, iron, and aging, it has a number of limitations. First, only a limited supply of mature and aged mice was available. Based upon this, we were unable to obtain data on a) the effects of a high iron diet on mature Hfe−/− mice with or without sepsis, and b) apoptosis or cytokine levels in aged, septic WT mice. We have previously demonstrated that aged, septic C57Bl/6 mice have a disproportionate increase in gut and splenic apoptosis [31], but cannot definitively prove that aged C57Bl/6 x 129/SvJ mice respond to a septic insult like young or mature animals with elevated intestinal and lymphocyte apoptosis. Similarly, although iron loading did not produce any alterations in apoptosis in aged septic or sham Hfe−/− mice, this does not prove that iron loading would not alter cell death in mature animals, which we did not examine in this study. We also do not know the effect of iron loading prior to CLP on apoptosis in aged WT animals although this has not been shown to alter mortality in younger animals. Since the apoptosis data on the mature animals is based upon a small number of animals, this increases the risk of overestimating apoptosis levels in septic mice based upon sample size. However, it is clear that no apoptotic response was seen in any aged Hfe−/− mice regardless of iron loading (n=28 aged animals total), and the number of apoptotic cells was higher in each septic mature animal (n=4) than every septic aged animal (n=14). Further, we cannot know the functional significance of our results since we do not know the 1-week survival of aged Hfe−/− mice or aged WT mice of this genetic background subjected to CLP and therefore cannot know whether the lack of apoptotic response affects mortality. Finally, while Hfe−/− mice are functionally deleted in the gene responsible for hemochromatosis and have documented abnormalities in their iron handling, people with the disorder typically have a mutation (rather than a deletion) in the gene. As such, the mice used in this study may not fully mimic human hemochromatosis, and our results must be interpreted with caution.
Despite these limitations, this study demonstrates that the combination of sepsis, chronic iron dysregulation and aging yields an apoptotic response markedly different from what would be expected based upon each variable in isolation or a combination of two. Further in vivo studies are necessary to determine if other pre-existing illness superimposed upon acute critical illness affects the host’s pathobiological response to overwhelming infection as well as to clarify the effect of the combination of iron loading after sepsis in a host who already has chronic abnormalities in iron regulation.
Acknowledgments
Supported by National Institutes of Health Grants GM 66202, GM00709 (to CMC), GM48095 (to TGB), GM08795 GM 44118, GM 55194 (to RSH) and P30 DK52574 (Washington University Digestive Diseases Research Core).
We thank Kevin Tinsley for outstanding technical assistance.
References
- 1.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 2.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock. 1997;7:247–253. doi: 10.1097/00024382-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. 1996;87:4261–4275. [PubMed] [Google Scholar]
- 4.Chung CS, Wang W, Chaudry IH, Ayala A. Increased apoptosis in lamina propria B cells during polymicrobial sepsis is FasL but not endotoxin mediated. Am J Physiol Gastrointest Liver Physiol. 2001;280:G812–G818. doi: 10.1152/ajpgi.2001.280.5.G812. [DOI] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002;30:195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 7.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 8.Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, Zollner KM, Buchman TG, Korsmeyer SJ, Karl IE. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. 1999;162:4148–4156. [PubMed] [Google Scholar]
- 9.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S, Roy S, Black C, Grimm E, Aspiotis R, Han Y, Nicholson DW, Karl IE. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- 11.Patteril MV, Davey-Quinn AP, Gedney JA, Murdoch SD, Bellamy MC. Functional iron deficiency, infection and systemic inflammatory response syndrome in critical illness. Anaesth Intensive Care. 2001;29:473–478. doi: 10.1177/0310057X0102900504. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez RM, Corwin HL, Gettinger A, Corwin MJ, Gubler D, Pearl RG. Nutritional deficiencies and blunted erythropoietin response as causes of the anemia of critical illness. J Crit Care. 2001;16:36–41. doi: 10.1053/jcrc.2001.21795. [DOI] [PubMed] [Google Scholar]
- 13.Shander A. Anemia in the critically ill. Crit Care Clin. 2004;20:159–178. doi: 10.1016/j.ccc.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Shapiro MJ, Corwin MJ, Colton T. Efficacy of recombinant human erythropoietin in critically ill patients: a randomized controlled trial. JAMA. 2002;288:2827–2835. doi: 10.1001/jama.288.22.2827. [DOI] [PubMed] [Google Scholar]
- 15.Javadi P, Buchman TG, Stromberg PE, Husain KD, Dunne WM, Woolsey CA, Turnbull IR, Hotchkiss RS, Karl IE, Coopersmith CM. High-dose exogenous iron following cecal ligation and puncture increases mortality rate in mice and is associated with an increase in gut epithelial and splenic apoptosis. Crit Care Med. 2004;32:1178–1185. doi: 10.1097/01.ccm.0000124878.02614.4c. [DOI] [PubMed] [Google Scholar]
- 16.Hover AR, McDonnell SM, Burke W. Changing the clinical management of hereditary hemochromatosis: translating screening and early case detection strategies into clinical practice. Arch Intern Med. 2004;164:957–961. doi: 10.1001/archinte.164.9.957. [DOI] [PubMed] [Google Scholar]
- 17.Ajioka RS, Kushner JP. Clinical consequences of iron overload in hemochromatosis homozygotes. Blood. 2003;101:3351–3353. doi: 10.1182/blood-2002-11-3453. [DOI] [PubMed] [Google Scholar]
- 18.Beutler E. The HFE Cys282Tyr mutation as a necessary but not sufficient cause of clinical hereditary hemochromatosis. Blood. 2003;101:3347–3350. doi: 10.1182/blood-2002-06-1747. [DOI] [PubMed] [Google Scholar]
- 19.Wizorek JJ, Turnbull IR, Buchman TG. Iron overload before cecal ligation and puncture increases mortality. Shock. 2003;20:52–55. doi: 10.1097/01.shk.0000065770.72937.fd. [DOI] [PubMed] [Google Scholar]
- 20.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 22.Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS, Coopersmith CM, Buchman TG. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19:310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. 2003;124:1047–1058. doi: 10.1016/j.mad.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Schindowski K, Leutner S, Muller WE, Eckert A. Age-related changes of apoptotic cell death in human lymphocytes. Neurobiol Aging. 2000;21:661–670. doi: 10.1016/s0197-4580(00)00171-8. [DOI] [PubMed] [Google Scholar]
- 25.Phelouzat MA, Laforge T, Arbogast A, Quadri RA, Boutet S, Proust JJ. Susceptibility to apoptosis of T lymphocytes from elderly humans is associated with increased in vivo expression of functional Fas receptors. Mech Ageing Dev. 1997;96:35–46. doi: 10.1016/s0047-6374(97)01883-6. [DOI] [PubMed] [Google Scholar]
- 26.Telford WG, Miller RA. Aging increases CD8 T cell apoptosis induced by hyperstimulation but decreases apoptosis induced by agonist withdrawal in mice. Cell Immunol. 1999;191:131–138. doi: 10.1006/cimm.1998.1422. [DOI] [PubMed] [Google Scholar]
- 27.Pagliara P, Chionna A, Panzarini E, De Luca A, Caforio S, Serra G, Abbro L, Dini L. Lymphocytes apoptosis: young versus aged and humans versus rats. Tissue Cell. 2003;35:29–36. doi: 10.1016/s0040-8166(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 28.Ciccocioppo R, Di Sabatino A, Luinetti O, Rossi M, Cifone MG, Corazza GR. Small bowel enterocyte apoptosis and proliferation are increased in the elderly. Gerontology. 2002;48:204–208. doi: 10.1159/000058351. [DOI] [PubMed] [Google Scholar]
- 29.Holt PR, Moss SF, Heydari AR, Richardson A. Diet restriction increases apoptosis in the gut of aging rats. J Gerontol A Biol Sci Med Sci. 1998;53:B168–B172. doi: 10.1093/gerona/53a.3.b168. [DOI] [PubMed] [Google Scholar]
- 30.Martin K, Kirkwood TB, Potten CS. Age changes in stem cells of murine small intestinal crypts. Exp Cell Res. 1998;241:316–323. doi: 10.1006/excr.1998.4001. [DOI] [PubMed] [Google Scholar]
- 31.Turnbull IR, Buchman TG, Javadi P, Woolsey CA, Hotchkiss RS, Karl IE, Coopersmith CM. Age disproportionately increases sepsis-induced apoptosis in the spleen and gut epithelium. Shock. 2004;22:364–368. doi: 10.1097/01.shk.0000142552.77473.7d. [DOI] [PubMed] [Google Scholar]
- 32.Fleming RE, Migas MC, Zhou X, Jiang J, Britton RS, Brunt EM, Tomatsu S, Waheed A, Bacon BR, Sly WS. Mechanism of increased iron absorption in murine model of hereditary hemochromatosis: increased duodenal expression of the iron transporter DMT1. Proc Natl Acad Sci U S A. 1999;96:3143–3148. doi: 10.1073/pnas.96.6.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- 34.Coopersmith CM, Stromberg PE, Davis CG, Dunne WM, Amiot DM, Karl IE, Hotchkiss RS, Buchman TG. Sepsis from Pseudomonas aeruginosa pneumonia decreases intestinal proliferation and induces gut epithelial cell cycle arrest. Crit Care Med. 2003;31:1630–1637. doi: 10.1097/01.CCM.0000055385.29232.11. [DOI] [PubMed] [Google Scholar]
- 35.Trentzsch H, Stewart D, Paidas CN, De Maio A. The combination of polymicrobial sepsis and endotoxin results in an inflammatory process that could not be predicted from the independent insults. J Surg Res. 2003;111:203–208. doi: 10.1016/s0022-4804(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 36.Stewart D, Fulton WB, Wilson C, Monitto CL, Paidas CN, Reeves RH, De Maio A. Genetic contribution to the septic response in a mouse model. Shock. 2002;18:342–347. doi: 10.1097/00024382-200210000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Schulman AM, Claridge JA, Ghezel-Ayagh A, Johnson O, III, Young JS. Differential local and systemic tumor necrosis factor-alpha responses to a second hit of lipopolysaccharide after hemorrhagic shock. J Trauma. 2003;55:298–307. doi: 10.1097/01.TA.0000028970.50515.A0. [DOI] [PubMed] [Google Scholar]
- 38.van Griensven M, Kuzu M, Breddin M, Bottcher F, Krettek C, Pape HC, Tschernig T. Polymicrobial sepsis induces organ changes due to granulocyte adhesion in a murine two hit model of trauma. Exp Toxicol Pathol. 2002;54:203–209. doi: 10.1078/0940-2993-00247. [DOI] [PubMed] [Google Scholar]
- 39.Eissner B, Matz K, Smorodchenko A, Roschmann A, Specht BU. Chronic porcine two-hit model with hemorrhagic shock and Pseudomonas aeruginosa sepsis. Eur Surg Res. 2002;34:61–67. doi: 10.1159/000048889. [DOI] [PubMed] [Google Scholar]
- 40.Claridge JA, Weed AC, Enelow R, Young JS. Laparotomy potentiates cytokine release and impairs pulmonary function after hemorrhage and resuscitation in mice. J Trauma. 2001;50:244–252. doi: 10.1097/00005373-200102000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Zhao M, Laissue JA, Zimmermann A. Hepatocyte apoptosis in hepatic iron overload diseases. Histol Histopathol. 1997;12:367–374. [PubMed] [Google Scholar]