

Abstract
We describe a novel role for the ARF6 GTPase in the regulation of adherens junction (AJ) turnover in MDCK epithelial cells. Expression of a GTPase-defective ARF6 mutant, ARF6(Q67L), led to a loss of AJs and ruffling of the lateral plasma membrane via mechanisms that were mutually exclusive. ARF6-GTP-induced AJ disassembly did not require actin remodeling, but was dependent on the internalization of E-cadherin into the cytoplasm via vesicle transport. ARF6 activation was accompanied by increased migratory potential, and treatment of cells with hepatocyte growth factor (HGF) induced the activation of endogenous ARF6. The effect of ARF6(Q67L) on AJs was specific since ARF6 activation did not perturb tight junction assembly or cell polarity. In contrast, dominant-negative ARF6, ARF6(T27N), localized to AJs and its expression blocked cell migration and HGF-induced internalization of cadherin-based junctional components into the cytoplasm. Finally, we show that ARF6 exerts its role downstream of v-Src activation during the disassembly of AJs. These findings document an essential role for ARF6- regulated membrane traffic in AJ disassembly and epithelial cell migration.
Keywords: adherens junctions/ARF6/cell–cell adhesion/epithelia/membrane traffic
Introduction
Epithelial cells contain apical and basolateral plasma membrane domains that are separated by distinct junctional complexes, such as the apical tight junctions, and the basolaterally distributed adherens junctions (AJs), desmosomes and gap junctions (Drubin and Nelson, 1996). These junctional complexes obstruct the lateral diffusion of proteins and lipids between the two membrane domains, and also regulate the establishment of adhesive contacts between neighboring cells in polarized epithelia. During tumorigenesis, epithelial cells lose these contacts and acquire a motile or mesenchymal phenotype (Birchmeier and Birchmeier, 1993). Similar changes in morphology and behavior also occur during normal developmental processes, such as neural crest formation (Thiery et al., 1982). During these epithelial to mesenchymal transitions, the stability of the adherens type junctions (zonulae adherens) is markedly compromised (Birchmeier and Birchmeier, 1993; Schmidt et al., 1993). The AJs are specialized forms of cadherin-based adhesive contacts that are important for tissue organization in a developing embryo and the adult organism. Cadherins are type I transmembrane proteins that complex with cytosolic proteins, the catenins, which in turn provide anchorage to the actin cytoskeleton to form stable cell–cell contacts (Yap et al., 1997). The occurrence of mutated forms of cadherins and catenins in several epithelium-derived invasive carcinomas affirms the inverse relationship between the establishment of these junctions and the acquisition of a motile or invasive phenotype (Berx et al., 1995; Vermeulen et al., 1995; Birchmeier et al., 1996; Handschuh et al., 1999). Thus, the regulation of AJ assembly is an area of intense investigation.
Newly synthesized E-cadherin is targeted to the basolateral cell membrane (Chen et al., 1999) and is probably incorporated into AJs via lateral diffusion (Rajasekaran et al., 1996). At the basolateral end, cadherin molecules are constitutively endocytosed and recycled back to the plasma membrane (Le et al., 1999). While such cycling is minimal in confluent epithelial cell monolayers, a loss of cell–cell contact is accompanied by a concomitant increase in cadherin cycling. It has been speculated that endocytic traffic of E-cadherin is regulated by cell–cell adhesion and that this recycling pool serves as a mechanism for regulating the availability of E-cadherin for junction assembly, thereby allowing the cell to undergo rapid changes in morphology in response to extracellular stimuli (Kamei et al., 1999). At present, little is known about the regulation of E-cadherin traffic.
ARF6 (ADP-ribosylation factor 6), a member of the ARF family of Ras-related GTPases, cycles between its active GTP-bound and inactive GDP-bound forms in cells. The ARF6 GTPase cycle has been shown to regulate membrane movement between the plasma membrane and endosomal compartments in non-polarized cells (Chavrier and Goud, 1999). In addition, ARF6–GTP initiates cortical actin rearrangements at the cell periphery, accompanied by a depletion of stress fibers (D’Souza-Schorey et al., 1997). Both these processes, i.e. endosome recycling and actin remodeling, may impinge upon cell motility and the maintenance of cell polarity. Little is known about the signaling pathways and upstream regulatory molecules that modulate ARF6 function. Recent studies showed that extracellular agonists, such as bombesin and epidermal growth factor (EGF), trigger nucleotide exchange and activation of ARF6, and that the effect of bombesin was mediated by Gq (heterotrimeric GTP-binding protein q) (Boshans et al., 2000). An important breakthrough in ARF6 signaling was the finding that ARF6 could activate PIP 5-kinase type I, an enzyme that catalyzes the formation of phosphatidylinositol-4,5-bisphosphate (PIP2) from PI-4-P (Honda et al., 1999). Sustained activation of ARF6, by expression of a constitutively activated mutant, led to the recruitment of PIP 5-kinase to the cell surface and the generation of actin-rich ruffles. The synthesis and turnover of PIP2 have been implicated in a variety of events, including actin polymerization and membrane traffic, making this molecule an important component of signal transduction pathways (Martin, 1998).
Here we have investigated the role of the ARF6 GTPase cycle in differentiated and polarized Madin–Darby canine kidney (MDCK) epithelial cells. A previous study of the role of ARF6 in MDCK cells showed that ARF6 regulates endocytosis at the apical plasma membrane (Altschuler et al., 1999). In this study, we describe a novel role for ARF6 in the regulation of AJ turnover in epithelial cells. We show that by modulating the spatial distribution and traffic of cadherin-based junction components, the ARF6 GTPase regulates the acquisition of a migratory phenotype in epithelia and may function as a critical determinant of epithelial to mesenchymal transitions.
Results
Expression of ARF6–GTP promotes the disassembly of cadherin-based cell junctions
To investigate the role of ARF6 in epithelial cells, we first examined the effect of a constitutively activated GTP-bound mutant of ARF6, ARF6(Q67L), on actin filament distribution in MDCK epithelial cells. For these initial studies we used the pLZRS-IRES-GFP (where GFP is green fluorescent protein) based retroviral expression system, which allowed for the simultaneous transient expression of ARF6(Q67L) and GFP. A major advantage of the retrovirus expression system was that it allowed for expression of ARF6 mutants at levels that were barely above endogenous protein expression (see Materials and methods).
First, we examined the effect of expressing the constitutively activated, GTPase-defective ARF6 mutant, ARF6(Q67L), on actin filament distribution in MDCK cells. For these studies, confluent layers of MDCK cells were infected with retrovirus encoding ARF6(Q67L). Post-infection, actin filament distribution was assessed by staining cells with rhodamine–phalloidin, followed by confocal immunofluorescence microscopy. As shown in Figure 1A and B, a loss of actin staining was observed at the cell junctions between adjacent cells expressing ARF6(Q67L). This observation prompted us to investigate the effect of ARF6 on the distribution of components of the adherens and tight junctions. To this end, we first examined the effect of ARF6(Q67L) expression on the distribution of E-cadherin. Proteins of the cadherin and catenin families are essential components of the AJ (Yap et al., 1997). In epithelial cells, E-cadherins constitute a major class of adhesion molecules that support homophilic calcium-dependent cell–cell adhesion, whereas α and β catenins link E-cadherin to the actin cytoskeleton. Labeling of cells expressing the activated ARF6 mutant with anti-E-cadherin antibodies exhibited a loss of cadherin from AJs. Instead, E-cadherin was localized to the perinuclear compartments in the cytoplasm (Figure 1C and D) and at the ruffled edges of the lateral membrane (shown in Figure 2). This distribution pattern was in marked contrast to the localization of these proteins in untransfected monolayers of MDCK cells (Figure 1C, D, G and H, arrows) or cells infected with retrovirus alone (data not shown), in which the majority of the cadherin/catenin label was present at the cell junctions. We also examined the distribution of ZO-1, a component of tight junctions (Stevenson et al., 1986). In contrast to its effect on AJs, ARF6(Q67L) expression had no effect on the distribution of ZO-1 (Figure 1E and F), indicating that tight junctions were unperturbed.
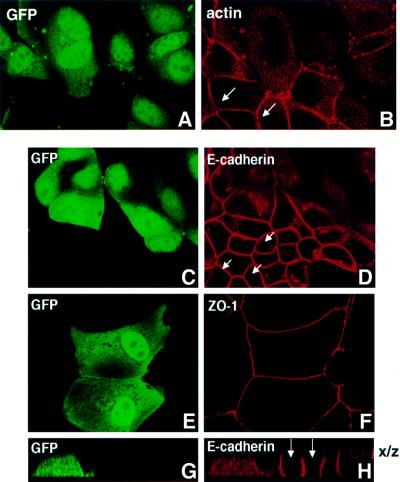
Fig. 1. Selective loss of AJs by expression of constitutively activated ARF6. MDCK cells grown on coverslips were infected with retrovirus encoding ARF6(Q67L) and GFP. Cells were labeled for actin (A and B), E-cadherin (C, D, G and H) and ZO-1 (E and F). Cells were viewed by confocal microscopy and optical sections at the plane of the cell junctions are shown (A–F). Vertical (x/z) sections of E-cadherin-labeled cells from a separate field are also shown (G and H). Infected cells transiently expressing ARF6(Q67L) exhibited diffuse green GFP staining (A, C, E and G). Infected cells exhibit a loss of actin and E-cadherin label at the cell junctions, while the AJs of untransfected cells (arrows) are intact. Also, tight junctions are intact on ARF6(Q67L)-expressing cells.
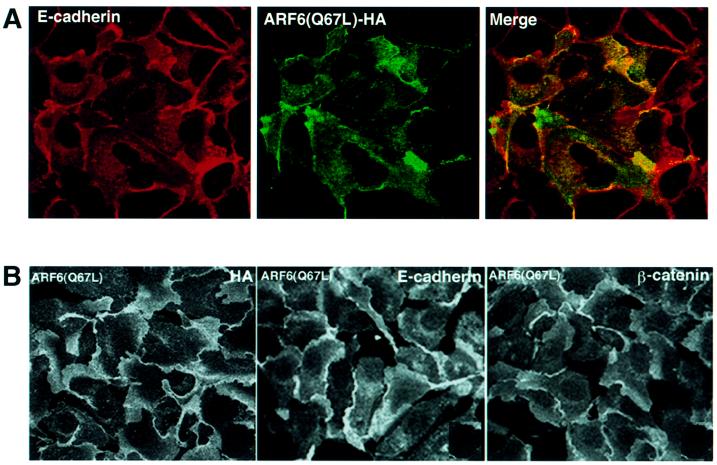
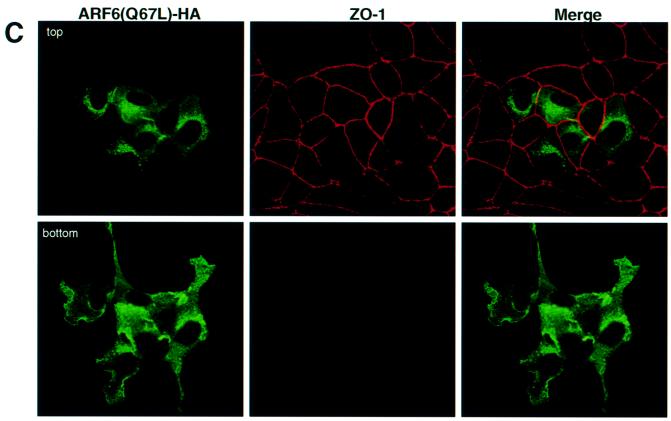
Fig. 2. Distribution of ARF6(Q67L) in MDCK cells. (A) Cells transiently expressing HA-tagged ARF6(Q67L) were labeled for E-cadherin (red) and HA (green). Coincident staining appears yellow in the merged image. (B) MDCK cells stably expressing HA-tagged ARF6(Q67L) were labeled for HA (left), E-cadherin (middle) and β-catenin (right). ARF6(Q67L), E-cadherin and β-catenin localize predominantly to the perinuclear cytoplasm and at the cell periphery in areas of membrane ruffling. (C) Cells transiently expressing HA-tagged ARF6(Q67L) were labeled for HA and ZO-1. Cells were viewed by confocal microscopy and optical sections at two different confocal planes are shown. ARF6(Q67L)-expressing cells have intact tight junctions.
Next, we utilized a retroviral expression system in which the GFP was replaced with neomycin. In order to determine the distribution of ARF6, the protein was epitope tagged with hemagglutinin (HA). This sytem allowed us to select for a population of cells stably expressing HA-tagged ARF6(Q67L). We observed that expression of HA-tagged protein had the same effect on AJ assembly as untagged ARF6–GTP. Furthermore, the majority of E-cadherin and activated ARF6 exhibited predominantly overlapping distributions at plasma membrane ruffles and at perinuclear membrane vesicles (Figure 2A). Noticeably, cell populations expressing ARF6(Q67L) formed rather loose colonies compared with the compact colonies formed by normal MDCK cells in cell culture. Figure 2B shows labeling for ARF6–GTP, E-cadherin and β-catenin at regions of membrane ruffling in cell lines stably expressing ARF6(Q67L). Although ARF6–GTP-expressing cells exhibited a loss of cadherin-based junctions and surface ruffles, the tight junctions were intact. In Figure 2C, a group of ARF6–GTP-expressing cells that exhibit membrane ruffling, but intact tight junctions, is shown.
The effect of ARF6–GTP on AJ disassembly is independent of actin cytoskeleton remodeling and phospholipid metabolism
To investigate whether the effect of ARF6 on AJ disassembly could be attributed to its regulatory role in membrane traffic or its effect on actin organization, we utilized an ARF6 triple mutant, ARF6(Q67L:Q37E:S38I), which contained additional mutations in the effector-binding domain of the protein. This effector domain mutant was shown to be incapable of actin remodeling, but did not interfere with ARF6–GTP-mediated membrane cycling (Al-Awar et al., 2000). It is likely that the mutations in the effector loop prevent interaction with key downstream targets required for ARF6-induced peripheral actin remodeling. Similarly to previous observations in HeLa cells, MDCK cells transfected with ARF6(Q67L: Q37E:S38I) did not exhibit prominent actin-rich membrane ruffles at the peripheral edges (data not shown). However, expression of ARF6(Q67L:Q37E:S38I) mimicked the effect of ARF6(Q67L) on AJ disassembly, with the majority of the cadherin label present in perinuclear vesicles in the absence of membrane ruffles (Figure 3). These findings suggest that ARF6-mediated regulation of AJ stability and turnover is independent of its effect on the actin cytoskeleton, and probably occurs by mobilization of AJ components via vesicle traffic.
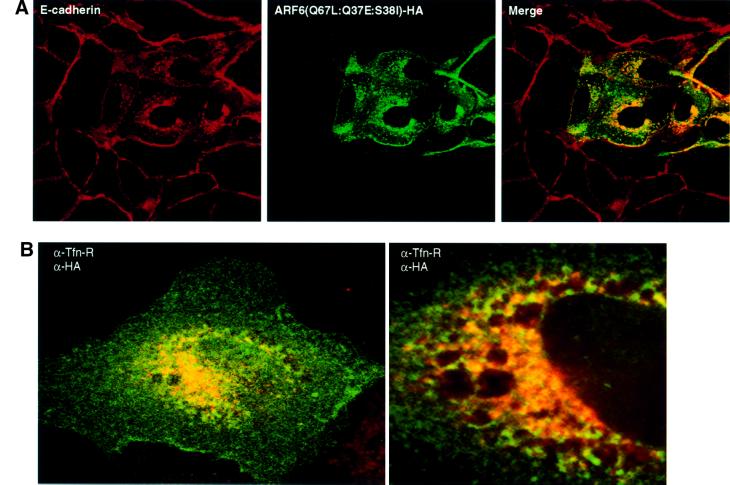
Fig. 3. ARF6(Q67L:Q37E:S38I), an effector domain ARF6 mutant incapable of membrane ruffling, can induce AJ disassembly. (A) Cells transiently expressing HA-tagged ARF6(Q67L:Q37E:S38I) were labeled for E-cadherin (red) and HA (green). Coincident staining appears yellow in the merged image. Transfected cells exhibited a loss of E-cadherin label at the cell junctions with a redistribution of the protein to the perinuclear vesicles in the cytoplasm. (B) Cells were labeled for HA (red) and for transferrin receptor (Tfn-R; red). Coincident staining appears yellow in the merged image. A stacked image of optical sections along the z-axis is shown (left). A magnified image across a single confocal plane at the perinuclear region is shown on the right.
We examined the nature of the perinuclear structures that acquired E-cadherin. Double-labeling experiments revealed substantial co-localization of E-cadherin/ARF6-positive vesicles with endosomal markers, such as the transferrin receptor (Tfn-R), in perinuclear compartments (Figure 3B). No overlapping distribution was observed with antibodies against Golgi, endoplasmic reticulum or lysosomal markers (data not shown). The redistribution of E-cadherin into transferrin receptor-containing endosomes is particularly relevant since cell surface E-cadherin has been shown to be trafficked constitutively along endocytic recycling pathways in MDCK cells (Le et al., 1999).
ARF6(Q67L) stimulates actin remodeling by increasing levels of PIP2 (Honda et al., 1999). Thus, we examined whether the effect of ARF6–GTP on AJs was mediated by its ability to induce locally elevated levels of PIP2. For these studies, we utilized an expression plasmid encoding the pleckstrin homology domain of PLC-δ tagged with GFP (PH–GFP), which has previously been characterized as a marker of PIP2 distribution in cells (Varnai and Balla, 1998). We transfected plasmid encoding PH–GFP into MDCK cells expressing ARF6(Q67L). As shown in the montage in Figure 4A, we found that PH–GFP labeled ARF6–GTP-induced surface ruffles, but did not localize to ARF6–GTP-generated internal endosomal vesicles. Double labeling for E-cadherin and PIP2 also revealed no overlap in E-cadherin and PIP2 distribution in endosomes (Figure 4B). In normal MDCK cells, GFP labeling was diffuse in the cytoplasm with minimal labeling at the cell surface in endogenous protrusions (Figure 4C). These latter findings revealed that the two phenotypes, i.e. the internalization of AJ components and membrane ruffling, can be uncoupled and are probably mediated by distinct downstream effectors and signaling pathways.
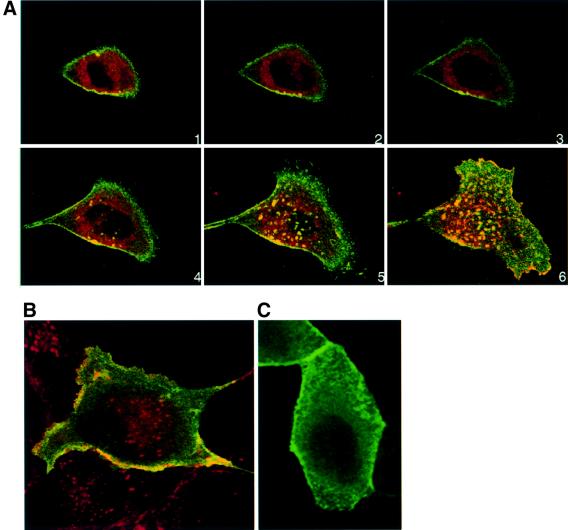
Fig. 4. ARF6–GTP-generated endosomal vesicles are devoid of PIP2. (A) HA-tagged ARF6(Q67L)-expressing cells were transfected with plasmid encoding PH–GFP, and 24 h post-transfection cells were fixed and labeled with anti-HA monoclonal antibody to determine ARF6 distribution (red). Images were merged and coincident staining appears yellow. Merged images of horizontal x/y sections along the z-axis are shown [1 (top)–6 (bottom)]. An overlapping distribution of PH–GFP (i.e. PIP2) and ARF6(Q67L) was detected in surface ruffles, but not in ARF6(Q67L)-positive perinuclear vesicles. (B) ARF6(Q67L)-expressing cells were transfected with plasmid encoding PH–GFP and labeled for E-cadherin. Coincident staining for E-cadherin (red) and PH–GFP (green) appears yellow. Merged images of a single optical section at the perinuclear plane are shown. (C) Cells infected with retrovirus alone were transfected with plasmid encoding PH–GFP, and 24 h post-transfection, cells were fixed and viewed under a confocal microscope. PH–GFP label was mainly diffuse in the cytoplasm and some label was seen at the cell surface.
Expression of ARF6–GTP has no effect on cell polarity
We investigated whether ARF6(Q67L)-induced disassembly of AJs was accompanied by a loss of cell polarity. To this end, cells infected with retrovirus encoding HA-tagged ARF6(Q67L) were grown on transwell filter units, and localization of ARF6–GTP was compared with the distribution of other apical and basolateral membrane proteins. Images along the x/z-axis showed that ARF6(Q67L) localized to the basolateral membrane and cytoplasm, since its distribution partially overlapped with transferrin receptors, a basolateral marker (Figure 5A), and not with GP-135, an apical membrane marker (Figure 5B and D), or with ZO-1, a component of the apical tight junctions (Figure 5C). The cells were polarized and exhibited microvilli at the apical end. No labeling for ARF6–GTP was observed in the tips of the microvilli (Figure 5D). The localization of ARF6–GTP to the basolateral membrane and cytoplasm may reflect a specific role for ARF6 in the constitutive recycling of E-cadherin from AJs to the basolateral membrane, as has been described previously (Le et al., 1999). These studies also revealed that despite the loss of AJs, cells expressing ARF6(Q67L) retained their polarized distribution of apical and basolateral membrane proteins.
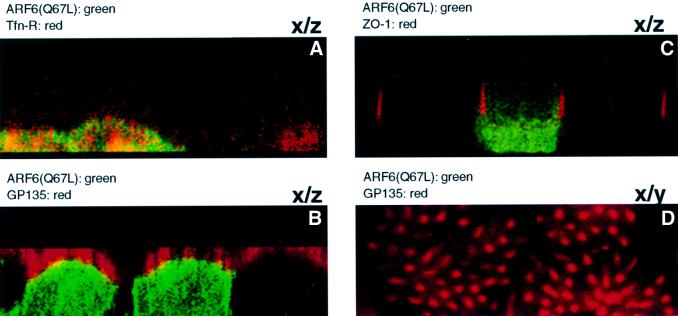
Fig. 5. ARF6 activation does not perturb cell polarity. MDCK cells infected with retrovirus encoding HA-tagged ARF6(Q67L) for transient protein expression were grown on transwell filters, fixed, double labeled as indicated below, and processed by immunofluorescence microscopy. Cells were viewed using a confocal imaging system. Cells were double labeled with anti-HA rabbit polyclonal antibody (green) and with mouse monoclonal antibodies (red) to either Tfn-R (A), gp135 (B and D) or ZO-1 (C). Merged images of vertical (x/z) sections are shown (A–C). A horizontal x/y section at a single confocal plane at the apical microvilli is also shown (D). Coincident red and green staining appears yellow.
While the above data clearly demonstrate the basolateral distribution of ARF6–GTP, an earlier study by Altschuler et al. (1999) reported that ARF6–GTP localized exclusively to the apical plasma membrane of MDCK cells. At this point, the reason for this disparity is still unclear. However, in addition to the MDCK cell line used in this study, we have found that ARF6–GTP localizes to the lateral plasma membrane in other epithelial cell lines including MCF-7 and T-47D (data not shown).
ARF6(T27N), the dominant-negative ARF6 mutant, enhances cell–cell adhesion
To assess further the importance of the ARF6 GTPase cycle in cell–cell adhesion, we analyzed cells expressing HA-tagged ARF6(T27N), the dominant-negative ARF6 mutant defective in GTP binding, and cells expressing HA-tagged wild-type ARF6. Transient and stable expression of ARF6(T27N) and wild-type ARF6 in MDCK cells was achieved using the neomycin-based retroviral system described above. We observed that ARF6(T27N)-expressing cells formed tight colonies that appeared to be even more compact than those formed by cells infected with retrovirus alone. Confocal images of these cells along the x/y-and x/z-axis revealed that the majority of ARF6(T27N) localized predominantly to the AJs (Figure 6A and B). A small fraction of the label was also seen at subapical membrane compartments.
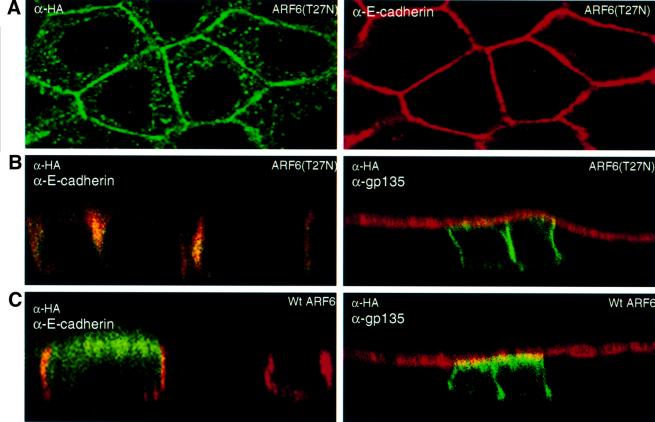
Fig. 6. Subcellular distribution of ARF6(T27N) and wild-type ARF6. (A) Cells expressing HA-tagged ARF6(T27N) were double labeled with anti-HA rabbit polyclonal antibody (green) and anti-E-cadherin mouse monoclonal antibody (red). An image across a single confocal plane at the cell junctions is shown to emphasize the localization of ARF6(T27N) at the cell junctions. (B) Cells expressing HA-tagged ARF6(T27N) were double labeled for HA (green) and for either E-cadherin (red, shown on the left) or gp135 (red, shown on the right). Coincident red and green staining appears yellow. Merged images taken along the x/z-axis are shown. ARF6(T27N) localizes to the AJs of MDCK cells; a small fraction of label was also seen at the lateral membrane and subapical region. (C) Cells transiently expressing HA-tagged wild-type (wt) ARF6 were labeled for HA (green) and for either E-cadherin (red, left) or gp135 (red, right). Coincident red and green staining appears yellow. Merged images taken along the x/z-axis are shown. Wild-type ARF6 localizes to the AJs, the lateral membrane and subapical membranes.
Expression of wild-type ARF6 had no effect on cell morphology and was identical to normal untransfected MDCK cells in cell culture (not shown). Images of ARF6 distribution along the x/z-axis revealed that the wild-type protein was localized to the AJs and to the subapical region of the cell (Figure 6C). Thus, the distinct and unique localization patterns of ARF6 and its mutants defective in GTP binding and hydrolysis suggest that the ARF6 GTPase shuttles between the AJs and vesicular compartments in the cytoplasm, and probably regulates the traffic of proteins between these compartments.
To substantiate our findings obtained by morphological investigations, we determined whether the detergent-solubility properties of β-catenin (an AJ component) were altered in cell lines expressing wild-type ARF6 or its mutants, ARF6(Q67L) and ARF6(T27N). For these experiments, we used the Triton X-100 solubility assay, in which cells were extracted with ‘CSK’ buffer containing 0.5% Triton X-100, and the fraction of β-catenin that partitioned into the Triton-insoluble (cytoskeleton-associated) fraction was monitored using immunoblot analysis. As seen in Figure 7, in the case of cells expressing wild-type ARF6, approximately half of the cellular β-catenin was present in the insoluble fraction. A similar distribution was observed in untransfected MDCK cells (not shown). However, in cells expressing ARF6(T27N), most of the β-catenin was cytoskeleton associated (resistant to Triton X-100 solubilization), whereas in cells expressing the ARF6–GTP mutant, the majority of β-catenin was detergent soluble. Thus, the differences in subcellular distribution and phenotype of wild-type ARF6 and its mutants defective in GTP hydrolysis and binding were reflected in the detergent-solubility properties of β-catenin in cell lines expressing the various mutant forms of ARF6. Moreover, the increased association of β-catenin with the cytoskeletal fraction in ARF6(T27N)-expressing cell lines indicates that an inhibition of nucleotide exchange on ARF6 prevents the turnover of AJs, and thereby stabilizes the epithelial phenotype.
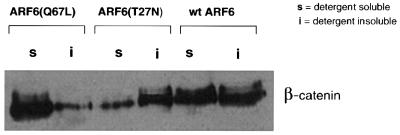
Fig. 7. Effect of the ARF6 GTPase cycle on the detergent-solubility properties of β-catenin. Cells stably expressing either ARF6, ARF6(Q67L) or ARF6(T27N) were lysed in CSK-A buffer containing 0.5% Triton X-100, centrifuged, and Triton-soluble (s) and -insoluble (i) fractions were analyzed for β-catenin distribution using immunoblotting procedures.
ARF6 regulates hepatocyte growth factor/scatter factor-induced AJ disassembly
Since the studies described above suggested that the ARF6 GTPase cycle regulates AJ turnover, we investigated the importance of ARF6 activation in a physiologically relevant disassembly of AJs. To this end, we examined the effect of dominant-negative ARF6 expression in cells treated with hepatocyte growth factor (HGF)/scatter factor. HGF is involved in the invasive behavior of several tumor cell types, and treatment of epithelial cells with HGF leads to the activation of the receptor tyrosine kinase c-met. HGF-induced signaling induces the disruption of cell–cell adhesion by promoting the internalization of E-cadherin (Stella and Comoglio, 1999). As stated above, E-cadherin is constitutively internalized and then recycled back to the basolateral surface (Le et al., 1999). For our investigations, untransfected MDCK cells and those stably transfected with ARF6(T27N) were incubated with HGF for varying time intervals, and E-cadherin distribution was monitored. In untransfected cells, consistent with previously reported findings, E-cadherin was internalized upon HGF treatment; there was a dose-dependent increase in internalization with increased exposure to HGF (not shown). Figure 8A shows the distribution of E-cadherin in normal MDCK cells and in cells expressing ARF6(T27N), respectively, after 4 h of HGF treatment. As shown, expression of the dominant-negative ARF6 mutant ARF6(T27N) completely blocked HGF-induced cadherin internalization and breakdown of AJs. Furthermore, by phase-contrast microscopy we found that cells stably expressing ARF6(T27N) abolished HGF-mediated cell scatter (data not shown). These findings suggest that ARF6 plays a critical role in the internalization of E-cadherin and mediates HGF-induced AJ disassembly.
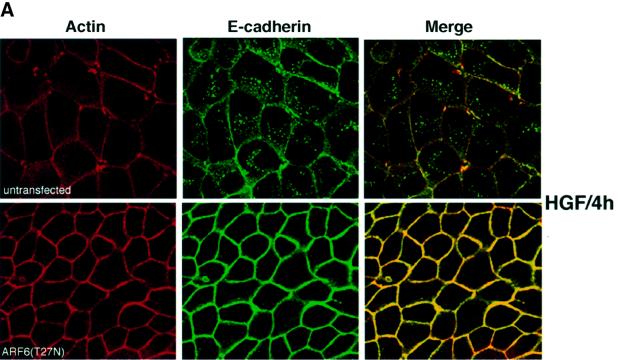
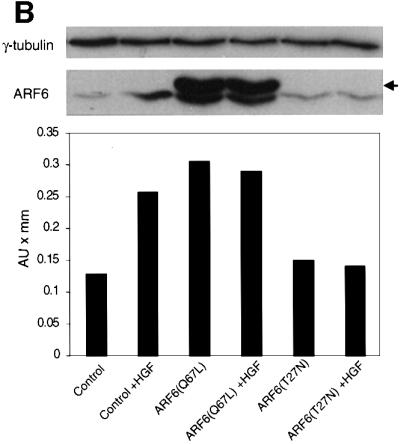
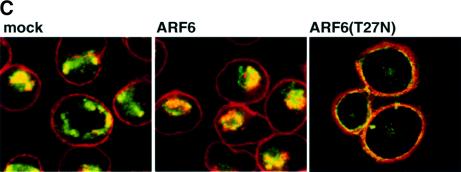
Fig. 8. Effect of dominant-negative ARF6 on HGF- and Ca2+ depletion-mediated AJ disassembly. (A) Untransfected MDCK cells or cells stably expressing ARF6(T27N) were grown on coverslips and incubated with 5 ng/ml HGF for 4 h. Cells were fixed and labeled for E-cadherin (green), and stained for actin (red). As shown, ARF6(T27N) expression blocks HGF-induced internalization of E-cadherin. (B) HGF induces the subcellular redistribution of endogenous ARF6. Normal MDCK cells or cells stably expressing either ARF6(Q67L) or ARF6(T27N) were lysed in CSK-A buffer, resolved on 14% SDS gels and the Triton-soluble fraction was analyzed for endogenous ARF6 distribution with anti-ARF6 monoclonal antibody using immunoblotting procedures. Band density was measured using the enhanced UltroScan XL Laser Densitometer (Pharmacia). As a loading control, the membrane was also probed for γ-tubulin expression. HGF treatment led to a marked increase in soluble ARF6. In cells expressing ARF6(Q67L), similar amounts of soluble ARF6 were detected in the presence and absence of HGF, while expression of ARF6(T27N) blocked the redistribution of ARF6 into the soluble pool. Exogenous ARF6(Q67L) runs at a higher molecular weight than the endogenous protein (arrow). Exogenous ARF6(T27N) was not detected on the blot since most of the ARF6(T27N) was present in the insoluble fraction (not shown). The data shown are representative of four independent experiments. (C) Normal MDCK cells (left) and those expressing wild-type ARF6 (center) or ARF67(T27N) (right) were incubated with EDTA (2.5 mM) at 37°C for 15 min. Cells were fixed and labeled for E-cadherin (green) and stained for actin (red). Merged images are shown; coincident staining appears yellow. ARF6(T27N) expression blocks the Ca2+ depletion-induced collapse of E-cadherin into the cytoplasm.
Next we asked whether we could assess the activation profile of endogenous ARF6 in response to HGF treatment using the detergent-solubility assay described above. We asked whether HGF treatment would increase the soluble pool of endogenous ARF6. To this end, untransfected MDCK cells were exposed to HGF for 1 h, after which cells were subjected to detergent-based fractionation as described above and the endogenous ARF6 protein associated with the detergent-soluble fraction was determined. As shown in Figure 8B, we observed a marked increase in soluble ARF6 after HGF treatment. When the assay was performed in cell lines stably expressing the dominant-negative ARF6 mutant ARF6(T27N), HGF-induced redistribution of endogenous ARF6 into the soluble pool was inhibited. In contrast, a majority of the endogenous ARF6 pool was soluble in cells expressing ARF6(Q67L); the latter is likely to be a reflection of the ‘activated ARF6’ phenotype. These results suggest that HGF induced the translocation of endogenous ARF6 protein to the detergent-soluble fraction, a process that was blocked by dominant-negative ARF6. Thus, ARF6 activation lies downstream of c-met receptor activation.
Further support for the role of ARF6 activation in E-cadherin internalization came from the demonstra tion that expression of dominant-negative ARF6, ARF6(T27N), blocked the redistribution of E-cadherin into the cytoplasm upon disassembly of the AJs induced by Ca2+ depletion (Figure 8C). In untransfected cells and in cells expressing wild-type ARF6, E-cadherin was redistributed to the perinuclear cytoplasm upon depletion of extracellular Ca2+. In contrast, in cells expressing ARF6(T27N), a majority of the E-cadherin remained at the cell surface despite the loss of Ca2+-dependent, E-cadherin-based cell–cell adhesion.
Effect of the ARF6 GTPase cycle on epithelial cell migration
Taken together, the studies described above suggest that ARF6 activation might serve to promote the migratory potential of epithelial cells. Thus, we examined the migratory capacity of cells stably expressing either wild-type ARF6, ARF6(Q67L), ARF6(Q67L:Q37E:S38I) or ARF6(T27N), compared with untransfected MDCK cells, using a transwell filter migration assay. For these experiments, cells were seeded on top of transwell filters and migration of cells toward chemoattractant in the lower chamber was monitored. Assessment of the number of cells that migrated revealed that ARF6(Q67L)-expressing cells exhibited a dramatic increase in cell migration compared with control untransfected cells or cells expressing wild-type ARF6. The effect of the ARF6–GTP mutant on cell migration was observed only 12–16 h after cells were seeded on filters. No change in migratory capacity was observed at earlier time points (within 6–8 h). This observation is significant because it implies that the effect of ARF6–GTP on epithelial cell migration is probably not due to a direct effect on the ‘migration machinery’ per se (such as integrin receptors or other focal adhesion components), but rather due to its effect on AJs. In contrast to the effect of ARF6–GTP, expression of dominant-negative ARF6(T27N) blocked the basal migratory capacity of MDCK cells (Figure 9), suggesting a requirement for ARF6 activation in MDCK cell migration. Finally, cells expressing the activated mutant of ARF6, incapable of membrane ruffling, did not lead to increased migratory capacity of MDCK cells (Figure 9). This result implies that AJ disassembly is not sufficient for increased cell migration, and that other factors, such as cytoskeletal remodeling, are also required.
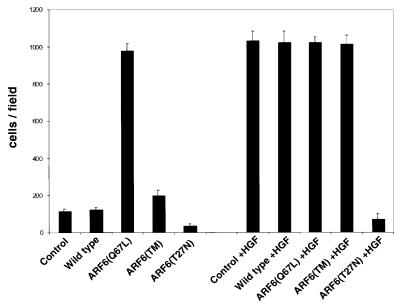
Fig. 9. The ARF6 GTPase cycle modulates epithelial cell migration. Cells stably expressing either wild-type ARF6, ARF6(Q67L), ARF6 triple mutant [ARF6(TM)] or ARF6(T27N) were grown on transwell filters and their migratory potential was assessed in the presence or absence of HGF as indicated. The number of cells/field that migrated through the filter after 16 h was counted. The mean of six separate fields is shown. The data are representative of four independent experiments.
We also examined the effect of the ARF6 mutants described above on HGF-induced increase in migratory potential. As shown in Figure 9, HGF enhanced the migratory potential of MDCK cells to a similar extent to ARF6(Q67L). In contrast to the lack of enhanced migratory capacity of cells expressing the ARF6 effector domain mutant, HGF treatment of these cells promoted cell migration to the same extent as ARF6(Q67L), suggesting that HGF may stimulate other signaling events that function in concert with ARF6–GTP to promote cell migration. However, expression of ARF6(T27N) abrogated HGF-induced cell migration. Taken together, the above studies indicate a requirement for ARF6 activation during ‘basal and stimulated’ epithelial cell migration.
ARF6 functions downstream of pp60v-src activation during AJ turnover
As stated earlier, HGF-induced disassembly of AJs is accompanied by tyrosine phosphorylation and activation of c-met, a receptor tyrosine kinase (Bottaro et al., 1991). c-met activation, in turn, stimulates pp60v-src-mediated tyrosine phosphorylation of the catenins, a process that has been shown to promote AJ disassembly and cell scattering (Behrens et al., 1993). pp60v-src activity has also been shown to promote disassembly of tight junctions via the phosphorylation of ZO-1 (Takeda and Tsukita, 1995). To determine whether the effect of ARF6–GTP on AJs was coupled to tyrosine phosphorylation events that accompany loss of cell–cell adhesion, we examined the effect of ARF6(Q67L) on AJ disassembly in the presence of genestein and herbimycin. We found that genestein, a general tyrosine kinase inhibitor, and herbimycin, a Src kinase inhibitor, had no effect on ARF6(Q67L)-induced redistribution of E-cadherin (data not shown). These results suggested that ARF6–GTP activity was either downstream of or parallel to pp60v-src-mediated signaling pathways. To investigate the possibility of a linear relationship between pp60v-src and ARF6, we utilized a previously characterized MDCK cell line stably transfected with a temperature-sensitive mutant of v-Src (Behrens et al., 1993). As described, growth of MDCK-pp60v-src cells at the permissive temperature (35°C) induces massive cell scattering, as opposed to compact colonies, which are formed at 41°C, the non-permissive temperature (see Figure 10A and B). As expected, addition of herbimycin blocked pp60v-src-mediated cell scattering (Figure 10C). Expression of ARF6(T27N) also completely abolished v-Src-mediated cell scattering at permissive temperatures (Figure 10D). Noticeably, the ARF6(T27N) colonies were less organized since the cells appeared somewhat less adherent (Figure 10, arrows). This may be due to v-Src-mediated phosphorylation of proteins at cell–cell junctions. Furthermore, using the detergent-solubility assay described earlier, we found increased levels of ‘soluble’ (i.e. activated) endogenous ARF6 in MDCK-pp60v-src cells at permissive temperatures compared with the soluble ARF6 pool at non-permissive temperature (data not shown). These investigations demonstrate that activation of ARF6 is downstream of v-Src activation during AJ disassembly, and that ARF6–GTP-mediated internalization of junctional components is a critical step during AJ disassembly and cell scattering.
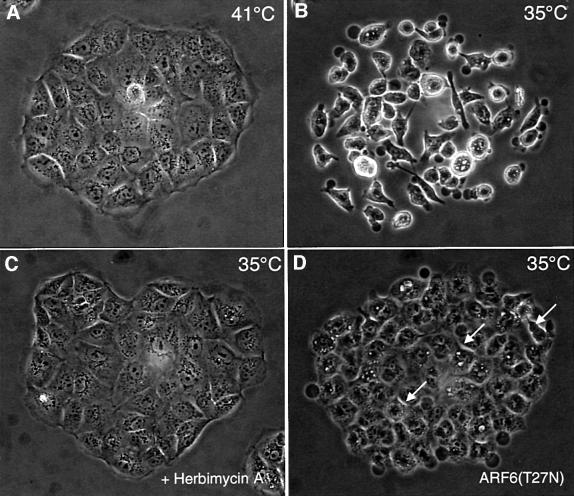
Fig. 10. Dominant-negative ARF6 blocks pp60v-src-induced cell scattering. Compact colonies of MDCK-pp60vsrc cell lines grown at 41°C, as visualized by phase-contrast microscopy, are shown (A). Alterations in colony morphology induced by temperature shift to 35°C, in the absence (B) or presence (C) of herbimycin, are shown. (D) Phase-contrast image of MDCK-pp60vsrc cell lines expressing ARF6(T27N) at permissive temperature (35°C). Both herbimycin and dominant-negative ARF6 block Src-induced cell scattering. Cells in the latter colonies are less organized and appear to have ruffled edges (see arrows).
Discussion
In this study, we have investigated the role of the ARF6 GTPase cycle in epithelia. We have shown that sustained activation of ARF6 by expression of a GTPase-defective mutant, ARF6(Q67L), in MDCK cells resulted in the disassembly of AJs and ruffling of the lateral plasma membrane. Our data also revealed that these processes, namely AJ disassembly and membrane ruffling, are mutually exclusive. While ARF6–GTP-induced membrane ruffling is mediated by PIP2 generation, the effect of ARF6–GTP on AJs was independent of its effects on actin remodeling and phospholipid metabolism. In addition, ARF6–GTP-induced disassembly of AJs correlated with increased migratory capacity of cells. In marked contrast to the phenotypes generated by constitutively activated ARF6, dominant-negative ARF6, ARF6(T27N), localized almost exclusively to AJs and appeared to enhance the epithelial phenotype. HGF promoted activation of endogenous ARF6, and expression of dominant-negative ARF6 blocked HGF-mediated internalization of cadherin-based junctional components into the cytoplasm and cell migration. Finally, we showed that ARF6 exerts its role downstream of v-Src activation during the disassembly of cell–cell adhesive contacts. The data presented here demonstrates that the ARF6 GTPase cycle is a critical regulator of the acquisition of the migratory phenotype in epithelia and documents an essential role for ARF6-regulated membrane traffic in the disassembly of cell–cell junctions.
ARF6–GTP-induced AJ disassembly and actin remodeling: effects uncoupled
In addition to its well characterized role in actin remodeling, PIP2 has also been shown to have a role in various membrane traffic events (Martin, 1998). For instance, endosomal vesicles accumulate in nerve terminals lacking synaptojanin, an inositol-5-phosphatase that releases phosphate and metabolizes PIP2 (Cremona and De Camilli, 2001). Consistent with previously reported findings in other cell types (Honda et al., 1999), we observed elevated levels of PIP2 in regions of membrane ruffles in MDCK cells expressing activated ARF6. However, there was no accumulation of PIP2 on ARF6–GTP vesicles that contained internalized AJ components. Thus, the accumulation of endocytic vesicles probably occurs independently of the effects of ARF6 on phospholipid metabolism. Further support for this contention is our finding that cadherin internalization and AJ disassembly can occur by expression of an activated ARF6 mutant incapable of membrane ruffling. Interestingly, labeling for PIP2 in these transfected cells was similar to the staining observed in untransfected cells (F.Palacios and C.D’Souza-Schorey, unpublished observations). The above findings demonstrate that the ARF6–GTP effector pathways that mediate AJ disassembly and ruffling of the lateral membrane are distinct in epithelial cells.
An obligate role for ARF6–GTP-induced membrane internalization during AJ disassembly
A depletion of extracellular Ca2+, phosphorylation of the c-met receptor, v-Src-mediated phosphorylation of various junctional components, and the depolymerization of the actin cytoskeleton have all been implicated in AJ disassembly (Gumbiner, 2000). ARF6–GTP-induced disassembly of AJs appears to be independent of v-Src-mediated phosphorylation events. Moreover, the demonstration that expression of the actin remodeling-defective ARF6–GTP mutant was able to induce AJ disassembly rules out actin remodeling as a potential mechanism for the effect of ARF6 on AJs. However, the inhibition of E-cadherin internalization upon HGF treatment or Ca2+ depletion, by expression of dominant-negative ARF6, indicates that ARF6 regulates AJ assembly via its effect on membrane transport from the AJs to the perinuclear cytoplasm.
AJ assembly and turnover is a highly dynamic process
E-cadherin molecules at the cell surface are constitutively endocytosed and then recycled back to the lateral membrane (Le et al., 1999). This pool of recycling E-cadherin allows the cell to undergo rapid changes in morphology in response to several stimuli, such as HGF and EGF. The inhibition of HGF-induced internalization of E-cadherin from the cell surface, by dominant-negative ARF6, indicates that ARF6 activation is required for the cytoplasmic redistribution of E-cadherin during AJ disassembly. Furthermore, the block in HGF- and v-Src-induced cell scattering, by dominant-negative ARF6, points toward an obligatory role of ARF6-regulated membrane traffic in AJ disassembly and turnover. Taken together, our findings indicate that the ARF6 GTPase cycle modulates the trafficking of E-cadherin between the lateral membrane and intracellular compartments, and hence can dictate the availability of cycling E-cadherin molecules for assembly into AJs.
The ARF6 GTPase cycle has an essential role in epithelial cell migration
The ability of ARF6–GTP to induce AJ disassembly and membrane ruffling points toward ARF6 as a potential regulator of epithelial cell migration. Consistent with this contention, we have demonstrated that sustained activation of ARF6, by expression of a GTP-bound ARF6 mutant, leads to a dramatic stimulation in the migratory potential of epithelia using transwell filter assays. Expression of dominant-negative ARF6 abolished HGF-induced cell migration. From these studies, we conclude that there is an obligatory requirement for ARF6 activation during HGF migration. As indicated earlier (see Results), the effect of the ARF6–GTP mutant on cell migration was not acute and was observed no earlier than 12 h after cells were seeded on filters. A likely explanation for these observations is that the ARF6–GTP-induced increase in migration is due to its effect on AJ disassembly. The disassembly of AJs may cause a breakdown of other cell–cell junctions, since the assembly of other junctional complexes, such as desmosomes and tight junctions, depends on activation of the cadherin-based system (Gumbiner, 1988; Wheelock and Jensen, 1992; Marrs et al., 1995). This would lead to a marked increase in migratory capacity over a more prolonged time period. The observation that the activated ARF6 mutant incapable of membrane ruffling had no significant effect on migratory capacity indicates that AJ disassembly alone is not sufficient for ARF6-induced migration. Peripheral membrane and cytoskeletal rearrangements may also be required for an overall increase in migratory capacity. However, we cannot rule out the possibility that this ARF6-effector domain mutant is incapable of cellular functions other than surface ruffling which may also be required for cell migration. Interestingly, cells expressing the ARF6-effector domain mutant showed enhanced migratory potential in response to HGF treatment. Thus, HGF probably activates ARF6-independent pathways that act synergistically with the ARF6-effector domain mutant to promote cell migration.
ARF6–GTP in polarized and non-polarized cells: parallel roles?
Figure 11 describes our working model for the regulation of AJ turnover by the ARF6 GTPase cycle in epithelial cells. We propose that nucleotide exchange on ARF6 at the AJs occurs in response to extracellular stimuli and downstream of Src activation. Activation of ARF6 promotes AJ disassembly and ruffling of the lateral membrane, although these processes are mediated by distinct effector molecules. GTP hydrolysis on ARF6 (by ARF6 GAPs) at the perinuclear compartment and at the lateral membrane would then promote the redistribution of E-cadherin to reassemble AJs. In fact, plasma membrane and cytoplasmic ARF6 GAPS, ACAP1 and ACAP2, respectively, have been described recently (Jackson et al., 2000). In addition, ARF6 may be sorted to subapical compartments (since wild-type ARF6 and its mutants were detected at the subapical region). At the apical end, ARF6 may regulate endocytic events, as described previously (Altschuler et al., 1999), or may serve as a membrane reservoir for delivery of additional cargo back to the basolateral membrane. The subapical membrane route through which ARF6 traffics in MDCK cells is still unclear and will require additional investigation.
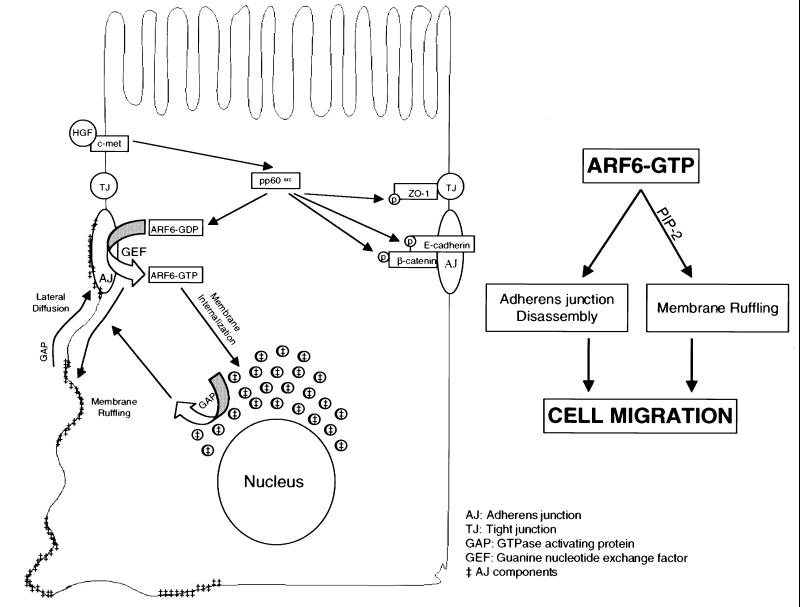
Fig. 11. Working model for the regulation of AJ turnover by the ARF6 GTPase cycle. Activation on ARF6 at the AJs (by GEFs) occurs in response to extracellular stimuli and downstream of Src activation. This, in turn, promotes AJ disassembly and ruffling of the lateral membrane, although these processes are mediated by distinct effector molecules. GTP hydrolysis on ARF6 (by GAPS) at perinuclear compartments and the lateral membrane results in redistribution of E-cadherin to and at the lateral membrane to reassemble AJs. The scheme alongside shows that the concerted effect of ARF6–GTP on AJ disassembly and membrane ruffling is required for epithelial cell migration.
It is tempting to draw comparisons between the role of ARF6 in epithelial cells described above and its previously characterized role in other cell types, such as CHO and HeLa cell lines (Radhakrishna and Donaldson, 1997; D’Souza-Schorey et al., 1998). In the latter cells, nucleotide exchange on ARF6 directs the outward flow of recycling endosomal membrane to the leading edge and elicits peripheral actin rearrangements. In turn, GTP hydrolysis triggers the recruitment of ARF6 back to recycling endosomes. In epithelia, nucleotide exchange on ARF6 induces the recycling of AJ components to perinuclear compartments and ruffling of the lateral plasma membrane. GTP hydrolysis triggers the redistribution of junctional components back to the lateral membrane and the reassembly of AJs. What then is the underlying function of ARF6 in different cell types? It is possible that the ARF6 GTPase cycle serves to promote the acquisition of migratory potential. In non-polarized cells, ARF6 directs the outward flow of endosomal membrane, thereby modulating the structural organization and composition of the leading edge. In epithelia, it induces the disassembly of AJs by its effect on membrane traffic. Thus, the ARF6 GTPase cycle is probably a critical determinant of the motile phenotype in different cell types.
The Rho family of GTPases have also been shown to regulate cell–cell adhesion, probably via their effects on junctional components and/or the actin cytoskeleton (Sander and Collard, 1999). For instance, activation of the Rac1 GTPase in MDCK cells has been shown to enhance cadherin-mediated cell–cell contacts (Hordijk et al., 1997). Analogous to previous studies in HeLa cells and CHO cells, wherein activation of ARF6 was shown to promote redistribution of endosomal Rac1 to the cell surface (Radhakrishna et al., 1999; Boshans et al., 2000), it is possible that, in epithelia, activation of ARF6 at the AJ will promote internalization of Rac1 into endosomal compartments and, as a result, downregulate the Rac1–GTP response. Investigations on how ARF6 function is coupled to the effects of the Rho family GTPases are under way. These studies will provide vital information toward the understanding of signaling pathways that regulate cell migration/invasion, a major challenge in cancer cell biology.
Materials and methods
Generation of retroviral expression plasmids and amphotropic retrovirus
The retroviral expression plasmids were constructed by subcloning cDNAs encoding ARF6, ARF6(Q67L) and ARF6(T27N) into the expression vector pLZRS-IRES-eGFP. The vector pLZRS-IRES-eGFP encodes a multicloning site, followed by an IRES (internal ribosome entry site) sequence and the enhanced GFP gene. For plasmid selection, eGFP was replaced by the neomycin resistance gene (pLZRS-IRES-neo). Expression plasmids encoding HA-tagged ARF6, ARF6(Q67L) and ARF6(T27N) were generated by PCR-based amplification of ARF6 cDNAs using a 5′ primer containing an EcoRI restriction site and the N-terminal sequence of ARF6, and a 3′ primer containing an XhoI restriction site, followed by complementary sequence of the HA epitope and the C-terminal sequence of ARF6. The PCR product was subcloned into the EcoRI and XhoI sites of pLZRS-IRES-neo. The plasmids were used for the generation of amphotropic retrovirus using procedures described elsewhere (Michiels et al., 2000). The expression plasmid encoding PH–GFP was a kind gift from Dr Tamas Balla, NIH.
Mutagenesis
ARF6(Q67L:Q37E:S38I) was generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. HA-tagged ARF6(Q67L) cDNA was used as template and the following oligonucleotides (5′-GTTGAAGCTGGGCGAGAT CGTGACCACCATTC-3′ and 5′-GAATGGTGGTCACGATCTCGCCCAGCTTCAAC-3′) encoding mutations (underlined) were used as primers. The sequence was verified, and ARF6(Q67L:Q37E:S38I) cDNA was subcloned into pLZRS-IRES-neo for the generation of retrovirus, as described above.
Cell culture, transfection and generation of stable cell lines
MDCK II cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, penicillin and streptomycin. For cell transfections, 1 ml of freshly thawed retrovirus-containing supernatant mixed with 1% DOTAP liposomal transfection reagent (Boehringer Mannheim) was layered onto the cells in 35 mm dishes, and infection was allowed to proceed for 4 h at 37°C. Cells were gently washed in phosphate-buffered saline and then incubated in normal growth media. Twenty-four hours post-incubation, cells were trypsinized and seeded onto glass coverslips or on 0.4 µm pore size clear polyester transwell filters (Corning-Costar), followed by incubation for another 12–24 h. Stable cell lines expressing ARF6 wild-type and mutant cDNAs were generated by retroviral transduction, followed by selection of transfected cells using G418. Expression of the ARF6 proteins was less than a fold above endogenous protein expression (Figure 12).
Fig. 12. Expression of wild-type ARF6, ARF6(Q67L) and ARF6(T27N) in MDCK cells using retroviral expression. Lysates of MDCK cells stably expressing either HA-tagged wild-type ARF6, ARF6(Q67L) or ARF6(T27N) were resolved on SDS gels, transferred to nitrocellulose membrane and probed using immunoblotting procedures for either total ARF6 expression or for exogenous ARF6 using anti-HA monoclonal antibody. Expression of ARF6 in transfected cells was barely a fold above endogenous protein expression. Note that unlike the data in Figure 8B, exogenous protein did not migrate at a higher molecular weight compared with endogenous ARF6 since a lower percentage SDS gel was used.
MDCK cell lines stably transfected with a temperature-sensitive mutant of v-src (Birchmeier and Birchmeier, 1993) were grown at 41°C (non-permissive temperature for pp60v-src activity) in DMEM supplemented with 10% fetal bovine serum. Cells were then switched to 35°C for 4 h for experimentation at permissive temperatures. For the generation of ARF6(T27N)-expressing stable cell lines, cells were then transfected using the retroviral transfection system and further selected using G418 as described earlier.
Immunofluorescent labeling
Procedures used for immunofluorescent staining and microscopy were conducted as described previously (Boshans et al., 2000). In the case of filter-grown cells, immunolabeling was conducted in a transwell chamber and both sides of the filter were labeled as indicated. Anti-E-cadherin mouse monoclonal antibody (3B8) was a kind gift from Dr Warren Gallin (University of Alberta). The anti-mouse monoclonal antibody against ZO-1 (R26.4C) was obtained from the hybridoma bank, University of Iowa. Anti-β-catenin mouse monoclonal antibody was obtained from Transduction Laboratories. Anti-mouse monoclonal and anti-rabbit polyclonal antibodies against HA were obtained from BabCo. Anti-transferrin receptor mouse monoclonal was from Zymed. Anti-gp135 (3F2) was a kind gift of Dr George Ojakian. Rhodamine–phalloidin was obtained from Molecular Probes.
Cell migration assays
MDCK cells stably expressing either ARF6, ARF6(Q67L), ARF6(T27N) or ARF6(Q67L:Q37E:S38I) were grown in normal growth medium on 8 µm pore size polycarbonate transwell filters (Costar). Migration of cells into the lower chamber containing conditioned medium (Kleinman and Jacob, 1998) was followed every 4 h. After incubation overnight, the cells in the lower chamber were fixed and counted by phase-contrast microscopy. In a separate set of experiments, migration assays were carried out as described, except that 20 ng/ml HGF (Calbiochem) was added to medium in the upper chamber, 2 h after seeding cells on filters.
Triton X-100 solubility assay
Untransfected MDCK cells or cells stably expressing ARF6, ARF6(Q67L) or ARF6(T27N) were grown in 35 mm tissue culture dishes. Cells were then lysed in CSK-A buffer containing 0.5% Triton X-100 (Tsukamoto and Nigam, 1999) for 20 min on ice. Cell lysates were centrifuged at 14 000 r.p.m., and the pellet and supernatant were resolved by SDS–PAGE. β-catenin (Transduction Labs) or ARF6 (3F8.1) (Yang et al., 1998) distribution was assessed by immunoblot analysis.
Acknowledgments
Acknowledgements
We thank Dr Peter J.Peters for helpful discussions and critical reading of the manuscript, Drs William Gallin and George Okajian for antibodies, Rob van der Kammen, Dr Jim Marrs and Dr Julie Donaldson for helpful suggestions, Dr W.Birchmeier for the gift of the MDCK-pp60v-src cell line and Dr Tamas Balla for the expression plasmid encoding PH–GFP. F.P. is a recipient of a predoctoral fellowship and J.S. is the recipient of a postdoctoral fellowship from the Walther Cancer Institute. This work was supported in part by a grant from the American Heart Association and interim funds from the University of Notre Dame to C.D.-S.
References
- Al-Awar O., Radhakrishna,H., Powell,N.N. and Donaldson,J.G. (2000) Separation of membrane trafficking and actin remodeling functions of ARF6 with an effector domain mutant. Mol. Cell. Biol., 20, 5998–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler Y., Liu,S., Katz,L., Tang,K., Hardy,S., Brodsky,F., Apodaca,G. and Mostov,K. (1999) ADP-ribosylation factor 6 and endocytosis at the apical surface of Madin–Darby canine kidney cells. J. Cell Biol., 147, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J., Vakaet,L., Friis,R., Winterhager,E., Van Roy,F., Mareel,M.M. and Birchmeier,W. (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol., 120, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G., Cleton-Jansen,A.M., Nollet,F., de Leeuw,W.J., van de Vijver,M., Cornelisse,C. and van Roy,F. (1995) E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J., 14, 6107–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C. and Birchmeier,W. (1993) Molecular aspects of mesenchymal–epithelial interactions. Annu. Rev. Cell Biol., 9, 511–540. [DOI] [PubMed] [Google Scholar]
- Birchmeier W., Behrens,J., Weidner,K.M., Hulsken,J. and Birchmeier,C. (1996) Epithelial differentiation and the control of metastasis in carcinomas. Curr. Top. Microbiol. Immunol., 213, 117–135. [DOI] [PubMed] [Google Scholar]
- Boshans R.L., Szanto,S., van Aelst,L. and D’Souza-Schorey,C. (2000) ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol. Cell. Biol., 20, 3685–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottaro D.P., Rubin,J.S., Faletto,D.L., Chan,A.M., Kmiecik,T.E., Vande Woude,G.F. and Aaronson,S.A. (1991) Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science, 251, 802–804. [DOI] [PubMed] [Google Scholar]
- Chavrier P. and Goud,B. (1999) The role of ARF and Rab GTPases in membrane transport. Curr. Opin. Cell Biol., 11, 466–475. [DOI] [PubMed] [Google Scholar]
- Chen Y.T., Stewart,D.B. and Nelson,W.J. (1999) Coupling assembly of the E-cadherin/β-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J. Cell Biol., 144, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O. and De Camilli,P. (2001) Phosphoinositides in membrane traffic at the synapse. J. Cell Sci., 114, 1041–1052. [DOI] [PubMed] [Google Scholar]
- Drubin D.G. and Nelson,W.J. (1996) Origins of cell polarity. Cell, 84, 335–344. [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C., Boshans,R.L., McDonough,M., Stahl,P.D. and Van Aelst,L. (1997) A role for POR1, a Rac1-interacting protein, in ARF6-mediated cytoskeletal rearrangements. EMBO J., 16, 5445–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza-Schorey C., van Donselaar,E., Hsu,V.W., Yang,C., Stahl,P.D. and Peters,P.J. (1998) ARF6 targets recycling vesicles to the plasma membrane: insights from an ultrastructural investigation. J. Cell Biol., 140, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. (1988) Cadherins: a family of Ca2+-dependent adhesion molecules. Trends Biochem. Sci., 13, 75–76. [DOI] [PubMed] [Google Scholar]
- Gumbiner B.M. (2000) Regulation of cadherin adhesive activity. J. Cell Biol., 148, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschuh G. et al. (1999) Tumour-associated E-cadherin mutations alter cellular morphology, decrease cellular adhesion and increase cellular motility. Oncogene, 18, 4301–4312. [DOI] [PubMed] [Google Scholar]
- Honda A. et al. (1999) Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell, 99, 521–532. [DOI] [PubMed] [Google Scholar]
- Hordijk P.L., ten Klooster,J.P., van der Kammen,R.A., Michiels,F., Oomen,L.C. and Collard,J.G. (1997) Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science, 278, 1464–1466. [DOI] [PubMed] [Google Scholar]
- Jackson T.R., Brown,F.D., Nie,Z., Miura,K., Foroni,L., Sun,J., Hsu,V.W., Donaldson,J.G. and Randazzo,P.A. (2000) ACAPs are arf6 GTPase-activating proteins that function in the cell periphery. J. Cell Biol., 151, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei T., Matozaki,T., Sakisaka,T., Kodama,A., Yokoyama,S., Peng,Y.F., Nakano,K., Takaishi,K. and Takai,Y. (1999) Coendocytosis of cadherin and c-Met coupled to disruption of cell–cell adhesion in MDCK cells—regulation by Rho, Rac and Rab small G proteins. Oncogene, 18, 6776–6784. [DOI] [PubMed] [Google Scholar]
- Kleinman H.K. and Jacob,K. (1998) Cell motility. In Bonifacino,J.S., Dasso,M., Harford,J.B., Lippincott-Schwartz,J. and Yamada,K.M. (eds), Current Protocols in Cell Biology. Vol. 1. John Wiley & Sons, Inc., New York, NY.
- Le T.L., Yap,A.S. and Stow,J.L. (1999) Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol., 146, 219–232. [PMC free article] [PubMed] [Google Scholar]
- Marrs J.A., Andersson-Fisone,C., Jeong,M.C., Cohen-Gould,L., Zurzolo,C., Nabi,I.R., Rodriguez-Boulan,E. and Nelson,W.J. (1995) Plasticity in epithelial cell phenotype: modulation by expression of different cadherin cell adhesion molecules. J. Cell Biol., 129, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T.F. (1998) Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation and membrane trafficking. Annu. Rev. Cell Dev. Biol., 14, 231–264. [DOI] [PubMed] [Google Scholar]
- Michiels F., van der Kammen,R.A., Janssen,L., Nolan,G. and Collard,J.G. (2000) Expression of Rho GTPases using retroviral vectors. Methods Enzymol., 325, 295–302. [DOI] [PubMed] [Google Scholar]
- Radhakrishna H. and Donaldson,J.G. (1997) ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol., 139, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna H., Al-Awar,O., Khachikian,Z. and Donaldson,J.G. (1999) ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci., 112, 855–866. [DOI] [PubMed] [Google Scholar]
- Rajasekaran A.K., Hojo,M., Huima,T. and Rodriguez-Boulan,E. (1996) Catenins and zonula occludens-1 form a complex during early stages in the assembly of tight junctions. J. Cell Biol., 132, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E.E. and Collard,J.G. (1999) Rho-like GTPases: their role in epithelial cell–cell adhesion and invasion. Eur. J. Cancer, 35, 1905–1911. [DOI] [PubMed] [Google Scholar]
- Schmidt J.W., Piepenhagen,P.A. and Nelson,W.J. (1993) Modulation of epithelial morphogenesis and cell fate by cell-to-cell signals and regulated cell adhesion. Semin. Cell Biol., 4, 161–173. [DOI] [PubMed] [Google Scholar]
- Stella M.C. and Comoglio,P.M. (1999) HGF: a multifunctional growth factor controlling cell scattering. Int. J. Biochem. Cell Biol., 31, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Stevenson B.R., Siliciano,J.D., Mooseker,M.S. and Goodenough,D.A. (1986) Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol., 103, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H. and Tsukita,S. (1995) Effects of tyrosine phosphorylation on tight junctions in temperature-sensitive v-src-transfected MDCK cells. Cell Struct. Funct., 20, 387–393. [DOI] [PubMed] [Google Scholar]
- Thiery J.P., Duband,J.L. and Delouvee,A. (1982) Pathways and mechanisms of avian trunk neural crest cell migration and localization. Dev. Biol., 93, 324–343. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T. and Nigam,S.K. (1999) Cell–cell dissociation upon epithelial cell scattering requires a step mediated by the proteasome. J. Biol. Chem., 274, 24579–24584. [DOI] [PubMed] [Google Scholar]
- Varnai P. and Balla,T. (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol., 143, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen S.J., Bruyneel,E.A., Bracke,M.E., De Bruyne,G.K., Vennekens,K.M., Vleminckx,K.L., Berx,G.J., van Roy,F.M. and Mareel,M.M. (1995) Transition from the noninvasive to the invasive phenotype and loss of α-catenin in human colon cancer cells. Cancer Res., 55, 4722–4728. [PubMed] [Google Scholar]
- Wheelock M.J. and Jensen,P.J. (1992) Regulation of keratinocyte intercellular junction organization and epidermal morphogenesis by E-cadherin. J. Cell Biol., 117, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.Z., Heimberg,H., D’Souza-Schorey,C., Mueckler,M.M. and Stahl,P.D. (1998) Subcellular distribution and differential expression of endogenous ADP-ribosylation factor 6 in mammalian cells. J. Biol. Chem., 273, 4006–4011. [DOI] [PubMed] [Google Scholar]
- Yap A.S., Brieher,W.M. and Gumbiner,B.M. (1997) Molecular and functional analysis of cadherin-based adherens junctions. Annu. Rev. Cell Dev. Biol., 13, 119–146. [DOI] [PubMed] [Google Scholar]