

Abstract
The transporter associated with antigen processing (TAP) is an ABC transporter formed of two subunits, TAP1 and TAP2, each of which has an N-terminal membrane-spanning domain and a C-terminal ABC ATPase domain. We report the structure of the C-terminal ABC ATPase domain of TAP1 (cTAP1) bound to ADP. cTAP1 forms an L-shaped molecule with two domains, a RecA-like domain and a small α-helical domain. The diphosphate group of ADP interacts with the P-loop as expected. Residues thought to be involved in γ-phosphate binding and hydrolysis show flexibility in the ADP-bound state as evidenced by their high B-factors. Comparisons of cTAP1 with other ABC ATPases from the ABC transporter family as well as ABC ATPases involved in DNA maintenance and repair reveal key regions and residues specific to each family. Three ATPase subfamilies are identified which have distinct adenosine recognition motifs, as well as distinct subdomains that may be specific to the different functions of each subfamily. Differences between TAP1 and TAP2 in the nucleotide-binding site may be related to the observed asymmetry during peptide transport.
Keywords: ABC transporter/antigen presentation/ATPase/TAP
Introduction
ATP-binding cassette (ABC) transporters are ubiquitous membrane proteins that transport a range of molecules, from small sugars to large polypeptides, across membranes (Higgins, 1992). They share a common domain organization with two membrane-spanning domains and two cytoplasmic ABC ATPase domains. The energy of ATP binding and hydrolysis is used to select and transport the substrates. A number of bacterial ABC transporters have been extensively studied, especially the histidine transporter (HisJQMP2) and the maltose transporter (MalEFGK2), which use a periplasmic substrate-binding protein (HisJ or MalE) to import their substrate (Nikaido and Hall, 1998). Mammalian ABC transporters, most of which are substrate exporters, include the multiple drug resistance protein P-glycoprotein (Pgp), the cystic fibrosis transmembrane conductance regulator (CFTR) and the transporter associated with antigen processing (TAP). TAP is an ABC transporter formed by heterodimerization of TAP1 and TAP2, each of which has an N-terminal membrane-spanning domain and a C-terminal ABC ATPase domain (Kelly et al., 1992). TAP is an endoplasmic reticulum (ER) resident protein, which transports cytosolic peptides generated by the proteasome to the ER lumen for loading onto MHC class I molecules (see Abele and Tampé, 1999; Karttunen et al., 1999; Lankat-Buttgereit and Tampé, 1999 for recent reviews). TAP is part of a peptide-loading complex composed of MHC class I molecules and their specific chaperone, tapasin, as well as the more general chaperones calnexin, calreticulin and the thiol oxidoreductase ERp57 (Lehner and Trowsdale, 1998; Cresswell et al., 1999).
A large number of ATP- and GTP-binding and hydrolyzing proteins, including helicases, myosins, DNA repair proteins and ABC transporters, contain two conserved sequences (Walker et al., 1982): the Walker A motif, also known as the P-loop, has a consensus sequence of GxxGxGKST, where x represents any amino acid, and the Walker B motif has four aliphatic residues followed by two negatively charged residues, generally aspartate followed by glutamate. Structures of nucleotide-binding proteins show that the Walker A motif forms a loop that binds to the α- and β-phosphates of di- and tri-nucleotides. The function of the Walker B motif, which forms a β-strand, in ABC ATPases is less clear. It may help coordinate the Mg2+ ion, possibly through a water molecule (Hopfner et al., 2000) or it may polarize the attacking water molecule (Hung et al., 1998). ABC ATPases are a subset of nucleotide hydrolases (see Holland and Blight, 1999; Jones and George, 1999 for a review), which share three other motifs: (i) the signature, or C, motif (consensus LSGGQ) that is well conserved but for which a function has not been unambiguously determined, although it may be a γ-phosphate sensor in the opposing molecule of the dimer (Hopfner et al., 2000) and/or signal to the membrane-spanning domains (Holland and Blight, 1999); (ii) the Q-loop (also referred to as the ‘lid’), which contains a glutamine that interacts with the γ-phosphate through a water, which may be the attacking nucleophile (Hung et al., 1998; Hopfner et al., 2000); and (iii) the switch region (Schneider and Hunke, 1998) that contains a conserved histidine, which has also been postulated to polarize the attacking water molecule for hydrolysis (Hopfner et al., 2000).
Using X-ray crystallography, we have determined the structure of the monomeric state of the C-terminal ABC ATPase domain of TAP1 (cTAP1) bound to ADP. This represents the first structure of a mammalian ABC transporter ATPase as well as the first structure of an ABC transporter ATPase in the ADP state. The differences in sequence between TAP1 and TAP2 within the cTAP1 nucleotide-binding site may be the basis for part of the asymmetry in their ATPase activities during peptide transport. Comparisons with the structures and sequences of other ABC ATPases from the ABC transporter proteins as well as the RAD50 and structural maintenance of chromatin ABC ATPases, and the MutS and MutS homolog (MSH) proteins allow the identification of key regions and residues that are specific to each family.
Results and discussion
Structure determination
The C-terminal domain of human TAP1 (cTAP1, residues 489–748) was expressed in soluble form in Escherichia coli, purified by nickel-chelating chromatography through its N-terminal His6 tag and crystallized in the presence of ATP and Mg2+. The structure was solved by molecular replacement using HisP (Hung et al., 1998) as a search model (see Table I and Materials and methods). The final model, with an R-factor of 19.8% (Rfree = 24.0%), includes residues 492–742 of TAP1 (489–491 and 743–748 were not visible), ADP–Mg2+ and 114 water molecules. The structure corresponds to the monomeric state of cTAP1. Although some pairs of molecules are formed by crystal lattice contacts in the unit cell of the P62 crystal, these interfaces are small (the largest being ∼650 Å2), bear no resemblance to previously observed ABC ATPase dimerization interfaces (Hung et al., 1998; Diederichs et al., 2000; Hopfner et al., 2000; Lamers et al., 2000; Obmolova et al., 2000), and are, therefore, unlikely to resemble the physiological interface between TAP1 and TAP2 ATPase domains.
Table I. Data collection and refinement statistics.
| Data processing | |
| resolution(Å) | 30.0–2.40 (2.49–2.40) |
| No. of reflections | 13 721 (1364) |
| redundancy | 3.7 (3.7) |
| I/σI | 15.9 (3.5) |
| completeness (%) | 99.7 (100.0) |
| Rsym (%)a | 5.3 (40.1) |
| Refinement | |
| data range (Å) | 30.0–2.40 (2.55–2.40) |
| reflections in working set | 13 235 (1859) |
| reflections in Rfree set | 1358 (219) |
| Rcrys (%)b | 19.8 (24.9) |
| Rfree (%)c | 24.0 (29.3) |
| No. of protein atoms | 1925 |
| No. of ADP atoms | 27 |
| No. of Mg2+ atoms | 1 |
| No. of solvent atoms | 114 |
| r.m.s. deviation bond length (Å) | 0.007 |
| r.m.s. deviation bond angles (°) | 1.3 |
| average B-factor | 52.1 |
aRsym = ∑h′<|Ih – Ih′|>/∑h′Ih′ where <|Ih – Ih′|> is the average of the absolute deviation of a reflection Ih′ from the average Ih of its symmetry and Friedel equivalents.
bRcrys = ∑||Fo| – |Fc||/∑|Fo| where Fc is the calculated structure factor.
cRfree is as for Rcrys but calculated for 2% of randomly chosen reflections that were omitted from the refinement.
cTAP1 overall structure
The C-terminal domain of TAP1 forms an L-shaped molecule, with two domains (Figure 1A and B). The larger domain is structurally related to RecA and other nucleotide-binding proteins. This RecA-like domain contains two β-sheets and six α-helices, which include the Walker A, Walker B, Q-loop, D-loop and switch regions, and forms the nucleotide-binding site. The second, helical domain is smaller, formed by four α-helices, and contains the signature motif. The overall structure of cTAP1 is most similar to that of HisP, the ATPase subunit of the bacterial histidine transporter, with an r.m.s. deviation of 2.39 Å over 223 Cα atoms (Hung et al., 1998), and MalK, the ATPase subunit of the bacterial maltose transporter (r.m.s. deviation of 3.32 Å over 223 Cα atoms; Diederichs et al., 2000). HisP and Malk are the two ABC transporter ATPases for which coordinates are available. Similarly to HisP, cTAP1 has a helix α2 linking strands β5 and β8. However, cTAP1 is missing strands β6 and β7, which precede helix α2 in HisP. In MalK, helix α2 is replaced by a loop.
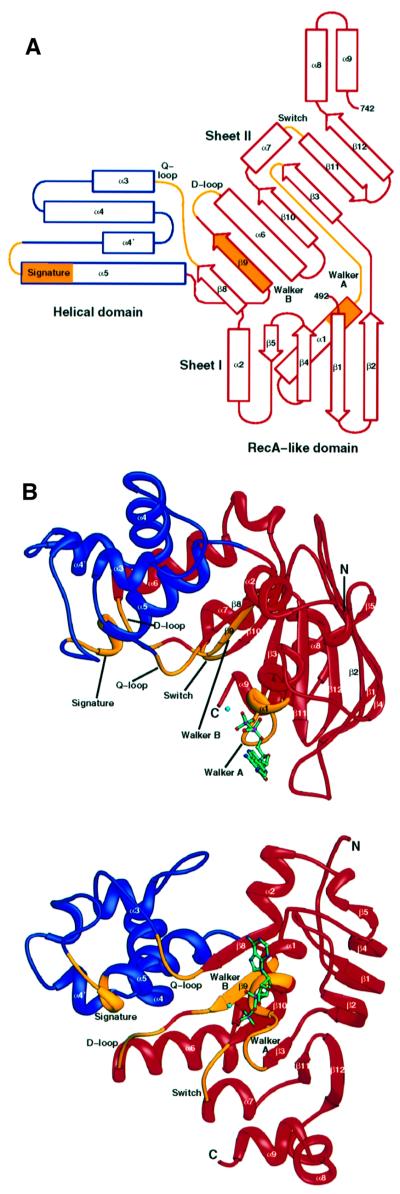
Fig. 1. Structure of cTAP1. (A) Topology diagram of cTAP1 color-coded as in (B). (B) Ribbon diagrams of cTAP1, highlighting the helical domain in blue, the RecA-like domain in red and the conserved sequence motifs in yellow. The top shows a ‘side’ view of cTAP1, whereas the bottom shows cTAP1 viewed from the bottom (90° rotation from the top view). The β-strands and α-helices are labeled according to the HisP topology. The ADP (green) and Mg2+ ion (cyan) are shown as ball-and-stick models.
Two of the conserved sequence motifs, the Q-loop as well as the loop preceding helix α5, which contains the signature motif, have high B-factors in this ADP-bound cTAP1 structure, suggesting that they are less well ordered than most of the structure. The Q-loop interacts with the γ-phosphate of ATP in the RAD50 structures with ATP or non-hydrolyzable analog ATPγS bound (Hopfner et al., 2000). Similarly, in the RAD50 dimer formed in the presence of ATP, the signature motif from the opposing monomer forms part of the dimerization interface and interacts with the γ-phosphate. Therefore, the cTAP1 Q-loop and signature motifs are likely to be stabilized and possibly altered in location by the presence of a γ-phosphate in the ATP-bound state, and by dimerization of the TAP1 and TAP2 ATPase domains.
Domain structure
As first seen in the HisP structure (Hung et al., 1998) and previously predicted (Hyde et al., 1990; Mimura et al., 1991), the cTAP1 ABC transporter ATPase consists of two domains, a RecA-like domain (Arm I in HisP) and a smaller helical domain (Arm II in HisP). The RecA structure superimposes onto the RecA-like domain, and shares most of its secondary structure, except for strand β5 and helices α2, α8 and α9. RecA also has an α-helix that superimposes onto helix α5 of the cTAP1 helical domain. The helical domain in cTAP1, HisP and MalK, as defined by the DOMID program (http://bioinfo1.mbfys.lu.se/Domid) and confirmed by the observed rigid body motion in MalK (Figure 2A), includes part of the Q-loop as well as the signature motif (Figure 1B). The two MalK molecules that form a dimer in the crystallographic asymmetric unit have different conformations related by a hinge motion (Diederichs et al., 2000), with the helical domain rotating ∼25° relative to the RecA-like domain (Figure 2A). Superposition of each monomer of the MalK dimer onto cTAP1 reveals that the cTAP1 structure is much more similar to the MalK B monomer (Figure 2C) than to the MalK A monomer (Figure 2B).
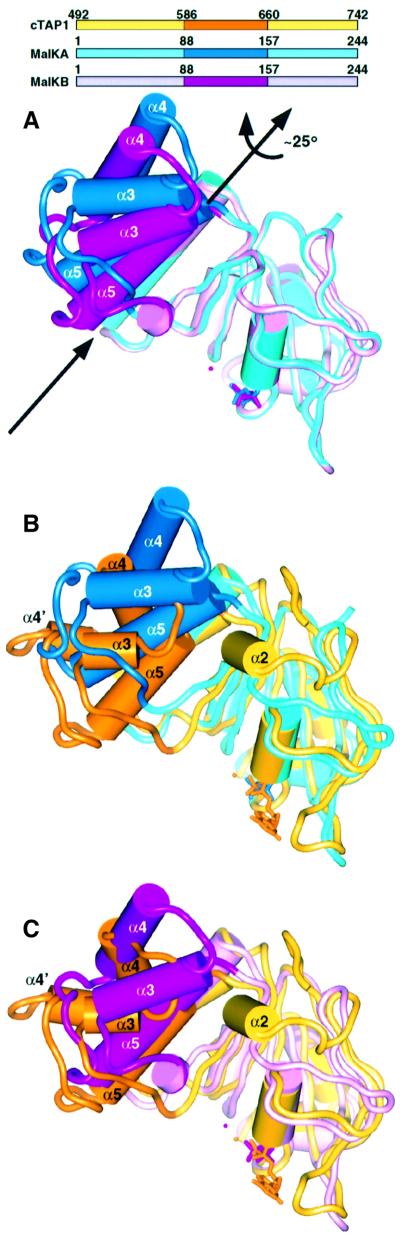
Fig. 2. Domain movements. (A) The two MalK monomers forming a dimer in the asymmetric unit (Diederichs et al., 2000) are superimposed using their RecA-like domain to show domain motion of the helical domain. cTAP1 is similarly superimposed onto the MalK A monomer (B) and the MalK B monomer (C) revealing that cTAP1 more closely resembles the MalK B monomer. The MalK A monomer is cyan (RecA-like domain, 1–88 and 158–244) and blue (helical domain, 89–157), the MalK B monomer is light pink (RecA-like domain, 1–88 and 158–244) and magenta (helical domain, 89–157), and cTAP1 is yellow (RecA-like domain, 492–586 and 661–742) and orange (helical domain, 587–660). The ions and nucleotides (ADP–Mg2+ for cTAP1, pyrophosphate for MalK A, and pyrophosphate–Mg2+ for MalK B) are represented in ball-and-stick in the same color as the corresponding helical domain.
The helical domain is characteristic of ABC transporters (see below and Schneider and Hunke, 1998; Holland and Blight, 1999) and, therefore, may be important in signaling between the membrane-spanning domains and ATPase domains of the ABC transporter. Hung et al. (1998) speculated that a motion of Arm II relative to Arm I in HisP could transmit information to the membrane- spanning domains of HisM/HisQ. Further evidence of flexibility of the helical domain is seen in the cTAP1 structure, where the average B-factor of the helical domain is 66.3 Å2, compared with 44.7 Å2 for the RecA-like domain. As mentioned in the previous section, the Q-loop and signature motif, both in the helical domain, have high B-factors (87.6 Å2 for the Q-loop and 79.7 Å2 for the signature motif and preceding loop) and are expected to be involved in binding the γ-phosphate. The observed flexibility of the helical domain may, therefore, transmit γ-phosphate-binding information to other parts of the transporter and/or store the free energy of ATP binding as strain energy to effect work as in other ATPases (Wang and Oster, 1998).
RAD50 also shows hinge motion of the structurally equivalent domain to the helical domain of transporter ATPases (referred to as lobe II, see below), upon ATP binding and dimerization (comparing a nucleotide-free monomer structure to an ATP-bound dimer structure). The RAD50 lobe II rotates ∼30° relative to lobe I (analogous to the cTAP1 RecA-like domain) between the two structures. Although the movement itself is different, it is the same structural region, including the signature motif that shows rigid body motion.
A TAP1 mutation, R659Q, was found in human lung cancer cells, which have a TAP deficient phenotype despite the presence of TAP1 and TAP2, although expression levels of TAP1 are low (Chen et al., 1996). The function of this mutant TAP1R659Q was recently analyzed in more detail (Saveanu et al., 2001). TAP1R659Q exhibits ∼50% of normal peptide transport when co-expressed with TAP2 in insect cells although it binds normally to ATP. These results were interpreted as likely to be an effect of reduced ATP hydrolysis (Saveanu et al., 2001). It is more likely that R659Q affects the coupling of hydrolysis to transport as this arginine residue is in the C-terminal linker between the helical domain and the RecA-like domain. The R659 side chain is located in the groove between helices α2 and α3 (Figure 1) and makes one hydrogen bond to the G601 carbonyl group. R659 is >20 Å away from the nucleotide-binding site. It would not be expected to form part of the TAP1–TAP2 interface in a RAD50-like dimer (Hopfner et al., 2000) although it may form part of the dimerization interface if TAP1 and TAP2 dimerize in a MalK-like conformation (Diederichs et al., 2000).
Comparison with non-transporter ABC ATPases
Superposition of the published ABC ATPase structures and the corresponding structure-based sequence alignment (Figure 3) show that there are several subtype-specific structural regions within the ATPase domain. The structures can be divided into three subfamilies: (i) the ABC transporter structures, including cTAP1, HisP and MalK, are representative of the ABC ATPase subfamily involved in interactions with the membrane-spanning domains and transport across membranes; (ii) the RAD50 and SMC structures show that they are close homologs, forming an ABC ATPase subfamily involved in various DNA repair and maintenance functions; and (iii) the MutS structures, and by sequence homology, the MutS homolog proteins (MSH) form another subfamily involved specifically in DNA mismatch repair.
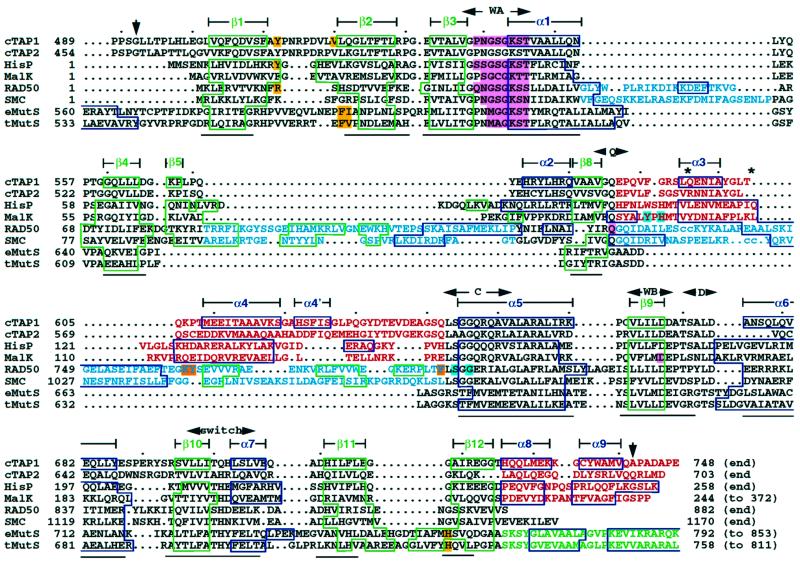
Fig. 3. Structure-based sequence alignment of all ABC ATPases for which structures have been published: human cTAP1, Salmonella typhimurium HisP crystallized with ATPγS (1b0u; Hung et al., 1998), Thermococcus litoralis MalK showing pyrophosphate in the active site (1g29; Diederichs et al., 2000), Pyrococcus furiosus RAD50 bound to ATP (1f2t and 1f2up; Hopfner et al., 2000), Thermotoga maritima SMC with no nucleotide bound (1e69; Lowe et al., 2001), E.coli MutS with ADP (1e3m; Lamers et al., 2000) and T.aquaticus MutS with ADP (1ewq and 1ewr; Obmolova et al., 2000). The sequence of human TAP2 was also aligned, based on homology to human TAP1. β-strands and α-helices are boxed green and blue, respectively, and numbered according to the cTAP1 terminology (derived from HisP) for simplicity. Nucleotide-interacting residues are highlighted (adenosine interactions in yellow, phosphate interactions in pink, adenosine interactions from the opposing monomer in the dimer in orange, and cross-dimer phosphate interactions in cyan). Subfamily-specific sequences are colored as in Figure 4. Conserved sequence motifs are labeled with side-to-side arrows: WA (Walker A), Q (Q-loop), C (signature or C motif), WB (Walker B), D (D-loop) and switch (switch region). Down-arrows indicate the beginning and end of the cTAP1 structure. The lines below the alignment identify regions that are structurally overlapping in all structures. The small dots above the alignment indicate increments of 10 in the TAP1 sequence. ‘cc’ in the RAD50 and SMC sequences (and * above it the alignment) shows the location of the coiled-coil domain inserted in RAD50 (150–734) and SMC (147–1022).
The RAD50/SMC subfamily has two insertions between the Walker A and Q-loop motifs, which lead to an extension of Sheet I of the RecA-like domain (blue in Figure 4B). The transporter subfamily and the RAD50/SMC subfamily both have extensions, absent in the MSH subfamily, between the Q-loop and the signature motif. In the ABC transporter subfamily, this extension forms most of the helical domain (red in Figure 4A). The extension in the RAD50/SMC subfamily is a mixed α/β structure and forms most of lobe II (blue in Figure 4B). This extension also includes a long (700–900 residue) coiled-coil region, which was omitted in the protein constructs from which the structure was determined. This coiled-coil region is thought to be important for the role of RAD50/SMC proteins in piecing back together large DNA fragments. The MSH family has a C-terminal extension (green in Figure 4C) of two α-helices, which are ‘swapped’ between the two monomers of a dimer. This partial ‘domain-swap’ may explain why the MSH subfamily can dimerize in the absence of nucleotide (Obmolova et al., 2000), whereas most other ABC ATPases do not (Hopfner et al., 2000; Lowe et al., 2001). Finally, the ABC transporter subfamily also has a C-terminal extension, which forms two α-helices (α8 and α9) that stack against the backside of Sheet II (Figure 4A). As this region seems to be conserved in length and in structure, but at the same time is specific to the transporter family of ABC ATPases, it may play a role in modulating transport or in interactions with the membrane-spanning domains of the transporter.
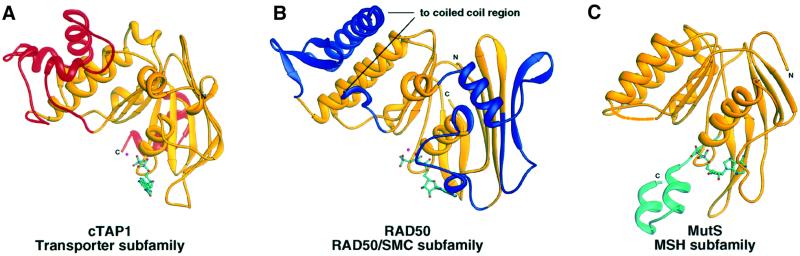
Fig. 4. (A) Ribbon diagram of cTAP1 highlighting in red the regions specific to the transporter subfamily of ABC ATPases, conserved in cTAP1, HisP (1b0u; Hung et al., 1998) and MalK (1g29; Diederichs et al., 2000). (B) Ribbon diagram of RAD50 (1f2u; Hopfner et al., 2000) highlighting in blue the regions structurally specific to the RAD50/SMC type ABC ATPase DNA repair proteins. (C) Ribbon diagram of E.coli MutS (1e3m; Lamers et al., 2000) with the region specific to the MSH subfamily of ABC ATPases colored green.
Nucleotide binding
Although the crystallization conditions contained ATP and Mg2+, the electron density maps show density corresponding to an ADP molecule in the nucleotide-binding site (Figure 5A). We have not measured the ATPase activity of this cTAP1 molecule, but Muller and co-workers were not able to show ATPase activity with a similar fragment (Muller et al., 1994). However, there may be some residual ATPase activity by cTAP1, or alternatively, the protein preparation may have contained a contaminating ATPase activity. We do not expect cTAP1 to have a wild-type ATPase activity level, as most current models require the presence of the TAP2 C-terminal domain to complete the active site. The conformation of the ADP molecule bound to cTAP1 is very similar to that of the ATPγS bound to HisP (Hung et al., 1998). Table II lists the nucleotide interactions with cTAP1. Residues from the P-loop (538-GPNGSGKST-546) are in close contact with the phosphate groups. Y512 stacks against the adenine base, while R515, V520 and G541 make interactions with the ribose (residues highlighted yellow in Figure 3). The lack of hydrogen-bonding interactions to the adenine base explains why TAP1 does not discriminate well between the different bases (Androlewicz et al., 1993; Wang et al., 1994; Hill et al., 1995; Russ et al., 1995; Knittler et al., 1999). The nucleotide-binding site of TAP2 is highly homologous to that of TAP1, with all residues contacting the ADP conserved between TAP1 and TAP2, and TAP2 also only weakly discriminates between different bases. Therefore, ADP is expected to interact in the same way with TAP2 as it does with TAP1.
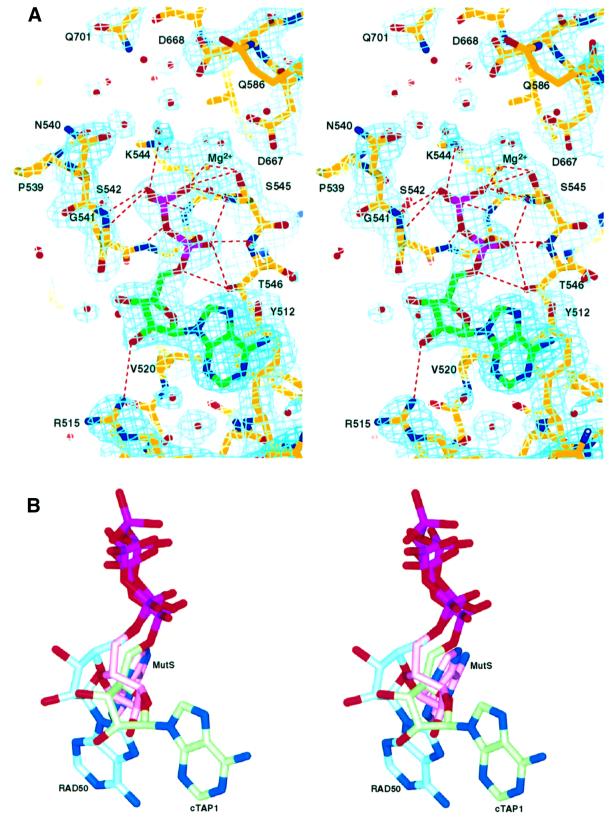
Fig. 5. The nucleotide-binding site. (A) Stereo representation of the σA-weighted 2Fo–Fc electron density map contoured at 1.2σ in the vicinity of the nucleotide-binding site showing clear density corresponding to ADP. Residues contacting the ADP, as well as other conserved residues in the vicinity of the active site are labeled. Hydrogen-bonding and salt-bridge interactions listed in Table II are marked by red dotted lines. (B) Stereo view of the three different adenosine conformations seen in the ABC ATPase structures. For reference, the cTAP1 ADP is shown in an orientation similar to that in (A). The cTAP1 ADP is in green, the RAD50 ATP in blue and the MutS ADP in pink.
Table II. Interactions between ADP–Mg2+ and cTAP1.
| Hydrogen bonds and salt bridges | ||
|---|---|---|
| Donor | Acceptor | Length (Å) |
| Mg2+ | ADP O2β | 2.1 |
| T546 Oγ1 | ADP O5* | 3.1 |
| T546 Oγ1 | ADP O2α | 2.8 |
| T546 N | ADP O2α | 2.6 |
| Mg2+ | S545 Oγ | 2.0 |
| S545 Oγ | ADP O2β | 2.9 |
| S545 N | ADP O2α | 3.0 |
| S545 N | ADP O2β | 2.9 |
| K544 Nζ | ADP O3β | 3.1 |
| K544 N | ADP O3β | 3.0 |
| G543 N | ADP O3α | 3.2 |
| G542 N | ADP O3β | 3.2 |
| G541 N | ADP O1β | 2.7 |
| R515 NH1 |
ADP O2* |
3.2 |
| van der Waals contacts (distance <4.0 Å) | ||
| ADP |
Protein |
|
| ADP adenosine base | Y512 | |
| ADP ribose | V520, G541 | |
Several other conserved residues are in close proximity to the cTAP1 ADP nucleotide, positioned so that they could interact with the γ-phosphate directly or through a water molecule in the ATP-bound form of cTAP1 (Figure 5A). These include Walker B residues D667 and D668, Q-loop residue Q586, which has poor side chain density and high B-factors in this ADP-bound structure, and switch region residue Q701. Q701 is most often a histidine in other transporters, and has been postulated to sense γ-phosphate binding (Diederichs et al., 2000) or modulate the polarization of the attacking water molecule (Hopfner et al., 2000). Mutations of this histidine disrupts transport in several ABC transporters, although its effects on ATP binding and hydrolysis vary (Schneider and Hunke, 1998). Due to crystal packing, residues T613 and V617 from a crystallographically related molecule make van der Waals contacts with the adenine base and the ribose of the nucleotide. There is no evidence that these interactions have any biological relevance.
Nucleotide triphosphate-binding proteins containing the Walker A motif (or P-loop) bind the triphosphate moiety of the nucleotide in a very similar orientation and conformation. However, different proteins of this P-loop-containing superfamily interact with the ribose and nucleotide base in very different ways. This is true even within the ABC ATPase family. Within the known ABC ATPase structures, three different adenosine conformations have been observed (Figures 4 and 5B), which correlate with the three ABC ATPase subfamilies, the transporter, RAD50/SMC and MSH subfamilies described above (Figure 4). Residues that bind adenosine are highlighted in yellow in Figure 3. The transporter subfamily has a conserved aromatic residue (Y512 in TAP1, F16 in HisP; Figure 3), which stacks against one side of the adenine ring. This aromatic residue is not conserved in the RAD50/SMC or MutS/MSH subfamilies. Instead, RAD50 has an arginine residue (R12), which stacks the hydrophobic part of its side chain against the adenine ring and hydrogen bonds to the ribose. It also has a phenylalanine (F791; highlighted orange in Figure 3), which comes from the adjacent monomer to stack with the adenine ring. This phenylalanine precedes the signature motif (Figure 3). It is not conserved in most other ABC ATPases (Figure 3). MutS binds adenosine through two motifs that are conserved only within the MutS/MSH family of proteins. Again, a phenylalanine (F596 in E.coli MutS and F561 in Thermus aquaticus MutS; Figure 3) stacks against the adenine ring. The backbone of the hydrophobic residue following this phenylalanine makes a hydrogen bond to the N6 of the adenine. The second motif stacks a histidine ring (H760 in E.coli MutS and H726 in T.aquaticus MutS) against the other side of the adenine ring. This histidine is very well conserved as either a histidine or an aromatic residue in MSH proteins (see sequence alignment in Obmolova et al., 2000), whereas the corresponding residue in other ABC ATPases is a well-conserved glycine (G718 in TAP1; Figure 3). Therefore, the three divergent subfamilies in the ABC ATPase family have evolved three different ways of recognizing the same nucleotide.
Asymmetry of the nucleotide-binding site in ABC transporters
The cTAP1 structure and the structure-based alignment of the ABC ATPases with known structures show that some sequence motifs are not conserved in TAP1. It is notable that TAP1 has a glutamine (Q701) in place of the highly conserved histidine in the switch region. In addition, TAP1 contains an aspartate instead of a glutamate at the end of the Walker B motif. Both the switch and Walker B motifs are thought to be important for γ-phosphate interaction and nucleotide hydrolysis. Furthermore, the signature motif of TAP2, which is expected to be involved in dimerization and hydrolysis at the TAP1 nucleotide-binding site, also contains non-conservative substitutions (LAAGQ versus the consensus LSGGQ). Therefore, three motifs expected to be involved in γ-phosphate binding and ATP hydrolysis have substitutions in the TAP1 site, whereas all the TAP2 nucleotide-binding site motifs contain the consensus sequences. This asymmetry of sequence conservation in the nucleotide-binding and active sites in TAP ATPases may be a reflection of the different roles of the two sites in peptide transport. Such asymmetry in TAP1 and TAP2 ATPase function has recently been postulated based on several biochemical studies (Knittler et al., 1999; Alberts et al., 2001; Hewitt et al., 2001; Karttunen et al., 2001; Lapinski et al., 2001).
Similar asymmetry has been observed at the biochemical level for several other transporters, including the multi-drug resistance transporter Pgp (Takada et al., 1998; Hrycyna et al., 1999), the cystic fibrosis chloride channel CFTR (Gadsby and Nairn, 1994; Senior and Gadsby, 1997), and the a-Factor pheromone transporter in Saccharomyces cerevisiae Ste6 (Proff and Kölling, 2001). Functional asymmetry has also been observed for HisP, which forms a homodimer that interacts with the membrane-spanning heterodimer HisM/HisQ (Kreimer et al., 2000). Similarly, the MalK homodimer structure is asymmetric, although the functional relevance of this asymmetry is unknown (Diederichs et al., 2000). The actual structural basis for functional asymmetry of the ABC ATPase domains in transporters awaits the determination of structures of ABC ATPase heterodimers, or of complete ABC transporters.
Materials and methods
Cloning and protein expression
The C-terminal domain of human TAP1 (residues 489–748) was cloned into the NdeI and BamHI sites of the pET15b vector (Novagen) by PCR amplification from the cDNA clone (gift of Peter Cresswell) using the following primers: forward primer, 5′-CGCGGCAGCCATATGCCACCCAGTGGTCTGTTG-3′; reverse primer, 5′-AGCCGGATCCTCATTCTGGAGCATCTGCAG-3′. The resulting expression cassette includes a 20-residue N-terminal extension with His6 tag and thrombin cleavage site, encoded by the pET15b vector. The protein was expressed in BL21(DE3) cells cultured in Luria–Bertani medium during an overnight induction period [induction at OD600 = 0.5 with 75 µM isopropyl-β-d-thiogalactopyranoside (IPTG)]. Cells were harvested and frozen in liquid nitrogen for storage at –80°C.
Protein purification and crystallization
All procedures were performed at 4°C. Cells were thawed, resuspended in 6 ml/g of cell paste of lysis buffer [20 mM Tris pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 0.1% Triton X-100, 5 mM MgCl2, 2 mM ATP, 1 mM phenylmethylsulfonyl (PMSF), 2.5 mM benzamidine, 0.2 mg/ml lysozyme] and lyzed by sonication. After centrifugation (8000 g, 45 min), the supernatant was loaded onto a Ni-NTA column (Qiagen) pre-equilibrated in 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 0.1% Triton X-100, 5 mM MgCl2. After a 15 column volume (c.v.) wash [20 mM Tris–HCl pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 5 mM MgCl2, 2 mM ATP, 1 mM PMSF, 7 mM β-mercaptoethanol (βME)], the protein was eluted using a l5 c.v. linear gradient (20–250 mM imidazole pH 7.0, in 20 mM Tris pH 8.0, 200 mM NaCl, 10% glycerol, 5 mM MgCl2, 2 mM ATP, 1 mM PMSF, 7 mM βME). The fractions containing cTAP1 protein were pooled, concentrated in a Centriprep-10 (Amicon), and further purified on a Superdex 200 HR16/60 (Amersham Pharmacia Biotech) size-exclusion column in 20 mM Tris pH 8.0, 100 mM NaCl, 10% glycerol, 5 mM MgCl2, 0.1 mM ATP, 1 mM PMSF, 7 mM βME, from which it eluted with an apparent molecular weight (based on the elution of standard proteins) of 33 kDa, corresponding with the size of a monomer (calculated mol.wt 30 398). The peak fractions were pooled, dialyzed against storage buffer [10 mM Tris pH 8.0, 50 mM NaCl, 10% glycerol, 5 mM MgCl2, 1 mM ATP, 1 mM dithiothreitol (DTT)] concentrated to ∼50 mg/ml in a Centriprep-10 and Centricon-10 (Amicon) and stored at 4°C. cTAP1 crystallized from Hampton Research Crystal Screen I condition 18 (20% PEG8000, 100 mM sodium cacodylate pH 6.5, 200 mM magnesium acetate). Drops were set up in a 1:1 ratio of precipitant to protein (at 10 mg/ml in storage buffer) at 4°C.
Data collection
Data were collected to 2.4 Å on a charged-coupled device Quantum 4 detector (ADSC) at the BioCARS BM-C beamline (Advanced Photon Source, Argonne National Laboratory) on a bipyramidal crystal (∼120 × 120 × 100 µM in size) which was flash-frozen in liquid nitrogen after brief soaking in 23% PEG8000, 100 mM sodium cacodylate pH 6.5, 200 mM magnesium acetate, 12.5% glycerol. Data were processed (Table I) using DENZO and SCALEPACK (HKL Research; Otwinowski and Minor, 1997). The crystal belongs to space group P62 with unit cell dimensions a = b = 87.57, c = 79.85, α = β = 90° and γ = 120°, with one cTAP1 molecule per asymmetric unit and ∼50% solvent content.
Structure determination and refinement
The structure was determined by molecular replacement with CNS (Crystallography & NMR System; Brünger et al., 1998) using HisP (1b0u; Hung et al., 1998) as a search model. The initial solution gave a poor R-factor (Rwork = 53%, Rfree = 53%) but displayed reasonable packing. Molrep (Vagin and Teplyakov, 1997) identified the same solution. Evidence that the solution was correct included the observation of strong electron density corresponding to the α- and β-phosphate groups as expected near the P-loop even though the nucleotide was omitted from the search model. Density modification with a solvent flipping protocol (Abrahams and Leslie, 1996), was performed on the molecular replacement phases to reduce model bias, using CNS (Brünger et al., 1998). The model was trimmed down to the regions having continuous electron density in the density-modified map (∼40% of the model). The side chains identical in HisP and cTAP1 were retained and the others replaced with Ala or Gly. After rigid body, positional and B-factor refinement, the R-factor was 49.0% (Rfree = 49.9%). More of the model was added as the density improved, through 15 cycles of manual rebuilding using O (Jones et al., 1991) and torsion angle, positional and B-factor refinement using CNS (Brünger et al., 1998). The final refinement statistics are listed in Table I. The final model contains residues 492–742 of TAP1 (residues 489–491 and 743–748 are not modeled although present in the protein crystallized), an ADP molecule, a Mg2+ ion and 114 water molecules. Figures were generated with RIBBONS (Carson, 1997) or SPOCK (http://quorum.tamu.edu/jon/spock/). Domain motions were determined using DOMOV (http://bioinfo1.mbfys.lu.se/cgi-bin/Domov/domov.cgi) and DynDom (Hayward and Berendsen, 1998). Coordinates and structure factors are available from the Protein Data Bank (entry 1JJ7).
Acknowledgments
Acknowledgements
We would like to thank P.Cresswell for the human TAP1 cDNA, A.Carfi as well as the staff of BioCARS beamline BM-C at APS for assistance during data collection, and members of the Wiley/Harrison Laboratory for useful discussions. This work was supported by the NIH and Howard Hughes Medical Institute (HHMI). R.G. is a postdoctoral fellow of the Damon Runyon-Walter Winchell Cancer Research Fund. D.C.W. is an investigator of the HHMI.
Note added in proof
Since this paper was submitted, two structures of Methanococcus jannaschii ABC transporter ATPases in the ADP-bound state have been published [Karpowich,N., Martsinkevich,O., Millen,L., Yuan,Y., Dai,P.L., MacVey,K., Thomas,P.J. and Hunt,J.F. (2001) Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure, 9, 571–586; and Yuan,Y.-R., Blecker,S., Martsinkevich,O., Millen,L., Thomas,P.J. and Hunt,J.F. (2001) The crystal structure of the MJ0796 ATP-binding cassette: implications for the structural consequences of ATP hydrolysis in the active site of an ABC-transporter. J. Biol. Chem., in press].
References
- Abele R. and Tampé,R. (1999) Function of the transport complex TAP in cellular immune recognition. Biochim. Biophys. Acta, 1461, 405–419. [DOI] [PubMed] [Google Scholar]
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Alberts P., Daumke,O., Deverson,E.V., Howard,J.C. and Knittler,M.R. (2001) Distinct functional properties of the TAP subunits coordinate the nucleotide-dependent transport cycle. Curr. Biol., 11, 242–251. [DOI] [PubMed] [Google Scholar]
- Androlewicz M.J., Anderson,K.S. and Cresswell,P. (1993) Evidence that transporters associated with antigen processing translocate a major histocompatibility complex class I-binding peptide into the endoplasmic reticulum in an ATP-dependent manner. Proc. Natl Acad. Sci. USA, 90, 9130–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson M. (1997) Ribbons. Methods Enzymol., 277, 493–505. [PubMed] [Google Scholar]
- Chen H.L., Gabrilovich,D., Tampé,R., Girgis,K.R., Nadaf,S. and Carbone,D.P. (1996) A functionally defective allele of TAP1 results in loss of MHC class I antigen presentation in a human lung cancer. Nature Genet., 13, 210–213. [DOI] [PubMed] [Google Scholar]
- Cresswell P., Bangia,N., Dick,T. and Diedrich,G. (1999) The nature of the MHC class I peptide loading complex. Immunol. Rev., 172, 21–28. [DOI] [PubMed] [Google Scholar]
- Diederichs K., Diez,J., Greller,G., Müller,C., Breed,J., Schnell,C., Vonrhein,C., Boos,W. and Welte,W. (2000) Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. EMBO J., 19, 5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby D.C. and Nairn,A.C. (1994) Regulation of CFTR channel gating. Trends Biochem. Sci., 19, 513–518. [DOI] [PubMed] [Google Scholar]
- Hayward S. and Berendsen,H.J.C. (1998) Systematic analysis of domain motions in proteins from conformational change; New results on citrate synthase and T4 lysozyme. Proteins, 30, 144–154. [PubMed] [Google Scholar]
- Hewitt E.W., Gupta,S.S. and Lehner,P.J. (2001) The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J., 20, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C.F. (1992) ABC transporters: from microorganisms to man. Annu. Rev. Cell. Biol., 8, 67–113. [DOI] [PubMed] [Google Scholar]
- Hill A., Jugovic,P., York,I., Russ,G., Bennink,J., Yewdell,J., Ploegh,H. and Johnson,D. (1995) Herpes simplex virus turns off the TAP to evade host immunity. Nature, 375, 411–415. [DOI] [PubMed] [Google Scholar]
- Holland I.B. and Blight,M.A. (1999) ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol., 293, 381–399. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Karcher,A., Shin,D.S., Craig,L., Arthur,L.M., Carney,J.P. and Tainer,J.A. (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell, 101, 789–800. [DOI] [PubMed] [Google Scholar]
- Hrycyna C.A., Ramachandra,M., Germann,U.A., Cheng,P.W., Pastan,I. and Gottesman,M.M. (1999) Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry, 38, 13887–13899. [DOI] [PubMed] [Google Scholar]
- Hung L.W., Wang,I.X., Nikaido,K., Liu,P.Q., Ames,G.F. and Kim,S.H. (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature, 396, 703–707. [DOI] [PubMed] [Google Scholar]
- Hyde S.C., Emsley,P., Hartshorn,M.J., Mimmack,M.M., Gileadi,U., Pearce,S.R., Gallagher,M.P., Gill,D.R., Hubbard,R.E. and Higgins, C.F. (1990) Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature, 346, 362–365. [DOI] [PubMed] [Google Scholar]
- Jones P.M. and George,A.M. (1999) Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett., 179, 187–202. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Karttunen J.T., Trowsdale,J. and Lehner,P.J. (1999) Antigen presentation: TAP dances with ATP. Curr. Biol., 9, R820–824. [DOI] [PubMed] [Google Scholar]
- Karttunen J.T., Lehner,P.J., Gupta,S.S., Hewitt,E.W. and Cresswell,P. (2001) Distinct functions and cooperative interaction of the subunits of the transporter associated with antigen processing (TAP). Proc. Natl Acad. Sci. USA, 98, 7431–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A. et al. (1992) Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature, 355, 641–644. [DOI] [PubMed] [Google Scholar]
- Knittler M.R., Alberts,P., Deverson,E.V. and Howard,J.C. (1999) Nucleotide binding by TAP mediates association with peptide and release of assembled MHC class I molecules. Curr. Biol., 9, 999–1008. [DOI] [PubMed] [Google Scholar]
- Kreimer D.I., Chai,K.P. and Ferro-Luzzi Ames,G. (2000) Nonequivalence of the nucleotide-binding subunits of an ABC transporter, the histidine permease and conformational changes in the membrane complex. Biochemistry, 39, 14183–14195. [DOI] [PubMed] [Google Scholar]
- Lamers M.H., Perrakis,A., Enzlin,J.H., Winterwerp,H.H., de Wind,N. and Sixma,T.K. (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G⋅T mismatch. Nature, 407, 711–717. [DOI] [PubMed] [Google Scholar]
- Lankat-Buttgereit B. and Tampé,R. (1999) The transporter associated with antigen processing TAP: structure and function. FEBS Lett., 464, 108–112. [DOI] [PubMed] [Google Scholar]
- Lapinski P.E., Neubig,R.R. and Raghavan,M. (2001) Walker A lysine mutations of TAP1 and TAP2 interfere with peptide translocation but not peptide binding. J. Biol. Chem., 276, 7526–7533. [DOI] [PubMed] [Google Scholar]
- Lehner P.J. and Trowsdale,J. (1998) Antigen presentation: coming out gracefully. Curr. Biol., 8, R605–608. [DOI] [PubMed] [Google Scholar]
- Lowe J., Cordell,S.C. and van den Ent,F. (2001) Crystal structure of the SMC head domain: an ABC ATPase with 900 residues antiparallel coiled-coil inserted. J. Mol. Biol., 306, 25–35. [DOI] [PubMed] [Google Scholar]
- Mimura C.S., Holbrook,S.R. and Ames,G.F. (1991) Structural model of the nucleotide-binding conserved component of periplasmic permeases. Proc. Natl Acad. Sci. USA, 88, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K.M., Ebensperger,C. and Tampe,R. (1994) Nucleotide binding to the hydrophilic C-terminal domain of the transporter associated with antigen processing (TAP). J. Biol. Chem., 269, 14032–14037. [PubMed] [Google Scholar]
- Nikaido H. and Hall,J.A. (1998) Overview of bacterial ABC transporters. Methods Enzymol., 292, 3–20. [DOI] [PubMed] [Google Scholar]
- Obmolova G., Ban,C., Hsieh,P. and Yang,W. (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature, 407, 703–710. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Proff C. and Kölling,R. (2001) Functional asymmetry of the two nucleotide binding domains in the ABC transporter Ste6. Mol. Gen. Genet., 264, 883–893. [DOI] [PubMed] [Google Scholar]
- Russ G., Esquivel,F., Yewdell,J.W., Cresswell,P., Spies,T. and Bennink,J.R. (1995) Assembly, intracellular localization and nucleotide binding properties of the human peptide transporters TAP1 and TAP2 expressed by recombinant vaccinia viruses. J. Biol. Chem., 270, 21312–21318. [DOI] [PubMed] [Google Scholar]
- Saveanu L., Daniel,S. and van Endert,P.M. (2001) Distinct functions of the ATP-binding cassettes of the transporters associated with antigen processing: a mutational analysis of Walker A and B sequences. J. Biol. Chem., 276, 22107–22113. [DOI] [PubMed] [Google Scholar]
- Schneider E. and Hunke,S. (1998) ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev., 22, 1–20. [DOI] [PubMed] [Google Scholar]
- Senior A.E. and Gadsby,D.C. (1997) ATP hydrolysis cycles and mechanism in P-glycoprotein and CFTR. Semin. Cancer Biol., 8, 143–150. [DOI] [PubMed] [Google Scholar]
- Takada Y., Yamada,K., Taguchi,Y., Kino,K., Matsuo,M., Tucker,S.J., Komano,T., Amachi,T. and Ueda,K. (1998) Non-equivalent cooperation between the two nucleotide-binding folds of P-glycoprotein. Biochim. Biophys. Acta, 1373, 131–136. [DOI] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov,A. (1997) MOLREP: and automated program for molecular replacement. J. Appl. Cryst., 30, 1022–1025. [Google Scholar]
- Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. and Oster,G. (1998) Energy transduction in the F1 motor of ATP synthase. Nature, 396, 279–282. [DOI] [PubMed] [Google Scholar]
- Wang K., Früh,K., Peterson,P.A. and Yang,Y. (1994) Nucleotide binding of the C-terminal domains of the major histocompatibility complex-encoded transporter expressed in Drosophila melanogaster cells. FEBS Lett., 350, 337–341. [DOI] [PubMed] [Google Scholar]