

Abstract
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is a potent developmental toxicant in most vertebrates. However, frogs are relatively insensitive to TCDD toxicity, especially during early life stages. Toxicity of TCDD and related halogenated aromatic hydrocarbons is mediated by the aryl hydrocarbon receptor (AHR), and specific differences in properties of the AHR signaling pathway can underlie differences in TCDD toxicity in different species. This study investigated the role of AHR in frog TCDD insensitivity, using Xenopus laevis as a model system. X. laevis, a pseudotetraploid species, expresses two distinct AHR1 genes, AHR1α and AHR1β. Sharing 86% amino acid identity, these likely represent distinct genes, both orthologous to mammalian AHR and paralogous to the AHR2 gene(s) in most fish. Both AHR1α and AHR1β exhibit TCDD-dependent binding of cognate DNA sequences, but they bind TCDD with at least 20-fold lower affinity than the mouse AHRb-1 protein, and they are similarly less responsive in TCDD-induced reporter gene induction in conjunction with the mouse CYP1A1 promoter. Furthermore, CYP1A6 and CYP1A7 induction by TCDD in cultured X. laevis A6 cells appears much less responsive than CYP1A induction in cell lines derived from more sensitive animals. Taken together, these data suggest that low affinity binding by X. laevis AHRs plays an important mechanistic role in the insensitivity of frogs to TCDD. An understanding of these molecular mechanisms should aid amphibian ecotoxicology and refine the use of frog embryos as a model [e.g. in FETAX (Frog Embryo Teratogenesis Assay-Xenopus)] for determining developmental toxicity of samples containing dioxin-like compounds.
INTRODUCTION
Halogenated aromatic hydrocarbons (HAH), including polychlorinated dibenzo-p-dioxins, dibenzofurans and biphenyls, are widespread environmental contaminants. The ubiquitous distribution of these compounds makes exposure of many species commonplace, and their lipophilic nature causes accumulation in animal and human tissues (Jensen 1987; Rappe et al. 1991). Many HAH compounds cause developmental toxicity (reviewed in Peterson et al. 1993). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is typically the most potent HAH for most species and experimental endpoints (reviewed in van den Berg et al. 1998). In rodents, prenatal TCDD exposure is associated with numerous teratogenic effects, including cleft palate, hydronephrosis, subcutaneous edema, hemorrhage, and mortality (reviewed in Birnbaum 1991; Couture et al. 1990; Peterson et al. 1993). Recent studies in fish (Belair et al. 2001; Elonen et al. 1998; Henry et al. 1997; Johnson et al. 1998; Teraoka et al. 2002; Zabel et al. 1995) and birds (Ivnitski et al.2001; Walker and Catron 2000) highlight the importance of cardiovascular toxicity, manifested as pericardial edema, cardiac malformations, reduced cardiac function, and inhibited definitive erythropoiesis.
Compared to other vertebrate groups, HAH effects in amphibians are poorly characterized. However, several studies suggest that ranid frogs exhibit substantial insensitivity to TCDD toxicity during both early development (Beatty et al. 1976; Jung and Walker 1997) and adult life stages (Beatty et al. 1976). Jung and Walker (Jung and Walker 1997) estimated that embryos and tadpoles of green frogs (Rana clamitans), leopard frogs (Rana pipiens) and American toads (Bufo americanus) are 100 to 1000-fold less sensitive to TCDD-induced lethality than most fish species. Like ranid frogs, embryos of Xenopus laevis (African clawed frog; family Pipidae) suffer little mortality following acute exposure to TCDD (Dell'Orto et al. 1998; Jung and Walker 1997) or polychlorinated biphenyl (PCB) mixtures (Gutleb et al. 2000; Gutleb et al. 1999). Some studies report measurable, HAH-induced changes in sublethal endpoints, including increased edema (Sakamoto et al. 1995), anemia, and erythrocyte apoptosis (Sakamoto et al. 1997), as well as delayed increases in mortality (Gutleb et al. 1999), reduced rate of metamorphosis, and increased incidence of tail deformities (Fisher et al. 2003). However, the most frequent and severe effects resulted only from long-term, high level exposures, beginning at least two weeks after fertilization, consistent with reduced overall sensitivity to HAH exposure relative to fishes.
Most (if not all) biological effects of dioxin-like HAH compounds are mediated by the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor from the basic-helix-loop-helix/PAS family of proteins (Gu et al. 2000). Following ligand binding in the cytosol, the AHR protein translocates to the nucleus, dissociates from a complex of chaperone proteins, and forms a heterodimer with the ARNT (aryl hydrocarbon receptor nuclear translocator) protein (Hoffman et al. 1991). This transcriptionally active complex binds cis-acting DNA elements (xenobiotic response elements; XREs) and alters the expression of target genes (reviewed in Hankinson 1995; Schmidt and Bradfield 1996). The AHR complex may also cause changes in gene expression patterns through complex interactions with other signaling pathways (reviewed in Carlson and Perdew 2002; Puga et al. 2002). AHR-mediated changes in gene expression are thought to play a mechanistic role in HAH toxicity. cDNA microarray studies have documented changes in the expression of hundreds of genes in cultured human hepatoma cells exposed to TCDD; these include mRNAs regulated both directly and indirectly by AHR signaling (Frueh et al. 2001; Puga et al. 2000b). The best-characterized AHR-regulated gene is cytochrome P4501A1 (CYP1A1), which is strongly induced (reviewed in Hankinson 1995; Schmidt and Bradfield 1996). Numerous studies in animals (including mammals, birds, and fish) and cell lines emphasize that properties of the AHR signaling pathway—specifically the expression or functional properties of the AHR itself—often underlie the wide variations in HAH sensitivity observed in different animal groups (reviewed in Hahn 1998).
We are using X. laevis as a model system for probing the mechanistic role of AHR function in the HAH insensitivity of developing frogs. X. laevis is of particular interest because of its widespread use as a general model of vertebrate development. It is also used in FETAX (Frog embryo teratogenesis assay-Xenopus) and similar bioassays of the developmental toxicity of chemicals, mixtures, and environmental samples (Bantle 1996; ASTM 1998). X. laevis is known to have an active AHR signaling pathway, including two CYP1A genes (Fujita et al. 1999), two ARNT genes (Bollerot et al. 2001; Rowatt et al. 2003), and an AHR (Ohi et al. 2003). We report the identification of a second AHR paralog (AHR1α) and characterize the expression patterns and TCDD-responsiveness of both AHRs. Understanding the molecular mechanisms of TCDD insensitivity in developing frogs is important for determining the human health relevance of frog embryo toxicity assays such as FETAX. Moreover, the unique features of frog AHR signaling—including gene number and orthology, expression patterns, and function during embryogenesis and metamorphosis -- may ultimately provide a novel perspective on the relationship between the mechanisms of TCDD toxicity and the endogenous functions of AHRs during vertebrate development.
MATERIALS AND METHODS
Animals and RNA Isolation
X. laevis and X. tropicalis frogs were purchased from Xenopus Express (Plant City, FL) and Nasco (Fort Atkinson, WI). Adults were injected with human chorionic gonadotropin (Sigma) and allowed to breed in plastic chambers as described (Dawson et al. 1992). Embryos were recovered, de-jellied in 2% cysteine, sorted for viability, and maintained in FETAX solution at 24° C throughout the collection period (Materials 1998). Developing animals were collected at different developmental stages (Nieuwkoop and Faber 1994). Total RNA was isolated from using RNA STAT-60 (Tel-Test, Inc.).
Oligonucleotide Primers
Primers were synthesized by Qiagen/Operon and used as described below.
cDNA cloning and plasmid construction
Initially, partial cDNAs encoding both X. laevis and X. tropicalis AHRs were amplified from stage 46 total RNA using RT-PCR with degenerate primers Qf and AHR-B1 as described previously (Hahn et al. 1997). The 5′ and 3′ end sequences of the X. laevis cDNAs were determined by RACE PCR (Frohman et al. 1988) using the SMART RACE cDNA amplification kit and the Advantage HF-2 PCR (Clontech). For 5′ RACE of AHR1α, the gene specific primer sequence was: 5′-CAGATTGCTGGAAACCCAGGTAG-3′; for 3′ RACE: 5′-AGAAAGGGAAAGATGGGTCCACG′-3′. For 5′ RACE reactions of AHR1β, the primer sequences was 5′-AGCTAACACCTGAGTCTAAGCACG-3′; and for 3′ RACE, 5′-GCAGAGCAAGACAGATGGTAACGGC-3′. Finally, cDNAs containing the entire open reading frames of the X. laevis AHRs were amplified using Platinum Pfx DNA polymerase (Invitrogen) and cloned into pCMVTNT (Promega). A single clone corresponded to the amino acid sequence encoded by each AHR contiguous sequence, which was verified by sequencing each position in at least three individual clones.
Sequence alignment and phylogenetic analysis
Multiple alignment of the indicated amino acid sequences was performed by using CLUSTAL X (Thompson et al. 1997). Aligned amino acid sequences comprising the well conserved PAS domain were used to construct phylogenetic trees by maximum parsimony [PAUP 4.0b10 (Swofford 1998)] and the Neighbor-Joining (NJ) algorithm (Saitou and Nei 1987). Alignment positions with gaps were excluded. Bootstrap analysis (Felsenstein 1985) was performed to assess relative confidence in the topologies.
Semi-quantitative RT-PCR
Expression of AHR, ARNT, and CYP1A mRNAs was assessed in adult organs, at various developmental stages, and in A6 cells via RT-PCR, essentially as described previously (Powell et al. 2000; Rowatt et al. 2003). Total RNA was treated with DNase I (DNA-free; Ambion) to eliminate contamination by genomic DNA and reverse transcribed to cDNA using Omniscript reverse transcriptase (Qiagen) primed by random hexamers (2.5 μM). Aliquots of the reverse transcription reactions (cDNA from 225ng total RNA) were used as templates for amplification in PCR with specific primers for (0.15 μM each). The linear range of detection for the various PCR products was determined by varying the cycle number from 25 to 45 in 3-cycle increments and measuring relative band intensities on 2% agarose gels using a ChemImager 4000 low light imaging system (Alpha Innotech) with automatic background subtraction (data not shown). Cycling conditions were: 94 ° C, 15 sec; 50°, 30 sec; 68°, 1 min for 28 cycles. Primer sequences for AHR1α were 5′-CCCTTCAATCCTGGAGATACGAA-3′ and 5′-GGCTTTCTCCATTCCTTGTGCTTC-3′; for AHR1β, 5′-TCTACGGCGAGAAAAAGGAGC-3′ and 5′-GAGGCAACCACCAAGACAAATCC-3′; and for β-actin, 5′-GCACCCCTGAATCCTAAAGC-3′ and 5′-CAATGATGAAGAAGAGGCAGC-3′. Primer sequences for amplifying CYP1A6 were 5′-CAGTATGGACTAACAATG-3′ and 5′-GGTAGAGAGACAATGATC-3′; for CYP1A7, 5′-CAGTATGGACTAACAATG-3′ and 5′-CAATGATGAAGAAGAGGCAGC-3′. In experiments to detect CYP1A transcripts, only 25 cycles were employed.
In vitro protein synthesis
TnT Quick Coupled Reticulocyte Lysate Systems (Promega) were used to synthesize unlabeled or 35S-labeled proteins in 25 μl reactions following manufacturer's directions. Aliquots of the TnT reactions were subjected to SDS-PAGE, followed by fluorography [using Amplify (Amersham)] and autoradiography. Mouse AHR (high affinity, b-1 allele; Burbach et al. 1992) and human ARNT were synthesized in the same fashion using pSPORTAHR and pSPORTARNT, gifts from Dr. C. A. Bradfield (University of Wisconsin).
Cytosolic extracts
Cytosolic extracts were prepared from pools of whole embryos or tadpoles (5 to 50 animals) using the method of Hahn et al. (1993). Briefly, flash-frozen tadpoles were powdered under liquid nitrogen, dissolved in MEDMG buffer (25 mM MOPS, pH 7.5, 1 mM EDTA, 5 mM EGTA, 20 mM Na2MoO4, 0.02% NaN3, 10% glycerol 1 mM DTT) containing protease inhibitors (20 μM tosyl-L phenylalanine chloromethyl ketone), 5 μg/ml leupeptin, 100 U/ml aprotinin, 7 μg/ml pepstatin A, and 0.1 mM phenylmethylsulfonyl fluoride) and homogenized. Homogenates were centrifuged at 750g, 12,000g, and 100,000g, and the final supernatant was frozen in liquid nitrogen.
Western Blotting
25 μg of cytosolic protein or 2 μl of a TnT reaction were subjected to SDS-PAGE and blotted to nitrocellulose. Blots were probed with dilution of a monoclonal antibody SA210 (Biomol; 300 μg/ml), directed against the N-terminal half of mouse AHR (Pollenz et al. 1994).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays were performed as described previously (Karchner et al. 1999; Powell et al. 1999), using proteins synthesized in TnT reactions Prior to protein synthesis, TnT lysates were extracted with dextran-coated charcoal [DCC; 1.0 mg/ml Norit N decolorizing charcoal (Fisher); 0.1 mg/ml dextran (Sigma) in MEDG] as described previously to reduce specific, background binding to the xenobiotic response element (XRE) probe (Karchner et al. 1999; Powell et al. 1999).
Velocity sedimentation analysis
Specific TCDD binding was detected by velocity sedimentation on sucrose gradients in a vertical tube rotor using 1,6-[3H]TCDD [33.1 Ci/mmol; >99% radiopurity; Chemsyn (Lenexa, KS)] as described previously (Karchner et al. 1999). Mouseb-1 and frog AHRs were synthesized in TnT reactions, diluted 1:2 in MEDMG buffer (25 mM MOPS, pH 7.5, 1 mM EDTA, 5 mM EGTA, 20 mM Na2MoO4, 0.02% NaN3, 10% glycerol 1 mM DTT), split into two 100 μl aliquots, and incubated for 18 hr at 4° C with 4 nM [3H]TCDD. Nonspecific binding was determined by reactions containing an empty vector [unprogrammed lysate (UPL)].
Saturation Binding Analysis
The binding affinity of X. laevis AHRs was measured in DCC-based saturation binding assays modified from Poland et al. (Poland et al. 1976) and Jensen and Hahn (Jensen and Hahn 2001). Mouseb-1 and frog AHRs were synthesized in TnT reactions, diluted 1:4 in MEDG buffer (25 mM MOPS, pH 7.5, 1 mM EDTA, 5 mM EGTA, 0.02% NaN3, 10% glycerol 1 mM DTT), and incubated with graded concentrations of [3H]TCDD in DMSO for 2.5 hr at 4° C in glass test tubes. 5 μl aliquots were taken from each mixture to measure the actual concentrations of [3H]TCDD in each tube. Duplicate 30 μl aliquots were then mixed with 30 μl of DCC in polypropylene tubes. Tubes were vortexed briefly three times and incubated on ice for 5 min between each vortexing. DCC was pelleted by centrifugation for 5 min at 12,000 x g. Bound [3H]TCDD was measured in 50 μl of each supernatant. Total and bound radioactivity were measured directly with a Beckman LS6500 scintillation counter. The concentration of free [3H]TCDD was determined by subtracting the bound concentration of [3H]TCDD from the total concentration.
Non-specific binding was measured in unprogrammed TnT lysates, plotted against the concentration of free [3H]TCDD, and fit to a linear equation. This equation was used to calculate the predicted non-specific binding in reactions containing AHR proteins at each free [3H]TCDD concentration. Specific binding was determined by subtracting the calculated non-specific binding from the total binding measured in each reaction. Specific binding data were fit by nonlinear regression to the equation describing the Langmuir binding isotherm (Kenakin 1999). Curve fits and statistics were accomplished using GraphPad Prism version 4.
Transactivation Assays
TCDD-dependent transcriptional activity of each AHR was measured in luciferase reporter gene assays using COS-7 monkey kidney cells (ATCC; Manassas, VA) co-transfected with pGudLuc 6.1 (XRE-containing firefly luciferase reporter; Garrison et al. 1996; Long et al. 1998), pRL-TK (Renilla luciferase transfection control; Promega), and AHR and ARNT expression constructs, essentially as described previously (Karchner et al. 2002). Transfections were carried out 24 hr after plating 30,000 cells in triplicate wells of a 48-well plate. For each well, a total of 300 ng of DNA was complexed with 1 μl of Lipofectamine 2000 (Invitrogen). The amounts of transfected AHR and ARNT DNA were adjusted to optimize the fold-inducibility of pGudLuc6.1 over basal reporter expression. AHR1α and AHR1β cDNAs were in pCMVTNT, while mouse AHR and human ARNT were in pSPORT (gifts from Dr. C. Bradfield), all driven by the CMV promoter. Transfected DNA amounts were 50 ng of AHR, 50 ng of ARNT, 20 ng of pGudLuc 6.1, and 3 ng of pRL-TK. The total amount of transfected DNA was kept constant by addition of pCMVTNT vector with no insert. Cells were treated 5 hr after transfection with either DMSO or TCDD at 0.5% final DMSO concentration. 18 hr after dosing, cells were lysed and luminescence measured using the Dual Luciferase Assay kit (Promega) in a TD 20/20 Luminometer (Turner Designs; Sunnyvale, CA). Luminescence values were determined as a ratio of the firefly luciferase units to the Renilla luciferase units. The fractional response was then determined for each AHR at each TCDD concentration by subtracting the relative luminescence of vehicle-treated cells and determining the ratio of each value to the maximal responsiveness level in the concentration-response curve (Poland and Glover 1975).
X. laevis Cell Culture
X. laevis A6 kidney epithelial cells (ATCC; Manassas, VA; Rokaw et al. 1996) were grown under the recommended conditions (26° C; 5% CO2 atmosphere; NCTC 109 medium plus 10%; fetal bovine serum, and 200 mM L-glutamine/penicillin/streptomycin) in 25 cm2 flasks pre-treated with purified human fibronectin (BD Biosciences). At 85% confluence cells were exposed for 24 hr with graded concentrations of TCDD dissolved in DMSO. Control cultures were exposed to an equal volume of DMSO (0.5% Total). Total RNA was extracted using QIAshredder spin columns and RNeasy kits (Qiagen) prior to use in RT-PCR.
RESULTS
Two AHR1 paralogs in Xenopus laevis: AHR1α and AHR1β
To identify AHR cDNAs from X. laevis, we used RT-PCR with degenerate primers and RACE PCR (Frohman et al. 1988) as described previously for numerous vertebrate species (Hahn et al. 1997; Karchner et al. 2000; Karchner et al. 1999). These efforts resulted in the isolation of two distinct AHR cDNAs encoding proteins we call AHR1α (836 aa; 94.2 kDa) and AHR1β (834 aa; 93.6 kDa). AHR1β is identical to the X. laevis sequence reported previously by Ohi et al. (Ohi et al. 2003). Sharing 86% overall amino acid identity, both proteins contain readily identifiable sequence signatures of AHRs from other species, including the basic-helix-loop-helix domain, the PAS domains (Coumailleau et al. 1995; Fukunaga et al. 1995; Gu et al. 2000), nuclear localization (Ikuta et al. 1998) and export (Berg and Pongratz 2001; Ikuta et al. 1998) motifs, and the retinoblastoma protein binding motif (LXCXE; Elferink et al. 2001; Puga et al. 2000a; Fig. 1). Their designation as AHR1 orthologs is based on phylogenetic analyses (Fig. 2), which places both sequences in a clade containing mammalian AHRs and fish AHR1. Neither sequence is in the distinct AHR2 clade previously identified in fish.
Fig. 1. Alignment of AHR amino acid sequences.

Deduced amino acid sequences of from Xenopus laevis AHR1α and AHR1β, Xenopus tropicalis AHR1, and Fundulus heteroclitus AHR1 and AHR2 (Karchner et al. 1999) were aligned using Clustal X, version 1.8 (Thompson et al. 1997). Identities are boxed a shaded. Dashes indicate gaps in alignment. Several putative functional domains are indicated: Basic and Helix-loop-helix domains are denoted with solid lines above the sequence: PAS A and PAS B domains with hatched lines, and the ligand binding domain with brackets. (Coumailleau et al. 1995; Fukunaga et al. 1995). NLS, nuclear localization signal (Ikuta et al. 1998), NES1, nuclear export signal 1 (Ikuta et al. 1998), NES2, nuclear export signal 2 (Berg and Pongratz 2001), and LXCXE motif, a retinoblastoma protein binding motif (Elferink et al. 2001; Puga et al. 2000a) are indicated with boxes.
Fig. 2. Phylogenetic analysis of Xenopus AHR sequences.
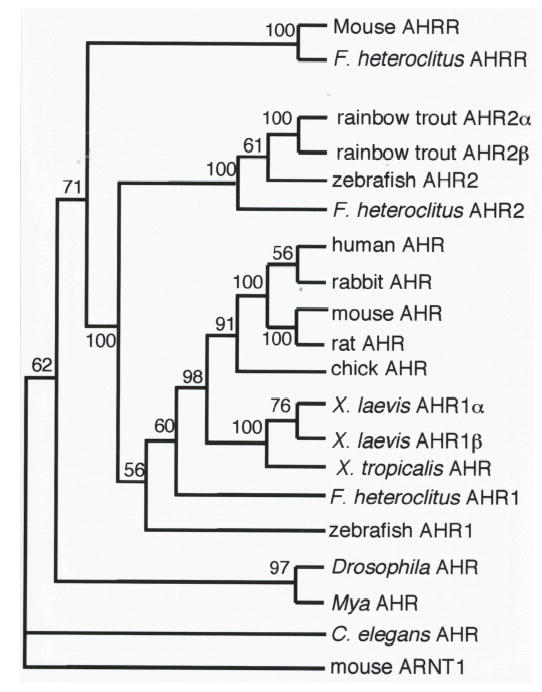
Amino acid sequences from the PAS domains of the indicated proteins were aligned, and the maximum parsimony tree was inferred with a branch-and-bound search using PAUP4.0b10 (Swofford 1998). Numbers at branch points are bootstrap values based on 100 samplings. This topology represents the bootstrap consensus tree. Mouse ARNT1 (Genbank accession number U14333) was used as the outgroup. Additional sequences include: C elegans AHR (aha-1; AF039570), Mya AHR (AAF70378), Drosophila AHR (spineless; AF0560630) Zebrafish AHR1 (AF258854), F. heteroclitus AHR1 (AF024591), chick AHR (AF260832), rat AHR (U09000), mouse AHR (M94623), rabbit AHR (D38226), human AHR (L19872), F. heteroclitus AHR2 (U29679), zebrafish AHR2 (AF063446), rainbow trout AHR2β (AF65138) and AHR2α (AF065137), F. heteroclitus AHRR (AF443441), and mouse AHRR (AB015140). The same evolutionary relationship between X. laevis AHR1α, AHR1β, and X. tropicalis AHR was apparent in a tree derived using the Neighbor-Joining approach (data not shown).
We suggest that the two sequences represent distinct, paralogous genes. The existence of closely related paralogs is consistent with the genomic history of the Xenopus genus, which includes several genome duplication events associated with speciation. One genome duplication occurred following the divergence of X. laevis (pseudotetraploid; 2n=36) and X. tropicalis (true diploid; 2n=20), approximately 30 mya (Hughes and Hughes 1993). If the AHR1 paralogs in X. laevis arose as a result of this genome duplication, then one would predict the existence of a single X. tropicalis AHR1 that is orthologous to both AHR1α and AHR1β from X. laevis. We tested this hypothesis by cloning partial AHR1 cDNAs from X. tropicalis. The sequences of ten different clones were essentially identical, while the same number of clones isolated from the same life stage of X. laevis (stage 47) readily yielded a number of sequences representing both AHR1α and AHR1β paralogs. Subsequent searches of the X. tropicalis genome revealed a single AHR gene that is identical to the cDNA reported here. This sequence, which includes nearly all of the PAS domain, was aligned with that of other AHRs and included in the phylogenetic analysis, which placed X. laevis AHR1α and AHR1β in their own clade orthologous to the X. tropicalis AHR1 (Fig. 2). This tree topology is entirely consistent with the hypothesis that the two paralogs arose in the X. laevis genome duplication (Hughes and Hughes 1993).
Expression of AHR1α and AHR1β
The relative mRNA expression of AHR1α and AHR1β was assessed by semi-quantitative RT-PCR using primers specific to each cDNA. The two AHRs exhibit similar expression patterns, both in the adult animal, where expression is widespread (Fig. 3a), and during development (Fig. 3b), when mRNAs were detectable in stage 12 and after but not at stage 8. One possible difference in expression may be in the adult brain and eye, where AHR1β mRNA appears to be more abundant than AHR1α. Notably, immunoreactive bands co-migrating with AHR1α and AHR1β proteins could be resolved and detected on western blots of cytosolic extracts derived from whole tadpoles, suggesting that both proteins are expressed in vivo (Fig. 3c).
Fig. 3. Expression patterns of X. laevis AHR1α and AHR1β.
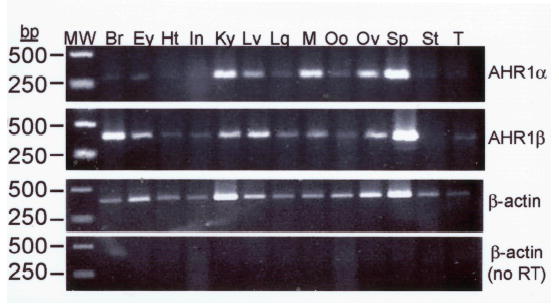
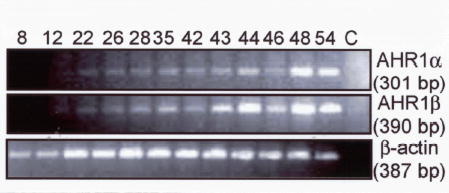

(A) mRNA in Adult organs. Expression of each transcript was assessed by RT-PCR using total RNA derived from the indicated organ from two adult frogs: Br, brain; Ey, eye; Ht, heart; In, intestines; Ky, kidney; Lv, liver; Lg, lung; M, leg muscle; Oo, immature oocyte; Ov, ovary; Sp, spleen; St, stomach; T, testis. Variability in efficiency is controlled by comparing each band density with that of β-actin from the same tissue. Positions of size markers (MW) are indicated at left (in base pairs, bp). Reactions in bottom panel omitted the addition of reverse transcriptase during cDNA synthesis reactions. (B) mRNA during different developmental stages. Specific transcripts were detected as in adult animals using total RNA from the Nieuwkoop and Faber (NF) stages (Nieuwkoop and Faber 1994) indicated at top. RNA was isolated from approximately 50 animals at each stage and pooled for RT-PCR. Reactions in lane C were performed with stage 54 RNA in the absence of reverse transcriptase. (C) AHR protein expression in different developmental stages. 25 μg of cytosolic protein isolated from the indicated NF stages and proteins synthesized in TNT reactions were subjected to SDS-PAGE and western blotting with antibody SA-210 (Biomol), directed against the N-terminal half of mouse AHR (Pollenz et al. 1994).
DNA Binding Activity of AHR1α and AHR1β
The strong sequence conservation of the DNA binding regions of both X. laevis AHRs suggests that both of these proteins may bind canonical XRE sequences. We assessed the DNA binding ability of these proteins synthesized in TnT reactions using electrophoretic mobility shift assays (Fig. 4a). Both proteins exhibited TCDD-stimulated DNA binding in conjunction with human ARNT1 (Fig. 4b). DNA binding was sequence-specific; the shifted band could be displaced by a 100-fold excess of unlabeled XRE but not by an XRE with a point mutation in the core binding sequence. Unprogrammed TnT lysate did not support specific XRE binding.
Fig. 4. TCDD stimulated DNA binding by X. laevis AHR proteins.
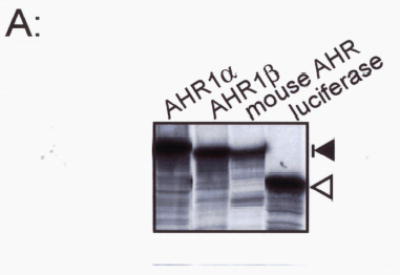
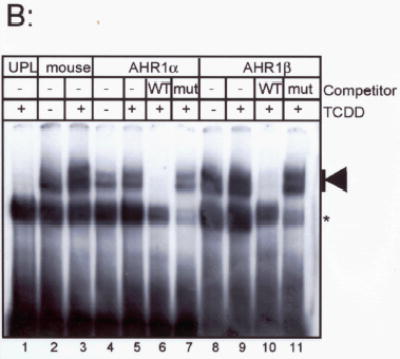
(A) Proteins were synthesized in TNT lysates. A fraction of each protein was synthesized in the presence of 35S-Methionine, subjected to SDS-PAGE, and visualized by autoradiography. Each lane contains 5 μl of TNT lysate. Mobility of AHR proteins (around 100 kDa) is indicated by the dark arrow, right. Mobility of firefly luciferase (54 kDa) is indicated by the open arrow. (B) Electrophoretic mobility shift assay. Each synthetic AHR protein was combined with an equal volume of synthetic human ARNT and incubated with DMSO (lanes 2, 4, 8) or 40 nM TCDD (lanes 1, 3, 4–7, 9–11). Lane 11 contained unprogrammed TNT lysate in lieu of AHR protein. Reactions in lanes 6, 7, 10, and 11 contained 100-fold excess unlabelled wild-type mouse DRE (WT) or mutant DREs (MUT). An arrow, right, indicates the position of TCDD-stimulated shifted bands. A nonspecifically shifted band is indicated by an asterisk.
As observed for other mammalian (Dolwick et al. 1993; Hirose et al. 1996) and non-mammalian AHRs (Abnet et al. 1999a; Karchner et al. 1999; Tanguay et al. 1999), X. laevis AHR1α and AHR1β exhibited some sequence-specific DNA binding in the absence of exogenous TCDD. The interaction could be reduced somewhat by pretreating the TnT lysates with DCC, suggesting that this DNA binding is not constitutive but rather due to a ligand intrinsic to the lysate. However, complete reduction of background binding could not be achieved, since the use of additional DCC eliminated the protein synthesis activity of the lysates. Further, it is possible that the promiscuous nature of the DNA binding in these assays relates to the use of heterologous components, including human ARNT and probe derived from the mouse CYP1A1 promoter.
Xenopus laevis AHRs exhibit low TCDD affinity and transcriptional responsiveness: Ligand Binding and Transactivation Properties of AHR1α and AHR1β
The ability of frog AHRs to bind TCDD specifically was directly demonstrated by velocity sedimentation analysis on sucrose density gradients. Both AHR1α and AHR1β exhibited detectable peaks of [3H]TCDD binding eluting from the gradient at a position similar to the mouse AHRb-1 (Fig. 5; Burbach et al. 1992). However, the degree of binding was much lower with the frog AHRs at the same concentration of TCDD, consistent with the electrophoretic mobility shift assays, in which DNA binding activity of frog AHRs exceeded background levels only at relatively high concentrations of TCDD (Fig. 4b and data not shown). Taken together with the low toxicity of TCDD in frogs, these data suggest that TCDD may bind AHR1α and AHR1β with low affinity. We tested this hypothesis by measuring the binding of affinity of each receptor for [3H] TCDD in saturation binding assays. AHR proteins were synthesized in TnT reactions (Fig. 4a), and non-specific binding was assessed using an equal volume of unprogrammed TnT lysate. X. laevisAHR1α exhibited an apparent Kd of 47.2 nM (Fig. 6a), while AHR1β was not saturable, even with free TCDD concentrations exceeding 25 nM, precluding ultimate quantification of TCDD affinity (Fig. 6b). In contrast, the mouse AHR exhibited an apparent Kd of only 2.4 nM (Fig. 6c). Even within the technical limitations presented by this in vitro approach (Bradfield et al. 1988; Ramadoss and Perdew 2004), it was clear that both X. laevis AHRs bound TCDD with substantially lower affinity than the mouse protein.
Fig. 5. Velocity sedimentation analysis of specific TCDD binding by X. laevis and mouse AHRs.
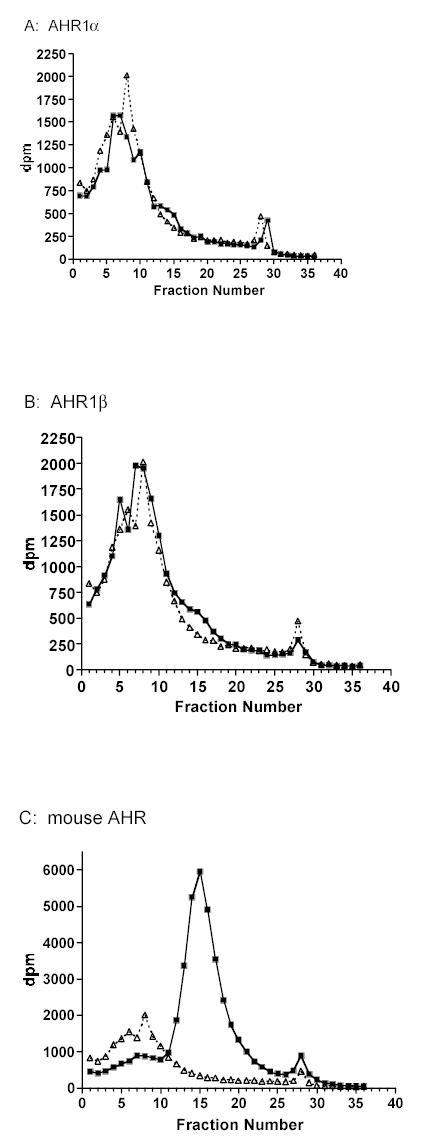
Synthetic AHR proteins (squares) or unprogrammed TnT lysates (UPL; triangles) were incubated with 4 nM 3H-TCDD and fractionated in a sucrose density gradient ligand-binding assay. Specific binding (indicated by arrows) is the difference between total binding (reactions containing an AHR) and non-specific binding (UPL). Sedimentation markers [14C]ovalbumin (3.6S) and [14C]catalase (11.3S) eluted at fractions 3–5 and 15–17, respectively.
Fig. 6. Saturation binding analysis of X. laevis and mouse AHRs.
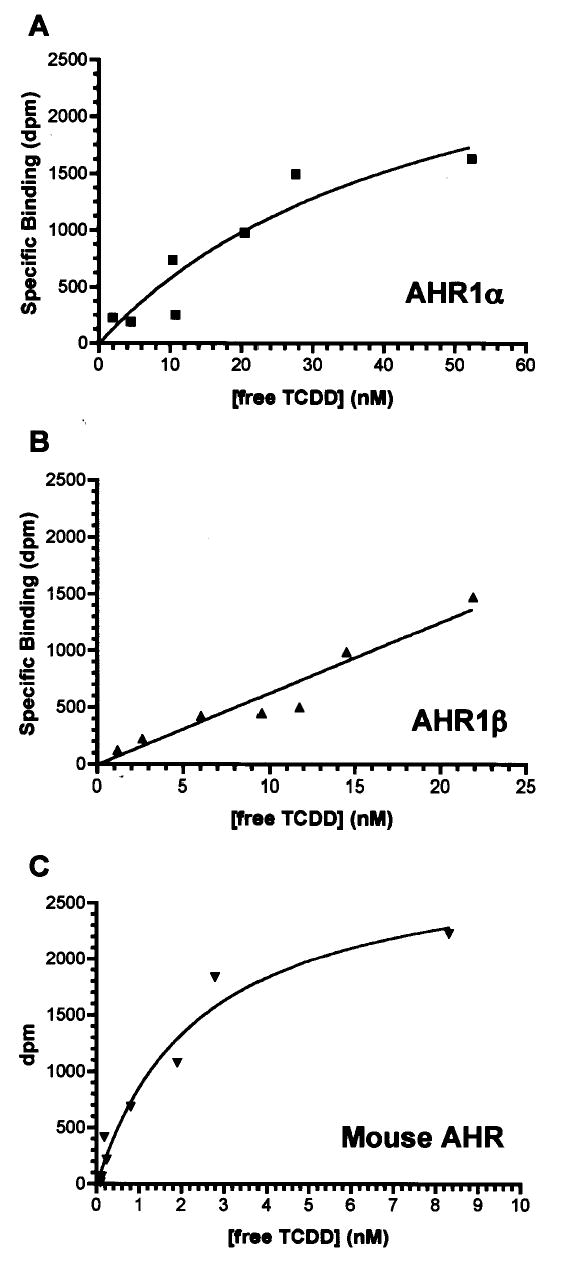
Panels show representative specific binding curves for (A) X. laevis AHR1α, (B) X. laevis AHR1β, and (C) mouse AHR, each synthesized in TnT lysates. Graded concentrations of [3H]TCDD were incubated with TnT reactions diluted in MEDMG buffer as described in Material and Methods. Unbound ligand was removed by extraction with dextran-coated charcoal and total binding quantified by liquid scintillation counting of the supernatant. Nonspecific binding was determined in parallel reactions containing unprogrammed TnT lysates. Curves were fit by nonlinear regression using GraphPad Prism version 4.0b. R2 (goodness of fit) values were 0.884 (AHR1α), 0.924 (AHR1β) and, 0.963 (mouse AHR).
Technical issues confounding the accurate estimation of equilibrium dissociation constants for AHR proteins in saturation binding assays have been well documented in previously published studies. To control for the potential effects of high lipophilic ligand concentration, high total protein concentration (Bradfield et al. 1988), and potential artifacts of cell-free protein synthesis (Ramadoss and Perdew 2004), we sought to confirm and further quantify results of the saturation binding analyses with transfection-based reporter gene assays. COS-7 cells were co-transfected with expression plasmids for a single AHR and human ARNT1 as well as pGudLuc6.1, a luciferase reporter gene containing a 480 bp portion of the mouse CYP1A1 regulatory region (Garrison et al. 1996; Long et al. 1998). The relative responsiveness of AHR1α, AHR1β, and mouse AHR was assessed using graded concentrations of TCDD (Fig. 7). Differences in transcriptional activity were readily apparent. Although the X. laevis AHRs activated greater overall luciferase activity than the mouse protein, the response mediated by mouse AHR was saturated at much lower levels (Fig. 7a). The relative potencies of TCDD are more readily visualized by plotting the fractional induction against TCDD concentration for each AHR, dissecting dose responsiveness from efficacy (Fig. 7b). Using non-linear regression analysis, we estimated the EC50 for mouse AHR at 0.13 nM, while EC50’s for X. laevis AHR1α and AHR1β were estimated at 3.3 nM and 7.4 nM, respectively. Thus, the EC50 values estimated in these experiments reflect the relationship between the Kd’s estimated by saturation binding analysis: TCDD exhibits at least 25-fold lower potency with either X. laevis AHR than with the mouse receptor.
Fig. 7. Transcriptional responsiveness of X. laevis and mouse AHRs in luciferase reporter gene assays.
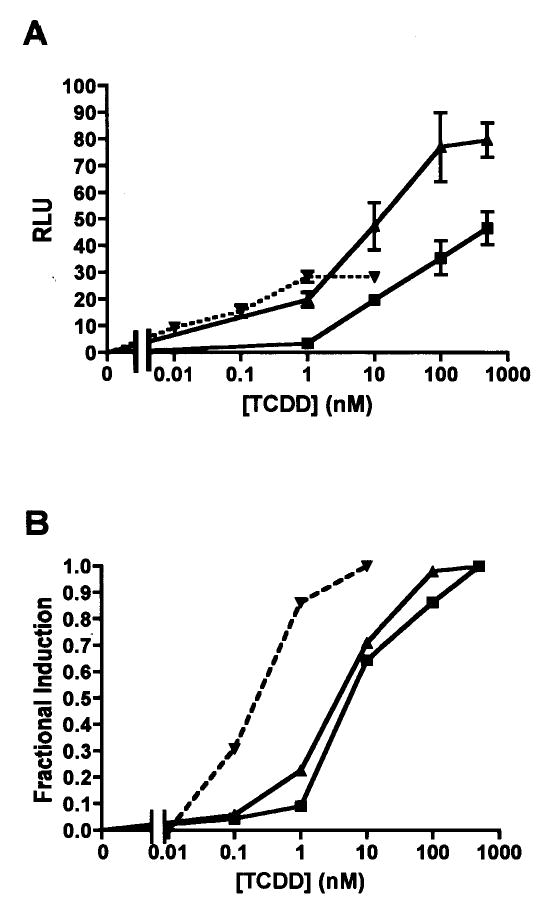
COS-7 cells were co-transfected with expression plasmids for a single AHR, human ARNT, pGudLuc6.1 (reporter construct), and pRL-TK (transfection control construct) as described in Materials and Methods. Cells were treated with DMSO or graded concentrations of TCDD, and luciferase activities measured after 18 hr. Relative luciferase units (RLU) were calculated by normalizing firefly luciferase activity to the activity of the transfection control, Renilla luciferase. Squares, X. laevis AHR1α; triangles, X. laevis AHR1β, inverted triangles with dotted line, mouse AHR. (A) Overall responsiveness. Relative luciferase expression is plotted against TCDD concentration. Each data point represents the mean of triplicate wells and error bars represent the standard error. Results are representative of three experiments. (B) Fractional induction. To more readily compare EC50s, mean RLU values from replicate experiments were pooled, and responsiveness at each TCDD concentration was normalized to the maximum response for each AHR.
TCDD responsiveness of X. laevis A6 cells
To confirm observations of X. laevis AHR function made in vitro and in a heterologous system, we sought to determine the TCDD responsiveness of endogenously expressed AHR proteins by measuring CYP1A expression, an endpoint mediated directly by AHR, in X. laevis in A6 cells (Rokaw et al. 1996). A6 cells were exposed for 24 hr to DMSO vehicle or graded concentrations of TCDD. RNA was extracted and semi-quantitative RT-PCR performed to determine which AHR and ARNT genes are expressed in these cells and to estimate the dose responsiveness of CYP1A6 and CYP1A7 (Fujita et al.1999) mRNA induction. A6 cells expressed both AHR1α and AHR1β as well as ARNT1 and ARNT2, although expression of ARNT2 mRNA was apparently much lower than the other transcripts (Fig. 8a). In TCDD-treated cells, the maximal response is not reached for either CYP1A even at a nominal concentration of 100 nM TCDD; the quantity of RT-PCR product increased substantially when cells were exposed to 500 nM (Fig. 8b). While it is possible that 500 nM TCDD induced a maximum level of CYP1A expression in these cells, there is no clear evidence for saturation of this response curve, and the maxima may yet require higher levels of TCDD exposure. Solubility concerns precluded using greater concentrations in these experiments. Nonetheless, CYP1A induction in X. laevis A6 cells appeared substantially less responsive to TCDD than other cell types, including zebrafish ZF-1 cells (EC50 = 590 pM; (Henry et al. 2001)), rainbow trout RTG-2 cells (EC50 = 35 pM; Pollenz and Necela 1998) and murine Hepa 1c1c7 cells (EC50 = 40 pM; Okey et al. 1994; Pollenz 1996). Because both AHR1α and AHR1β mRNAs were expressed in roughly comparable amounts, it is not clear which proteins actually participated in activation of CYP1A transcription in this system.
Figure 8. TCDD responsiveness in A6 cells.
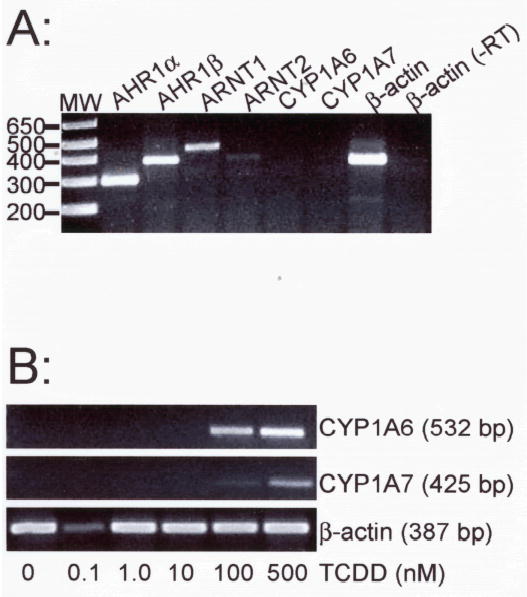
X. laevis A6 cells were grown to near confluence and treated with DMSO or graded concentrations of TCDD. CYP1A6 and CYP1A7 mRNA expression was detected by RT-PCR with total RNA. (A) Expression of mRNAs encoding components of the AHR signaling pathway. Target transcripts are indicated above each lane. Mobility of DNA size makers (MW) are indicated at left. One β-actin reaction omitted the addition of reverse transcriptase during the cDNA synthesis phase (-RT). (B) Cyp1A inducibility. Cells treated for 24 hr with graded concentrations of TCDD (indicated at bottom). Total RNA was isolated and subjected to RT-PCR with primers specific for CYP1A6 and CYP1A7. Controls lacking reverse transcriptase (not shown) were performed for each concentration and primer pair.
DISCUSSION
The overall objective of these studies was to investigate the role of AHR expression and function in frog HAH insensitivity. This phenomenon was first documented decades ago (e.g. Beatty et al. 1976) and subsequently in FETAX studies (reviewed in Bantle 1996), but mechanistic studies are less extensive. Jung and Walker (1997), using several North American ranid frogs, demonstrated the relatively short half life of TCDD in both embryos and tadpoles, suggesting that rapid elimination is a mechanistically important factor underlying the lack of TCDD-induced lethality in these species. However, they noted that the magnitude of difference in the TCDD elimination rate between the frogs and salmonid fish (~10-fold) may not adequately account for the much larger discrepancy in toxicity in these animals and suggested that differences in AHR expression or TCDD affinity might also contribute to frog insensitivity. Results from our study are consistent with a role for AHR function: In saturation binding assays using synthetic protein, in reporter gene assays using heterologously expressed protein, and in cultured cells expressing endogenous proteins, we show that X. laevis AHRs are consistently and substantially less responsive to TCDD than the mouse AHRb-1, a well characterized high affinity receptor from a sensitive strain of mice.
Variations in AHR affinity for TCDD are known to correlate with TCDD sensitivity differences in other species and strains of animals. For example, Sanderson and Bellward (1995) showed that pigeons, great blue herons, and cormorants exhibit TCDD affinities around 15 nM in hepatic cytosol, with EC50s for CYP1A induction ranging from 3–20 μg/kg egg. Chickens, which exhibit 10-fold higher affinity for TCDD (Kd=1.2 nM) showed an EC50 for CYP1A induction 10 to 100-fold lower than the less sensitive birds (0.2 μg/kg egg; Sanderson and Bellward 1995). In mice, a single point mutation (A375V) in the AHR is associated with a 4–5 fold reduction in TCDD affinity in the AHRd allele compared to the AHRb-1 allele (Ema et al. 1994; Poland et al. 1994; Ramadoss and Perdew 2004) and an 8 to 24-fold reduction in TCDD toxicity in AHRd/d animals compared with those homozygous for the b-1 allele (Birnbaum et al. 1990). Like AHRd, the lower affinity human AHR also contains valine at this position (Ema et al. 1994; Poland et al. 1994; Ramadoss and Perdew 2004) Notably, although they bind TCDD with low affinity, both X. laevis AHR1s resemble the high-affinity mouse allele with an alanine in the corresponding position. Given the great deal of divergence between the frog and mammalian proteins (~55% overall amino acid identity), the large differences in affinity and transcriptional activity seen between the frog and mouse AHRs likely result from a number of structural and functional variations in the proteins.
Saturation binding curves vs. AHR responsiveness in cells
The saturation binding assays performed in these studies (Fig. 6) likely underestimate the absolute affinity of the receptors for TCDD. In saturation binding assays with mouse and human AHRs, apparent binding affinities are known to vary inversely with overall protein concentration (Bradfield et al. 1988; Ramadoss and Perdew 2004). Detection of specific binding activity of the X. laevis AHRs required lower dilutions of the TnT lysates (and hence higher overall protein concentrations) than those used in previous studies (e.g Jensen and Hahn 2001), and the conditions for the mouse AHR binding assays were adjusted to match. Concomitantly, Kd estimates for the mouse AHRb-1 protein were 3 to 10-fold higher that those reported in previous studies of TnT proteins (Ema et al. 1994; Jensen and Hahn 2001; Poland et al. 1994), suggesting that the affinity of the frog AHRs for TCDD was also greater than measured. However, since assays were performed in parallel under identical conditions, the relative affinities determined here nonetheless provide compelling evidence for low affinity TCDD binding by X. laevis AHRs—a property that likely underlies low TCDD toxicity.
Importantly, the relative TCDD affinities of frog and mouse AHRs were reflected consistently in the activity of each protein in the luciferase reporter gene assays (Fig. 7). These transfection-based assays are perhaps a more relevant indication of the TCDD responsiveness of all three AHRs than the saturation binding curves, providing an integrated measure of ligand binding affinity and intrinsic efficacy. Recent studies comparing ligand binding by AHR in intact cells and in cell lysates suggest that receptors retain greater activity in intact cells, offering a more accurate reflection of their true ligand-binding properties (Ramadoss and Perdew 2004). Furthermore, in studies of several cell lines, the EC50 for CYP1A induction was typically well below the apparent Kd of the cytosolic receptor for TCDD (Hestermann et al. 2000; Pollenz 1996; Pollenz and Necela 1998), a phenomenon that relates to the existence of “spare receptors” in the system (Hestermann et al. 2000). We observed a similar relationship between in vitrobinding affinity and the dose response of luciferase reporter gene induction by X. laevis AHR1α and AHR1β, conceivably due to both technical and physiological factors.
Evolution and AHR gene multiplicity
The existence of multiple AHR genes in a single species is not without precedent. Many fish harbor two AHR genes, AHR1 and AHR2, that arose from an ancient gene duplication event predating the divergence of cartilaginous fish from the vertebrate lineage (Hahn et al. 1997). Phylogenetic analysis of X. laevis AHRs reveals that they arose from a much more recent gene duplication event, likely associated with a duplication of the entire genome, an common occurrence in members of the Xenopus genus . Consistent with this interpretation, phylogenetic analysis reveals that both AHR1α and AHR1β are orthologous to the AHR found in Xenopus tropicalis (Fig. 2), a true diploid species that diverged from the common Xenopus lineage prior to the genome duplication event associated with X. laevis. Numerous recently diverged paralogous genes have been documented in X. laevis (Hughes and Hughes 1993), including those encoding other nuclear receptors (Grun et al. 2002; Moore et al. 2002; Wu et al. 2003). The existence of AHR2 orthologs in X. laevis remains a possibility. However, BLAST searches of the recently re-annotated X. tropicalis genome revealed only one AHR gene, identical to the sequence we report here. This suggests that the more ancient AHR2 paralog may have been lost in the Xenopus lineage, as it apparently has in mammals (Hahn et al. 1997; Karchner et al. 1999).
X. laevis AHR1α and AHR1β are somewhat reminiscent of AHR2α and AHR2β, closely related AHR paralogs in rainbow trout (Oncorhyncus mykiss), another pseudotetraploid species (Abnet et al. 1999a), although the X. laevis AHR1 paralogs share fewer amino acid identities (86% vs. 97%). The rainbow trout AHR2 paralogs are both capable of binding TCDD, but they show distinct promoter and ligand preferences in reporter gene assays (Abnet et al. 1999b), a subtly different expression pattern, and differentially induced expression by TCDD exposure in some animal tissues (Abnet et al. 1999a), suggesting they have distinct physiological functions. Our studies reveal more similarities than differences in X. laevis AHR1α and AHR1β. They are very similar in size and sequence and share comparable expression patterns and responsiveness to TCDD. However, functional differences may well exist. The relative abilities of each protein to bind other AHR ligands, activate transcription in different promoter contexts, interact with X. laevis ARNT1 and ARNT2, and function in conjunction with each other are currently under investigation.
Significance of multiple, low affinity AHRs in X. laevis
In addition to their historical use as a model of vertebrate development, X. laevis embryos are used in FETAX, a standardized test of developmental toxicity (Bantle 1996; ASTM 1998). Substantial effort has been invested by research groups (e.g. Bantle et al. 1994; Bantle et al. 1996; Bantle et al. 1999; Bantle et al. 1990; Fort et al. 1989; Fort et al. 2001a; Fort et al. 2001b; Fort et al. 1998; Fort et al. 1995) and government panels (FETAX 2000) to validate this test and to adapt it for use in the screening of chemicals and environmental samples. With the discovery of two X. laevis AHRs with low affinity for TCDD, this study identifies an important mechanistic basis for the differences in HAH toxicity between the frog embryo model and other vertebrates, including humans, which should help evaluate and refine the use of FETAX in conjunction with HAH-containing samples. Low affinity of X. laevis AHRs for other ligands might also explain the low toxicity of polynuclear aromatic hydrocarbons in FETAX and underlie the historically reported low expression levels of Cytochromes P-450 during early frog development (Bantle 1996; Bantle et al. 1991; Fort et al. 1991; Fort et al. 2001a; Fort et al. 2001b; ASTM 1998).
Acknowledgments
We thank Dr. Mark E. Hahn, Dr. Sibel Karchner, and Diana Franks (Woods Hole Oceanographic Institution) for their generous sharing of advice, assistance, and materials in development of ligand binding assays and luciferase reporter gene assays. Blythe H. Philips (Kenyon College) provided expert technical assistance and animal husbandry support. Dr. C. A. Bradfield (University of Wisconsin) generously provided pSPORTAHR and pSPORTARNT plasmids. We thank Dr. Christopher M. Gillen (Kenyon College) for his critical reading of the manuscript. This work was supported by the Kenyon College Summer Science Scholars Program, Kenyon College Faculty Development Funds, and the National Institute of Environmental Health Sciences (R15 ES011130).
References
- Abnet CC, Tanguay RL, Hahn ME, Heideman W, Peterson RE. Two forms of aryl hydrocarbon receptor type 2 in rainbow trout (Oncorhynchus mykiss): Evidence for differential expression and enhancer specificity. J Biol Chem. 1999a;274:15159–15166. doi: 10.1074/jbc.274.21.15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abnet CC, Tanguay RL, Heideman W, Peterson RE. Transactivation activity of human, zebrafish, and rainbow trout aryl hydrocarbon receptors expressed in COS-7 cells: greater insight into species differences in toxic potency of polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners. Toxicol Appl Pharmacol. 1999b;159:41–1. doi: 10.1006/taap.1999.8719. [DOI] [PubMed] [Google Scholar]
- American Society for Testing and Materials [ASTM] (1998). Standard Guide for conducting the frog embryo teratogenesis assay--Xenopus (FETAX). In Annual Book of ASTM Standards, Vol. 11.05, pp. 826–836, Philadelphia, PA.
- Bantle, J. A. (1996). FETAX--A developmental toxicity assay using frog embryos. In Fundamentals of aquatic toxicology (G. M. Rand, ed., pp. 207–230. Taylor and Francis, Washington, DC.
- Bantle JA, Burton DT, Dawson DA, Dumont JN, Finch RA, Fort DJ, Linder G, Rayburn JR, Buchwalter D, Maurice MA, et al. Initial interlaboratory validation study of FETAX: phase I testing. J Appl Toxicol. 1994;14:213–23. doi: 10.1002/jat.2550140312. [DOI] [PubMed] [Google Scholar]
- Bantle, J. A., Dumont, J. N., Finch, R. A., and Linder, G. (1991). Atlas of abnormalities: A guide to the performance of FETAX Oklahoma State Publications Dept., Stillwater, OK.
- Bantle JA, Finch RA, Burton DT, Fort DJ, Dawson DA, Linder G, Rayburn JR, Hull M, Kumsher-King M, Gaudet-Hull AM, Turley SD. FETAX interlaboratory validation study: phase III--Part 1 testing. J Appl Toxicol. 1996;16:517–28. doi: 10.1002/(SICI)1099-1263(199611)16:6<517::AID-JAT385>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Bantle JA, Finch RA, Fort DJ, Stover EL, Hull M, Kumsher-King M, Gaudet-Hull AM. Phase III interlaboratory study of FETAX. Part 3 FETAX validation using 12 compounds with and without an exogenous metabolic activation system. J Appl Toxicol. 1999;19:447–72. doi: 10.1002/(sici)1099-1263(199911/12)19:6<447::aid-jat601>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Bantle JA, Fort DJ, Rayburn JR, DeYoung DJ, Bush SJ. Further validation of FETAX: evaluation of the developmental toxicity of five known mammalian teratogens and non-teratogens. Drug Chem Toxicol. 1990;13:267–82. doi: 10.3109/01480549009032286. [DOI] [PubMed] [Google Scholar]
- Beatty PW, Holscher MA, Neal RA. Toxicity of 2,3,7,8-tetrachloridibenzo-p-dioxin in larval and adult forms of Rana catespeiana. Bull Environ Contam Toxicol. 1976;16:578–581. doi: 10.1007/BF01685367. [DOI] [PubMed] [Google Scholar]
- Belair CD, Peterson RE, Heideman W. Disruption of erythropoiesis by dioxin in the zebrafish. Dev Dyn. 2001;222:581–94. doi: 10.1002/dvdy.1213. [DOI] [PubMed] [Google Scholar]
- Berg P, Pongratz I. Differential usage of nuclear export sequences regulates intracellular localization of the dioxin (aryl hydrocarbon) receptor. J Biol Chem. 2001;276:43231–8. doi: 10.1074/jbc.M105261200. [DOI] [PubMed] [Google Scholar]
- Birnbaum, L. S. (1991). Developmental toxicity of TCDD and related compounds: species sensitivities and differences. In Banbury Report 35: Biological Basis for Risk Assessment of Dioxins and Related Compounds (M. A. Gallo, R. J. Scheuplein and K. A. V. d. Heijden, eds.), pp. 51–67. Cold Spring Harbor Press.
- Birnbaum LS, McDonald MM, Blair PC, Clark AM, Harris MW. Differential toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57BL/6J mice congenic at the Ah locus. Fundam Appl Toxicol. 1990;15:186–200. doi: 10.1016/0272-0590(90)90175-j. [DOI] [PubMed] [Google Scholar]
- Bollerot K, Angelier N, Coumailleau P. Molecular cloning and embryonic expression of the Xenopus Arnt gene. Mechanisms of Development. 2001;108:227–231. doi: 10.1016/s0925-4773(01)00488-9. [DOI] [PubMed] [Google Scholar]
- Bradfield CA, Kende AS, Poland A. Kinetic and equilibrium studies of Ah receptor-ligand binding: use of [125I]2-iodo-7,8-dibromodibenzo-p-dioxin. Mol Pharmacol. 1988;34:229–237. [PubMed] [Google Scholar]
- Burbach KM, Poland A, Bradfield CA. Cloning of the Ah receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci USA. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson DB, Perdew GH. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol. 2002;16:317–25. doi: 10.1002/jbt.10051. [DOI] [PubMed] [Google Scholar]
- Coumailleau P, Poellinger L, Gustafsson JA, Whitelaw ML. Definition of a minimal domain of the dioxin receptor that is associated with hsp90 and maintains wild type ligand binding affinity and specificity. J Biol Chem. 1995;270:25291–25300. doi: 10.1074/jbc.270.42.25291. [DOI] [PubMed] [Google Scholar]
- Couture LA, Abbott BD, Birnbaum LS. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology. 1990;42:619–627. doi: 10.1002/tera.1420420606. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Schultz TW, Shroeder EC. Laboratory care and breeding of the African clawed frog. Lab Animal. 1992:31–36. [Google Scholar]
- Dell'Orto N, Cantelli D, Urani C. Cellular targets in response to dioxin exposure. Chemosphere. 1998;37:2809–2821. doi: 10.1016/s0045-6535(98)00323-3. [DOI] [PubMed] [Google Scholar]
- Dolwick KM, Schmidt JV, Carver LA, Swanson HI, Bradfield CA. Cloning and expression of a human Ah receptor cDNA. Mol Pharmacol. 1993;44:911–917. [PubMed] [Google Scholar]
- Elferink CJ, Ge NL, Levine A. Maximal aryl hydrocarbon receptor activity depends on an interaction with the retinoblastoma protein. Mol Pharmacol. 2001;59:664–73. doi: 10.1124/mol.59.4.664. [DOI] [PubMed] [Google Scholar]
- Elonen GE, Spehar RL, Holcombe GW, Johnson RD, Fernandez JD, Tietge JE, Cook PM. Comparative toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) to seven freshwater species during early-life-stage development. Environ Toxicol Chem. 1998;17:472–483. [Google Scholar]
- Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human aryl hydrocarbon receptors. J Biol Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- FETAX Expert Panel (2000). Minutes of the expert panel meeting on the frog embryo teratogenesis assay—Xenopus (FETAX): A proposed screening method for identifying the developmental toxicity potential of chemicals and environmental samples. The Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), Durham, NC.
- Fisher MA, Jelaso AM, Predenkiewicz A, Schuster L, Means J, Ide CF. Exposure to the polychlorinated biphenyl mixture Aroclor 1254 alters melanocyte and tail muscle morphology in developing Xenopus laevis tadpoles. EnvironmentalToxicology and Chemistry. 2003;22:321–8. [PubMed] [Google Scholar]
- Fort DJ, James BL, Bantle JA. Evaluation of the developmental toxicity of five compounds with the frog embryo teratogenesis assay: Xenopus (FETAX) and a metabolic activation system. J Appl Toxicol. 1989;9:377–88. doi: 10.1002/jat.2550090603. [DOI] [PubMed] [Google Scholar]
- Fort DJ, Rayburn JR, DeYoung DJ, Bantle JA. Assessing the efficacy of an Aroclor 1254-induced exogenous metabolic activation system for FETAX. Drug Chem Toxicol. 1991;14:143–60. doi: 10.3109/01480549109017873. [DOI] [PubMed] [Google Scholar]
- Fort DJ, Rogers RL, Paul RR, Stover EL, Finch RA. Optimization of an exogenous metabolic activation system for FETAX. II Preliminary evaluation. Drug Chem Toxicol. 2001a;24:117–27. doi: 10.1081/dct-100102605. [DOI] [PubMed] [Google Scholar]
- Fort DJ, Rogers RL, Stover EL, Finch RA. Optimization of an exogenous metabolic activation system for FETAX. I Post-isolation rat liver microsome mixtures. Drug Chem Toxicol. 2001b;24:103–15. doi: 10.1081/dct-100102604. [DOI] [PubMed] [Google Scholar]
- Fort DJ, Stover EL, Bantle JA, Rayburn JR, Hull MA, Finch RA, Burton DT, Turley SD, Dawson DA, Linder G, Buchwalter D, Dumont JN, Kumsher-King M, Gaudet-Hull AM. Phase III interlaboratory study of FETAX, Part 2: interlaboratory validation of an exogenous metabolic activation system for frog embryo teratogenesis assay--Xenopus (FETAX) Drug Chem Toxicol. 1998;21:1–14. doi: 10.3109/01480549809017846. [DOI] [PubMed] [Google Scholar]
- Fort DJ, Stover EL, Norton D. Ecological hazard assessment of aqueous soil extracts using FETAX. Frog Embryo Teratogenesis Assay--Xenopus. J Appl Toxicol. 1995;15:183–91. doi: 10.1002/jat.2550150308. [DOI] [PubMed] [Google Scholar]
- Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frueh FW, Hayashibara KC, Brown PO, Whitlock JPJ. Use of cDNA microarrays to analyze dioxin-induced changes in human liver gene expression. Toxicol Lett. 2001;122:189–203. doi: 10.1016/s0378-4274(01)00364-2. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Ohi H, Murayama N, Saguchi K, Higuchi S. Molecular cloning and sequence analysis of cDNAs coding for 3-methylcholanthrene-inducible cytochromes P450 in Xenopus laevis liver. Arch Biochem Biophys. 1999;371:24–28. doi: 10.1006/abbi.1999.1425. [DOI] [PubMed] [Google Scholar]
- Fukunaga BN, Probst MR, Reiszporszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem. 1995;270:29270–29278. doi: 10.1074/jbc.270.49.29270. [DOI] [PubMed] [Google Scholar]
- Garrison PM, Tullis K, Aarts JMMJG, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fund Appl Toxicol. 1996;30:194–203. doi: 10.1006/faat.1996.0056. [DOI] [PubMed] [Google Scholar]
- Grun F, Venkatesan RN, Tabb MM, Zhou C, Cao J, Hemmati D, Blumberg B. Benzoate X receptors alpha and beta are pharmacologically distinct and do not function as xenobiotic receptors. J Biol Chem. 2002;277:43691–7. doi: 10.1074/jbc.M206553200. [DOI] [PubMed] [Google Scholar]
- Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–61. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- Gutleb AC, Appelman J, Bronkhorst M, van dB JH, Murk AJ. Effects of oral exposure to polychlorinated biphenyls (PCBs) on the development and metamorphosis of two amphibian species (Xenopus laevis and Rana temporaria) The Science of the total environment. 2000;262:147–57. doi: 10.1016/s0048-9697(00)00598-2. [DOI] [PubMed] [Google Scholar]
- Gutleb AC, Appelman J, Bronkhorst MD, Van den Berg JHJ, Spenkelink A, Brouwer A, Murk AJ. Delayed effects of pre- and early-life time exposure to polychlorinated biphenyls on tadpoles of two amphibian species (Xenopus laevis and Rana temporaria) Envrionmental Toxicology and Pharmacology. 1999;8:1–14. doi: 10.1016/s1382-6689(99)00023-x. [DOI] [PubMed] [Google Scholar]
- Hahn ME. Mechanisms of innate and acquired resistance to dioxin-like compounds. Rev Toxicol. 1998;2:395–443. [Google Scholar]
- Hahn ME, Karchner SI, Shapiro MA, Perera SA. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc Natl Acad Sci USA. 1997;94:13743–13748. doi: 10.1073/pnas.94.25.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn ME, Lamb TM, Schultz ME, Smolowitz RM, Stegeman JJ. Cytochrome P4501A induction and inhibition by 3,3′,4,4′-tetrachlorobiphenyl in an Ah receptor-containing fish hepatoma cell line (PLHC-1) Aquat Toxicol. 1993;26:185–208. [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Henry TR, Nesbit DJ, Heideman W, Peterson RE. Relative potencies of polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners to induce cytochrome P4501A mRNA in a zebrafish liver cell line. Environ Toxicol Chem. 2001;20:1053–8. [PubMed] [Google Scholar]
- Henry TR, Spitsbergen JM, Hornung MW, Abnet CC, Peterson RE. Early life stage toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Zebrafish (Danio rerio) Toxicol Appl Pharmacol. 1997;142:56–68. doi: 10.1006/taap.1996.8024. [DOI] [PubMed] [Google Scholar]
- Hestermann EV, Stegeman JJ, Hahn ME. Relative contributions of affinity and intrinsic efficacy to aryl hydrocarbon receptor ligand potency. Toxicol Appl Pharmacol. 2000;168:160–72. doi: 10.1006/taap.2000.9026. [DOI] [PubMed] [Google Scholar]
- Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt) Mol Cell Biol. 1996;16:1706–1713. doi: 10.1128/mcb.16.4.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hughes MK, Hughes AL. Evolution of duplicate genes in a tetraploid animal, Xenopus laevis. Mol Biol Evol. 1993;10:1360–1369. doi: 10.1093/oxfordjournals.molbev.a040080. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem. 1998;273:2895–2904. doi: 10.1074/jbc.273.5.2895. [DOI] [PubMed] [Google Scholar]
- Ivnitski I, Elmaoued R, Walker MK. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary development is preceded by a decrease in myocyte proliferation and an increase in cardiac apoptosis. Teratology. 2001;64:201–12. doi: 10.1002/tera.1065. [DOI] [PubMed] [Google Scholar]
- Jensen AA. Polychlorobipneyls (PCBs), polychlorodibenzo-p-dioxins (PCDDs) and polychlorodibenzofurans (PCDFs) in human milk, blood, and adipose tissue. Sci Total Environ. 1987;64:259–293. doi: 10.1016/0048-9697(87)90250-6. [DOI] [PubMed] [Google Scholar]
- Jensen BA, Hahn ME. cDNA cloning and characterization of a high affinity aryl hydrocarbon receptor in a cetacean, the beluga, Delphinapterus leucas. Toxicol Sci. 2001;64:41–56. doi: 10.1093/toxsci/64.1.41. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Tietge JE, Jensen KM, Fernandez JD, Linnum AL, Lothenbach DB, Holcombe GW, Cook PM, Christ SA, Lattier DL, Gordon DA. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin to early life stage brook trout (Salvelinus fontinalis) following parental dietary exposure. Environ Toxicol Chem. 1998;17:2408–2421. [Google Scholar]
- Jung RE, Walker MK. Effects of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on development of anuran amphibians. Environ Toxicol Chem. 1997;16:230–240. [Google Scholar]
- Karchner SI, Franks DG, Powell WH, Hahn ME. Regulatory Interactions among Three Members of the Vertebrate Aryl Hydrocarbon Receptor Family: AHR Repressor, AHR1, and AHR2. J Biol Chem. 2002;277:6949–6959. doi: 10.1074/jbc.M110779200. [DOI] [PubMed] [Google Scholar]
- Karchner SI, Kennedy SW, Trudeau S, Hahn ME. Towards a molecular understanding of species differences in dioxin sensitivity: Initial characterization of Ah receptor cDNAs in birds and an amphibian. Mar Environ Res. 2000;20:51–56. doi: 10.1016/s0141-1136(00)00045-3. [DOI] [PubMed] [Google Scholar]
- Karchner SI, Powell WH, Hahn ME. Structural and Functional Characterization of two highly divergent aryl hydrocarbon receptors (AHR1 and AHR2) in the teleost Fundulus heteroclitus. Evidence for a novel class of ligand-binding basic helix-loop-helix Per-ARNT-Sim (bHLH-PAS) factors. J Biol Chem. 1999;274:33814–33824. doi: 10.1074/jbc.274.47.33814. [DOI] [PubMed] [Google Scholar]
- Kenakin, T. (1999). Pharmacologic Analysis of Drug-Receptor Interactions CRC/Raven Press, New York.
- Long WP, Pray-Grant M, Tsai JC, Perdew GH. Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol Pharmacol. 1998;53:691–700. doi: 10.1124/mol.53.4.691. [DOI] [PubMed] [Google Scholar]
- Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA, Lambert MH, Moore JT. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol. 2002;16:977–86. doi: 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop, P. D., and Faber, J. (1994). Normal Table of Xenopus Laevis (Daudin) Garland Publishing, Inc., New York and London.
- Ohi H, Fujita Y, Miyao M, Saguchi K, Murayama N, Higuchi S. Molecular cloning and expression analysis of the aryl hydrocarbon receptor of Xenopus laevis. Biochem Biophys Res Commun. 2003;307:595–9. doi: 10.1016/s0006-291x(03)01244-0. [DOI] [PubMed] [Google Scholar]
- Okey AB, Riddick DS, Harper PA. The Ah Receptor: Mediator of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-Dioxin (TCDD) and related compounds. Toxicol Lett. 1994;70:1–22. doi: 10.1016/0378-4274(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Peterson RE, Theobald HM, Kimmel GL. Developmental and reproductive toxicity of dioxins and related compounds - cross-species comparisons. CRC Crit Rev Toxicol. 1993;23:283–335. doi: 10.3109/10408449309105013. [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E. Genetic expression of aryl hydrocarbon hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: evidence for a receptor mutation in genetically non-responsive mice. Mol Pharmacol. 1975;11:389–398. [Google Scholar]
- Poland A, Glover E, Kende AS. Stereospecific, high-affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. J Biol Chem. 1976;251:4936–4946. [PubMed] [Google Scholar]
- Poland A, Palen D, Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- Pollenz RS. The aryl hydrocarbon receptor, but not the aryl hydrocarbon receptor nuclear translocator protein, is rapidly depleted in hepatic and nonhepatic culture cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1996;49:391–398. [PubMed] [Google Scholar]
- Pollenz RS, Necela B. Characterization of two continuous cell lines derived from Oncorhynchus mykiss for models of Ah-receptor mediated signal transduction. Direct comparison to the mammalian Hepa-1c1c7 cell line. Aquat Toxicol. 1998;41:31–49. [Google Scholar]
- Pollenz RS, Sattler CA, Poland A. The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence. Mol Pharmacol. 1994;45:428–438. [PubMed] [Google Scholar]
- Powell WH, Bright R, Bello SM, Hahn ME. Developmental and tissue-specific expression of AHR1, AHR2, and ARNT2 in dioxin-sensitive and -resistant populations of the marine fish, Fundulus heteroclitus. Toxicol Sci. 2000;57:229–239. doi: 10.1093/toxsci/57.2.229. [DOI] [PubMed] [Google Scholar]
- Powell WH, Karchner SI, Bright R, Hahn ME. Functional diversity of vertebrate ARNT proteins: Identification of ARNT2 as the predominant form of ARNT in the marine teleost, Fundulus heteroclitus. Arch Biochem Biophys. 1999;361:156–163. doi: 10.1006/abbi.1998.0992. [DOI] [PubMed] [Google Scholar]
- Puga A, Barnes SJ, Dalton TP, Chang C, Knudsen ES, Maier MA. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem. 2000a;275:2943–50. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- Puga A, Maier A, Medvedovic M. The transcriptional signature of dioxin in human hepatoma HepG2 cells. Biochem Pharmacol. 2000b;60:1129–42. doi: 10.1016/s0006-2952(00)00403-2. [DOI] [PubMed] [Google Scholar]
- Puga A, Xia Y, Elferink C. Role of the aryl hydrocarbon receptor in cell cycle regulation. Chemico-biological interactions. 2002;141:117–30. doi: 10.1016/s0009-2797(02)00069-8. [DOI] [PubMed] [Google Scholar]
- Ramadoss P, Perdew GH. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol Pharmacol. 2004;66:129–36. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- Rappe C, Bergqvist PA, Kjeller LO, Swanson S, Belton T, Ruppel B, Lockwood K, Kahn PC. Levels and patterns of PCDD and PCDF contamination in fish, crabs, and lobsters from Newark bay and the New York Bight. Chemosphere. 1991;22:239–266. [Google Scholar]
- Rokaw MD, West M, Johnson JP. Rapamycin inhibits protein kinase C activity and stimulates Na+ transport in A6 cells. J Biol Chem. 1996;271:32468–73. doi: 10.1074/jbc.271.50.32468. [DOI] [PubMed] [Google Scholar]
- Rowatt AJ, DePowell JJ, Powell WH. ARNT gene multiplicity in amphibians: Characterization of ARNT2 from the frog Xenopus laevis. J Exper Zool. 2003;300B:48–57. doi: 10.1002/jez.b.45. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sakamoto MK, Mima S, Takahashi KP, Tanimura T. Apoptotic cell death of erythrocytes in Xenopus larvae exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Pathol. 1997;25:398–402. doi: 10.1177/019262339702500409. [DOI] [PubMed] [Google Scholar]
- Sakamoto MK, Mima S, Tanimura T. A morphological study of liver lesions in Xenopus larvae exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with special reference to apoptosis of hepatocytes. J Environ Pathol Toxicol Oncol. 1995;14:69–82. [PubMed] [Google Scholar]
- Sanderson JT, Bellward GD. Hepatic microsomal ethoxyresorufin O-deethylase-inducing potency in ovo and cytosolic Ah receptor binding affinity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: Comparison of four avian species. Toxicol Appl Pharmacol. 1995;132:131–145. doi: 10.1006/taap.1995.1094. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Devel Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L. (1998). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, MA
- Tanguay RL, Abnet CC, Heideman W, Peterson RE. Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor. Biochim Biophys Acta (Gene Struct Express) 1999;1444:35–48. doi: 10.1016/s0167-4781(98)00252-8. [DOI] [PubMed] [Google Scholar]
- Teraoka H, Dong W, Ogawa S, Tsukiyama S, Okuhara Y, Niiyama M, Ueno N, Peterson RE, Hiraga T. 2,3,7,8-Tetrachlorodibenzo-p-dioxin toxicity in the zebrafish embryo: altered regional blood flow and impaired lower jaw development. Toxicol Sci. 2002;65:192–9. doi: 10.1093/toxsci/65.2.192. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg M, Birnbaum L, Bosveld BTC, Brunström B, Cook P, Feeley M, Giesy JP, Hanberg A, Hasegawa R, Kennedy SW, Kubiak T, Larsen JC, Leeuwen FXRv, Liem AKD, Nolt C, Peterson RE, Poellinger L, Safe S, Schrenk D, Tillitt D, Tysklind M, Younes M, Waern F, Zacharewski T. Toxic equivalency factors (TEFs) for PCBs, PCDDs, and PCDFs for humans and wildlife. Environ Health Persp. 1998;106:775–792. doi: 10.1289/ehp.98106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MK, Catron TF. Characterization of cardiotoxicity induced by 2,3,7, 8-tetrachlorodibenzo-p-dioxin and related chemicals during early chick embryo development. Toxicol Appl Pharmacol. 2000;167:210–21. doi: 10.1006/taap.2000.8992. [DOI] [PubMed] [Google Scholar]
- Wu KH, Tobias ML, Thornton JW, Kelley DB. Estrogen receptors in Xenopus: duplicate genes, splice variants, and tissue-specific expression. Gen Comp Endocrinol. 2003;133:38–49. doi: 10.1016/s0016-6480(03)00148-5. [DOI] [PubMed] [Google Scholar]
- Zabel EW, Cook PM, Peterson RE. Toxic equivalency factors of polychlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyl congeners based on early life stage mortality in rainbow trout (Oncorhynchus mykiss) Aquat Toxicol. 1995;31:315–328. [Google Scholar]