

Abstract
Appropriate nuclear membrane structure is important for all eukaryotic organisms as evidenced by the numerous human diseases and alterations in gene expression caused by inappropriate targeting of proteins to the inner nuclear membrane (INM). We report here the first genome-wide screen to identify proteins functioning in INM targeting. We transformed to near completion the 4850 members of the Saccharomyces cerevisiae deletion collection of unessential genes in the 96-well format with a plasmid encoding a reporter protein, Trm1-II-GFP, which normally resides at the INM. We found that deletion of genes encoding subunits of the N-terminal acetyltransferase, NatC, cause mislocation of Trm1-II-GFP from the INM to the nucleoplasm. Mass spectroscopic analysis indicates that Trm1-II-GFP is N-acetylated. N-terminal mutations of Trm1-II-GFP predicted to ablate N-acetylation cause nucleoplasmic location, whereas a variant with an N-terminal alteration predicted to allow N-acetylation by NatC is located at the INM, providing genetic support that Trm1p-II N-acetylation is necessary for its subnuclear INM location. However, because N-acetylation appears not to be sufficient for INM targeting, it may provide a necessary role for INM targeting by affecting Trm1-II-GFP structure and exposure of cis-acting INM targeting motifs. We also discovered that YIL090W/Ice2p, an integral membrane protein located in the endoplasmic reticulum, is necessary for efficient targeting of Trm1-II-GFP to the INM. YIL090W/Ice2p may serve as a tether for INM proteins or as a regulator of INM tethers. Our methodology can be extrapolated to obtain genome-wide perspectives of mechanisms necessary to achieve appropriate subcellular and/or suborganellar location for any resident protein.
THE nucleus is separated from the cytosol by a double membrane. Exchange of macromolecules between the nuclear interior and the cytoplasm occurs through nuclear pores, proteinaceous aqueous channels that connect the membranes. Appropriate nuclear membrane structure is very important for all organisms as evidenced by the numerous human diseases, such as Emery-Dreifuss muscular dystrophy and Hutchison-Gilford Progeria syndrome (reviewed in Gruenbaum et al. 2003), and by numerous alterations in gene expression in yeast (reviewed in Taddei et al. 2004) caused by inappropriate targeting of proteins that normally reside at the inner nuclear membrane (INM). In addition to the INM and outer nuclear membrane (ONM) and nuclear pore complexes (NPC), the nucleus contains other subnuclear structures including the nucleolus, cajal bodies, speckles, gems, and the nuclear matrix (reviewed in Lamond and Sleeman 2003). Because membranes do not separate the various subnuclear compartments from one another, their structure, biogenesis, and maintenance likely result from poorly described macromolecular interactions. Here we investigate the mechanisms that target/tether proteins to the INM.
The nuclear membrane is highly dynamic and has a complex structure. In higher eukaryotes it is disassembled and reassembled each cell division, but in organisms such as budding yeast the nuclear membrane remains intact during mitosis. This difference perhaps eliminates some of the dynamics that render analyses of nuclear membrane organization particularly difficult in higher eukaryotes. The INM is composed of numerous integral transmembrane proteins as well as peripherally associated proteins. It has been proposed that integral INM proteins achieve their location via a “diffusion retention” mechanism (reviewed in Holmer and Worman 2001). Accordingly, proteins transit from the endoplasmic reticulum (ER) to the contiguous ONM and then access the INM, likely via nuclear pore lateral channels. Integral INM proteins are prevented from returning to the ONM and ER by interactions with macromolecules such as the lamins and/or chromatin-binding proteins. Sorting of peripherally associated INM proteins is less well understood, but there is evidence that some of these proteins are tethered through protein-protein interactions with integral INM proteins (reviewed in Chu et al. 1998). Our focus is on the proteins that are peripherally associated with the INM.
The INM is complex and may be composed of functionally distinct domains since, in yeast, some actively transcribed genes are cotranscriptionally recruited to the part of the INM where NPCs are located (Casolari et al. 2004), whereas silenced DNA, such as telomeres and the transcriptionally quiescent HML and HMR mating-type loci, are preferentially located at pore poor areas (Taddei et al. 2004). NPCs contain ∼30 proteins, and all but 1 or 2 lack transmembrane domains (Rout et al. 2000; Cronshaw et al. 2002). Of INM peripheral proteins not associated with nuclear pores, the vertebrate lamins are the best characterized. Yeast homologs of the lamins have not been identified. However, numerous yeast proteins that are uniformly distributed around the INM are not pore associated (Huh et al. 2003).
Saccharomyces cerevisiae Trm1p is a tRNA-specific m22G methyltransferase catalyzing transfer of two methyl groups to a subset of G26 positions on tRNAs (Hopper et al. 1982). TRM1 encodes two proteins; Trm1p-I starts at the first AUG and modifies only mitochondrial tRNAs whereas Trm1p-II starts at the second AUG and modifies tRNAs encoded by both mitochondrial and nuclear genomes (Ellis et al. 1986; Li et al. 1989). Employing indirect immunofluorescence (IF) and a Trm1p-specific antibody, Rose et al. (1992) located Trm1p-II to the nuclear rim. Trm1p-II has the characteristics of a peripheral membrane protein because it is released from the nuclear membrane by treatment with salt and nonionic detergents (Rose et al. 1995) and because it lacks transmembrane domains. Although it is not possible to distinguish between the yeast ONM and the INM by light microscopy, Trm1p-II has been proposed to be located on the inner membrane because G26 modification occurs on precursor tRNAs restricted inside the nucleus by alteration of the tRNA export machinery (Etcheverry et al. 1979). Moreover, Trm1p-II contains a nuclear localization sequence (NLS) able to deliver passenger proteins to the nucleoplasm. Mutation of this NLS prevents endogenous Trm1p-II from associating with the nucleus and causes the protein to accumulate in the cytoplasm (Rose et al. 1992). Finally, as we show here, when Trm1p-II fails to interact with the nuclear envelope, it accumulates in the nucleoplasm.
As Trm1p-II is one of the best-studied members of the class of yeast proteins associated with the pore-free domain of the INM, we employed it as a reporter to conduct a genome-wide investigation to discover mechanisms involved in associating peripheral proteins with the INM. We uncovered two categories of gene products that affect the process: subunits of the N-terminal acetyltransferase (Nat)C heterotrimeric N-acetyltransferase and an ER-located integral membrane protein encoded by YIL090W/ICE2.
MATERIALS AND METHODS
Strains and media:
Yeast strains BY4741 (MATa his3Δ leu2Δ met15Δ ura3Δ) and BY4742 (MATα his3Δ leu2Δ lys2Δ ura3Δ) are the parents to the deletion collections (Winzeler et al. 1999; ResGen). The yeast collection with GST-ORF-containing plasmids was obtained from E. Phizicky (Martzen et al. 1999). Yeast strains were maintained on YEPD media with or without G418 (0.2 mg/ml) or on synthetic defined media lacking appropriate nutritional ingredients. Escherichia coli DH5α was used for propagation of recombinant DNA plasmids and was maintained in YT media with appropriate antibiotics.
Plasmids:
Most PCR reactions were carried out using Pfu DNA polymerase (Stratagene, La Jolla, CA). DNAs were ligated with T4 DNA ligase (New England Biolabs, Beverly, MA). The Pennsylvania State University (PSU) College of Medicine Macromolecular Core Facility generated all oligonucleotides. All constructs were sequenced by the same facility. pRS415-TRM1-II-GFP is a derivative of pGT554 (Li et al. 1989) in the pRS415 plasmid backbone (Sikorski and Hieter 1989). It encodes Trm1-II-GFP under the control of its endogenous regulatory sequences (details of the construction available upon request). pTRM1-II-GST: pGP54a is pRS416 containing a GAL1-GAL10 regulatory region (gift from J. Hopper, PSU College of Medicine). The sequence encoding Schistosoma japonicum GST was amplified by PCR using pYEX-GST (Martzen et al. 1999) as the template and inserted into pGP54a digested with SacI and XhoI to generate pGP54aGST. The TRM1 ORF beginning at the second AUG was amplified from genomic DNA (BY4741) with oligonucleotides introducing EcoRI and HindIII sites. The PCR product was cloned into the pTOPO-BluntII-TOPO cloning vector (Stratagene). EcoRI and HindIII were employed to transfer TRM1 into pGP54aGST. pGEMT-A1-4 and pGEMTA1-6 were generated to facilitate mutagenesis by subcloning a 1.4-kb TRM1 fragment spanned by two SpeI sites into pGEMT (Promega, Madison, WI), using Taq (Promega). The plasmids were utilized as templates for reverse PCR amplification and site-directed mutagenesis described below and then the mutant regions were exchanged with the corresponding fragment of pRS415-TRM1-II-GFP. pRS415-TRM1-II-ΔL2-GFP contains a deletion of the TRM1-II second codon. It was generated by reverse PCR using pGEMT-A1-4 as the template. pRS415-TRM1-II-K3 → E3-GFP and pRS415-TRM1-II-L2 → F2 were obtained using pGEMTA1-6 as the template. The mutations were introduced by employing the QuikChange II kit (Stratagene). pRS415-TRM1-II-(NatC)-GFP encodes Trm1p-II with an ectopic NatC modification sequence. It was generated by reverse PCR using pGEMT-TRM1-II-ΔL2 as the template, introducing four codons (underlined) of the PUP2 ORF N terminus at codon 2, encoding the sequence MFLTRK. pRS415-TRM1-II-(NatA)-GFP encodes Trm1p-II with an N-terminal ectopic NatA modification sequence. It was generated by reverse PCR using pGEMT-TRM1-IIΔL2 as the template introducing 4 N-terminal codons from RPS0A/YST1, a known NatA substrate, at codon 2, generating the sequence MSLPAK. To generate YIL090W-GST, YIL090W was amplified by PCR using BY4741 genomic DNA as template and oligonucleotide primers with flanking XmaI sites. The product was cloned into pCR-BluntII-TOPO (Invitrogen, San Diego) to obtain pTOPO-YIL. The YIL090W ORF was transferred from pTOPO-YIL to pGP-54aGST using XmaI to obtain an in-frame fusion with GST.
96-well transformation:
The procedure is based on the LiAc transformation protocol (Ito et al. 1983). Strains were grown in YEPD + G418 in 96-well plates at 23° for 2 days and then collected employing a Jouan CR412 centrifuge with an adapter accommodating 96-well plates. The media were aspirated and cells were resuspended in 100 μl transformation mix (41% PEG, 0.2 m LiAc, 0.1 m DDT, 0.5 μg boiled single-stranded sheared salmon sperm DNA) and 1 μg plasmid DNA. After heat shock at 45° for 45–60 min, transformants were selected by transferring aliquots to new plates containing selection media and grown at 23° for 3–4 days. By this method ∼90% of the wells contain transformed cells.
IF and microscopy:
In vivo localization of Trm1-II-GFP was assessed by visualization through the fluorescein-5-isothiocyanate (FITC) channel of a Nikon Microphot-FX microscope. IF studies were conducted as previously described (Li et al. 1989; Tolerico et al. 1999). Trm1-II-GST was located following induction by addition of galactose (2% final concentration) to cultures grown with 2% raffinose employing mouse monoclonal anti-GST [GST(12); Santa Cruz] at a 1:500 dilution. Mouse monoclonal anti-Nsp1p (Tolerico et al. 1999) was obtained from J. Aris (University of Florida) and used at a dilution of 1:20,000. Cy3-conjugated goat anti-mouse IgG (Jackson ImunoResearch Labs, West Grove, PA) was used at a 1:400 dilution to locate the primary antibodies. Cells were counterstained with DAPI (0.1 μg/ml) to locate DNA. Images were captured with a SenSys charge-coupled device camera (Photometrics, Tucson, AZ) using QED software and assembled using Adobe Photoshop 5.0.
Protein purification:
Trm1-II-GST was purified from BY4741 as published (Phizicky et al. 2002). Protein was eluted from Glutathione Sepharose-4B resin (Amersham Biosciences, Arlington Heights, IL) in 3 ml extraction buffer and concentrated to 0.4 μg/μl using a Millipore centrifugal filter device (50K). Protein concentration was determined by the Coomassie assay (Pierce, Rockford, IL). Protein purity was assessed using the NuPage 10% gels tricine gels (Invitrogen) followed by Coomassie blue or silver staining. Protein identity was verified by Western analysis employing monoclonal anti-GST (B-14; Santa Cruz) as primary antibody.
Mass spectrometry:
Analyses were performed in the Mass Spectrometry/Proteomics Core Facility at the Pennsylvania State College of Medicine. Two micrograms of Trm1-II-GST protein was digested with Glu-C endoproteinase (Roche, Indianapolis) with a protease-to-sample ratio of 1/100 (w/w). Digestions were carried out in 50 mm NH4H2PO4 (pH 7.8) with 10% (v/v) acetonitrile (AcN) for 10–15 hr at 25°. Samples were separated on an Eksigent NanoflowLC separation system using a Microm Magic C18 column 5-μm 0.1 × 150-mm nanoflow column in 2% AcN, 0.1% trifluoroacetic acid (TFA), and then eluted with a linear gradient over 34 min from 2% AcN, 0.1% TFA to 90% AcN, 0.1% TFA at a flow rate of 90 nl/min. α-Cyanohydroxycinnamic acid (5 mg/ml, 70% CH3OH, 0.1% TFA) was used as the MALDI matrix and loaded into the syringe on an LC Packings-Dionex ProBot and was added to the sample eluate through a postcolumn T-junction. The ProBot deposited samples (300 nl sample eluate, 500 nl matrix solution) every 20 sec onto Applied Biosystems MALDI target plates. After drying the plates were placed in the autoloader of an Applied Biosystems 4700 Proteomics Analyzer MALDI TOF-TOF, and 1000 laser shots per sample spot were automatically collected from 40 randomly chosen spots within each sample spot. Before sample spectra were taken masses were calibrated for each sample plate using 6 spots containing five known calibrants.
RESULTS
A genome-wide approach to identify yeast genes involved in targeting proteins to the INM:
Less than 20% of the ∼6200 genes in the yeast genome are essential (Winzeler et al. 1999). A collection of 4850 yeast strains, each with an unessential gene replaced by an ORF encoding a drug-resistance marker, has been assembled in an ordered array (Winzeler et al. 1999). Our goal was to employ the entire deletion collection to uncover genes involved in targeting proteins to the INM.
We needed to overcome two obstacles to reach this goal. First, we had to identify an easily detected reporter protein residing at the INM. Trm1p-II appeared to be an ideal candidate. We employed a fusion protein that contains GFP fused to Trm1p-II after the last amino acid (aa) codon. Trm1-II-GFP locates to the nuclear rim exactly as endogeneous Trm1p-II (Figure 1B; Rose et al. 1992, 1995) and its location is easily assessed in living cells when expressed from a low-copy plasmid.
Figure 1.—

Establishment of a system to conduct a genome-wide screen for defects in targeting proteins to the INM. (A) Transformation in the 96-well plate format. To show transformation efficiency yeast cells in wells were pinned to solid media: left, mock transformation; right, transformation with pRS415-TRM1-II-GFP. (B) Trm1-II-GFP associates with the nuclear rim. BY4741, parent to the MATa deletion collection, was transformed with pRS415-TRM1-II-GFP and live cells were viewed by fluorescence microscopy. Bar, 5 μm.
The second obstacle was a means to introduce the plasmid into ∼4850 members of the deletion collection. We adapted the one-step transformation protocol (Ito et al. 1983) to the 96-well format (materials and methods). Employing this procedure ∼90% of the wells contain transformed cells (Figure 1A, right) with no background growth of cells from mock transformations (Figure 1A, left). Strains not transformed in the first round were retransformed in a second round; nearly all the 4850 strains were transformed. The location of Trm1-II-GFP in each strain was monitored by fluorescence microscopy.
The NatC N-terminal acetyltransferase complex is essential for targeting Trm1p-II to the INM:
Defects in gene products involved in nuclear import, nuclear structure, and targeting/tethering to the INM could lead to Trm1-II-GFP mislocation. We predicted that defects in the latter would make Trm1-II-GFP nucleoplasmic. Deletion of MAK31 (mak31Δ) was the first mutation we found that caused Trm1-II-GFP to become nucleoplasmic (Figure 2A, top). We verified the nucleoplasmic location as the GFP signal completely overlaps with the chromosomal DNA signal as determined by DAPI costaining (Figure 2B). The data provide strong support for the previous belief that Trm1p resides at the INM, rather than at the ONM, because an ONM-located protein would locate in the cytoplasm when released from the nuclear membrane.
Figure 2.—
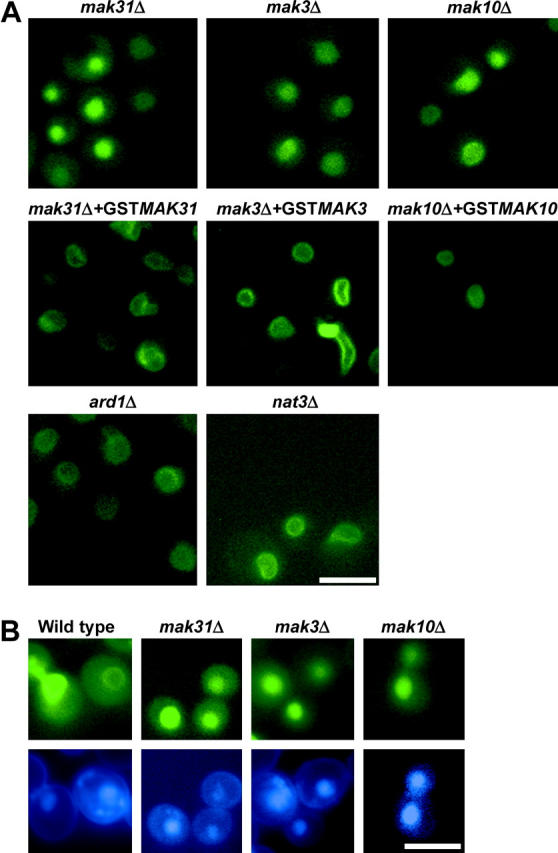
Localization of Trm1-II-GFP in mutant strains. (A) The NatC complex is required for Trm1-II-GFP INM localization, but NatA and NatB complexes are not. Top, deletions of the genes, mak31Δ, mak3Δ, and mak10Δ, encoding the NatC subunits. Middle, complementation of mak31Δ, mak3Δ, and mak10Δ with plasmids encoding GST-tagged versions restores Trm1-II-GFP rim localization. Bottom, ard1Δ and nat3Δ, encoding the catalytic subunits of the NatA and NatB complexes, respectively, do not affect Trm1-II-GFP localization. (B) Trm1-II-GFP is released into the nucleoplasm in mak31Δ, mak3Δ, and mak10Δ cells. Cells containing pRS415-TRM1-II-GFP were stained with DAPI (1 μg/ml) to show colocalization of Trm1p-II-GFP with nuclear DNA. Top, GFP; bottom, DAPI. Bars, 5 μm.
As a large number of strains in the deletion collection possess unintended genetic alterations (Hughes et al. 2000), we verified that mak31Δ causes Trm1-II-GFP mislocation by complementing mak31Δ with a plasmid encoding GST-Mak31p (Martzen et al. 1999). As a control the mak31Δ cells were also transformed with the vector alone. Trm1-II-GFP was nucleoplasmic in cells receiving vector (data not shown), but its INM location was restored in cells transformed with GST-MAK31 (Figure 2A, middle), complementing the phenotype and verifying that Mak31p is required to locate Trm1-II-GFP to the INM.
Mak31p is a subunit of the heterotrimeric NatC complex. NatC is an N-terminal acetyltransferase (NAT) and catalyzes N-acetyl addition to a subset of proteins. In addition to Mak 31p, Mak3p (the catalytic subunit) and Mak10p make up the complex and are required for activity (reviewed in Polevoda and Sherman 2003). This information led us to “jump ahead” in our systematic screen of the deletion collection to study the consequences of mak3Δ and mak10Δ upon the subnuclear distribution of Trm1-II-GFP. We found that deletion of MAK3 or MAK10 also mislocates Trm1-II-GFP to the nucleoplasm (Figure 2, A and B, top). We verified that the nucleoplasmic accumulation of Trm1-II-GFP is caused by deletion of MAK3 or MAK10 by complementation with plasmids encoding the corresponding genes using the yeast genomic GST-tagged collection (Figure 2A, middle). The data show that all three of the NatC subunits are necessary to target Trm1p-II to the INM. Trm1-II-GFP was also nucleoplasmic in the YPR050C deletion strain (data not shown). The putative YPR050C overlaps with MAK3 and causes a phenotype similar to that caused by NatC depletion. We surmise that inappropriate localization of Trm1-II-GFP in YPR050CΔ is actually due to MAK3 deletion.
There are four yeast NAT complexes each with different substrate specificity. Deletions of ARD1 and NAT3, the catalytic subunits of NatA and NatB complexes, respectively (Polevoda and Sherman 2003), do not cause nucleoplasmic location of Trm1-II-GFP (Figure 2A, bottom). Thus, the NatC complex may be specifically required for targeting/tethering Trm1-II-GFP to the INM.
Trm1-II-GFP could fail to associate with the INM in NatC-deficient cells because either NatC deficiency causes a general defect in nuclear membrane organization or NatC activity is required for tethering a subset of proteins to the INM. To differentiate between these two possibilities, we investigated whether NatC is important for subnuclear distribution of a different class of nuclear rim proteins, nucleoporins. Employing an Nsp1-specific antibody (Tolerico et al. 1999), we determined the location of this symmetrically distributed nucleoporin in wild-type and NatC-deficient cells. Although the NatC complex causes redistribution of the majority of the Trm1-II-GFP pool to the nucleoplasm, it has no apparent effect upon Nsp1p location (Figure 3; data shown for mak3Δ). Thus, there is no gross defect in nuclear membrane structure in NatC mutants.
Figure 3.—

mak3Δ does not affect Nsp1p localization. IF for Nsp1p shows punctate distribution surrounding DNA in wild-type and mak3Δ strains. Top, Nsp1p staining; bottom, merge of Nsp1p and DNA locations in the respective strains. Bar, 5 μm.
Trm1-II-GFP appears to be N-acetylated:
A requirement for NatC in targeting Trm1-II-GFP to the INM could be explained by at least three mechanisms. First, Trm1p-II could be a NatC substrate and its N-acetylation could function in INM targeting. Second, the Trm1p INM tether could be N-acetylated, enabling interaction with Trm1p. Finally, N-acetylation could act indirectly. Inspection of the Trm1p-II N-terminal sequence (all nomenclature here refers to aa starting at the second AUG; i.e., ORF codon 17 is referred to as aa1) revealed the sequence MLKA, which is a signature sequence of NatC substrates (Polevoda and Sherman 2003). We employed mass spectrometry (MS) to learn whether Trm1p-II is N-acetylated.
To purify Trm1p by affinity chromatography we created a novel construct because available constructs were either tagged at the N terminus, prohibiting N-acetylation, or predicted to encode only Trm1p-I, located in the mitochondria, rather than Trm1p-II. We generated a GAL-regulated version of Trm1p-II tagged at the C terminus with GST. Employing anti-GST and IF we determined that Trm1-II-GST is located predominately at the INM in wild-type cells (Figure 4A), but is nucleoplasmic in mak3Δ cells (data not shown), as is Trm1-II-GFP.
Figure 4.—

Trm1p-II is N-acetylated. (A) IF for wild-type cells expressing Trm1-II-GST. (B) SDS-PAGE of purified Trm1-II-GST (lanes 1 and 2) and marker proteins (lane 3). (C) N-terminal peptides identified by MS; arrows indicate identified Glu-C cut sites. Bar, 5 μm.
Each preparation of purified Trm1-II-GST contained primarily a single protein that migrated on SDS gels as predicted for the full-length fusion protein (Figure 4B) and confirmed to cross-react with anti-GST by Western analysis (data not shown). Previous studies showed that Trm1p has the same subnuclear location when overproduced (Rose et al. 1992, 1995), indicating that the INM tether for Trm1p is not limiting. Failure to copurify other proteins could mean that Trm1p is tethered to an integral INM protein that is not soluble under our purification conditions, or to a peripherally associated protein that is lost during purification, or that the Trm1p tether is not a protein.
Peptides obtained by proteolytic digestion of purified Trm1-II-GST were analyzed by MS. We did not detect the anticipated tryptic peptide Ac-MLK (or MLK), presumably due to its hydrophobicity. Therefore, we employed Glu-C, using conditions favoring cleavage after E and D. We detected a 2777-Da peptide that corresponds to the N-terminal 25 aa with an acetylated N terminus (Figure 4C), resulting from cleavage after D25. In addition to numerous other Trm1-II-GST-specific peptides, we detected a downstream peptide starting at F26, confirming that Glu-C cleaved Trm1-II-GST C-terminal to D25. Attempts to verify the 25-aa sequence by MS/MS failed to provide information. Data from protein sequencing were consistent with the presence of a blocked N terminus, presumably due to N-acetylation (data not shown). MS information for Trm1p-II-GST from mak3Δ cells was uninformative because we did not detect the 25-aa peptide, with or without the N-acetyl moiety, even though the adjacent downstream peptide starting at F26 was present. Instead, we detected peptides that could be accounted for by limited N-terminal proteolysis (data not shown), perhaps indicative of N-terminal protection by the N-acetyl moiety, either in vivo or in vitro during protein purification.
N-acetylation is necessary, but may not be sufficient, for targeting Trm1p-II to the INM:
Although the NatC complex is necessary for locating Trm1-II-GFP to the INM and Trm1-II-GST appears to possess an N-acetyl moiety, the fact that there are many potential NatC substrates raises the caveat that Trm1p-II N-acetylation may be merely coincidental. To address this, we generated Trm1-II-GFP variants that should not be capable of serving as NatC substrates and determined the consequences of these changes upon Trm1-II-GFP subnuclear distribution.
The NatC complex acetylates a subset of proteins possessing ML, MI, MF, or MW termini. A basic aa at the second position is inhibitory (Polevoda and Sherman 2003). We deleted codon 2, changing MLKA to MKA (Trm1-II-GFPΔL2). The resulting protein is nucleoplasmic in wild-type cells (Figure 5A, bottom). Studies of model substrates have shown that a positive aa at position 3, as found in Trm1p-II, promotes modification by NatC (Polevoda and Sherman 2003). Moreover, for L-A virus Gag protein, an authentic NatC substrate, changing R at position 3 to E prohibits its N-acetylation (Tercero et al. 1993). We changed Trm1-II-GFP K3 to E. When Trm1-II-GFP K3 → E3 was expressed in wild-type cells, it was located in the nucleoplasm (Figure 5A, bottom). Thus, both single-aa modifications predicted to change Trm1-II-GFP from an ideal substrate to an unlikely NatC substrate caused mislocation to the nucleoplasm. The data provide genetic support for the model that Trm1-II-GFP N-acetylation is essential for INM targeting; however, because these experiments also changed the N terminus of Trm1p it is possible that the N-terminal sequence, independent of its role in promoting acetylation, is also important for INM targeting.
Figure 5.—
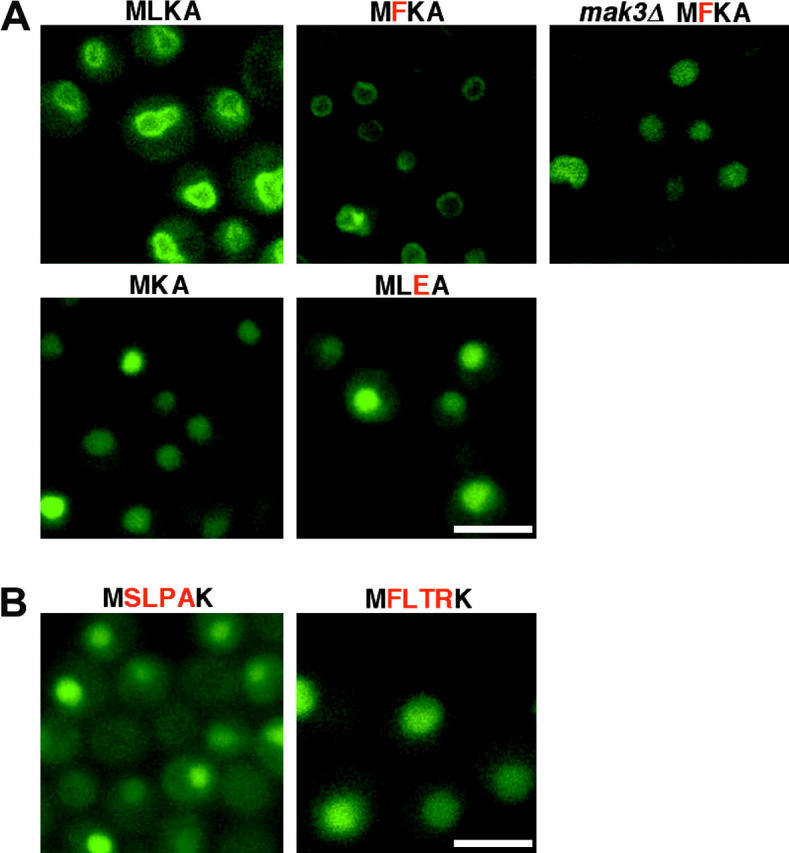
N-terminal amino acids important for Trm1-II-GFP INM targeting. (A) Only mutations predicted to disrupt N-acetylation cause mislocalization. MLKA, wild-type Trm1-II-GFP; MKA and MLEA, deletion of L2, and change of K3 to E3, respectively. Mutation predicted not to disrupt N-acetylation, MFKA, does not affect Trm1p-II-GFP association with the INM in wild-type cells; however, this protein is nucleoplasmic in mak3Δ cells. (B) Introduction of ectopic N-acetylation sequences at the N terminus does not restore INM location for Trm1-II-GFPΔL2. MSLPAK and MFLTRK, the NatA recognition sequence from Yst1p and the NatC sequence from Pup2p, respectively, appended to the N terminus of MKA. Bars, 5 μm.
To address whether the exact N-terminal Trm1p-II sequence is required for INM association, we changed L at position 2 to F. The resulting N-terminal sequence MFKA is a predicted substrate for NatC. When Trm1-II-GFP L2 → F2 was expressed in wild-type cells, it was located at the INM; however, when expressed in mak3Δ mutant cells, it was located in the nucleoplasm (Figure 5A, top). Thus, there is complete correlation between N-acetylation sequences that support N-acetylation and INM location and sequences predicted not to support N-acetylation that fail to locate at the INM, providing a genetic argument that N-terminal modification, rather than the exact sequence, is required for INM association.
If N-acetylation were the only requirement for INM association, then ectopic sequences predicted to provide this modification should direct proteins to the INM. To test this, our strategy was to provide a Trm1-II-GFP variant that does not associate with the INM by virtue of a single-aa change prohibiting modification by NatC (e.g., Trm1-II-GFPΔL2) with ectopic N-acetylation motifs. If N-acetylation is sufficient for INM association, then Trm1-II-GFP with ectopic N-acetylation motifs should reside at the INM. We chose two different 4-aa peptide sequences that are identical to the N termini of known substrates and added them to the Trm1-II-GFPΔL2 N terminus. This created MFLTRK, identical to the Pup2p N terminus, a known NatC substrate, and MSLPAK, identical to the Rps0Ap/Yst1p N terminus, a known NatA substrate (Polevoda and Sherman 2003). Neither Trm1-II-GFP variant located to the INM (Figure 5B). Either the variant Trm1p proteins are not N-acetylated as predicted, or, if they are, N-acetylation may not be sufficient for INM targeting, even though it is necessary. In support of the idea that N-acetylation may not be sufficient for INM targeting, it was previously reported that a Trm1p-II containing the first 197 of its 554 aa fused to β-galactosidase fails to associate with the INM (Li et al. 1989).
YIL090W/Ice2p, an integral membrane ER protein, is required for INM targeting:
Deletion of YIL090W/ICE2 resulted in a Trm1-II-GFP nucleoplasmic pool, although for this mutant there is residual Trm1-II-GFP at the nuclear rim (Figure 6A, top). To verify that ice2Δ is the cause of Trm1-II-GFP becoming nucleoplasmic, we transformed the Trm1-II-GFP-encoding plasmid into a strain with an independently generated deletion (MATα deletion collection) and found increased nucleoplasmic Trm1-II-GFP in the MATα YIL090WΔ/ice2Δ strain (Figure 6A, bottom, left). We also generated a new C-terminal GST-tagged Ice2p because the GST-tagged plasmid collection (Martzen et al. 1999) did not appear to contain a plasmid encoding full-length GST-YIL090W; INM location is restored by ICE2-GST (Figure 6A, bottom, right). The data confirm that Ice2p is important for appropriate distribution of Trm1-II-GFP to the INM.
Figure 6.—
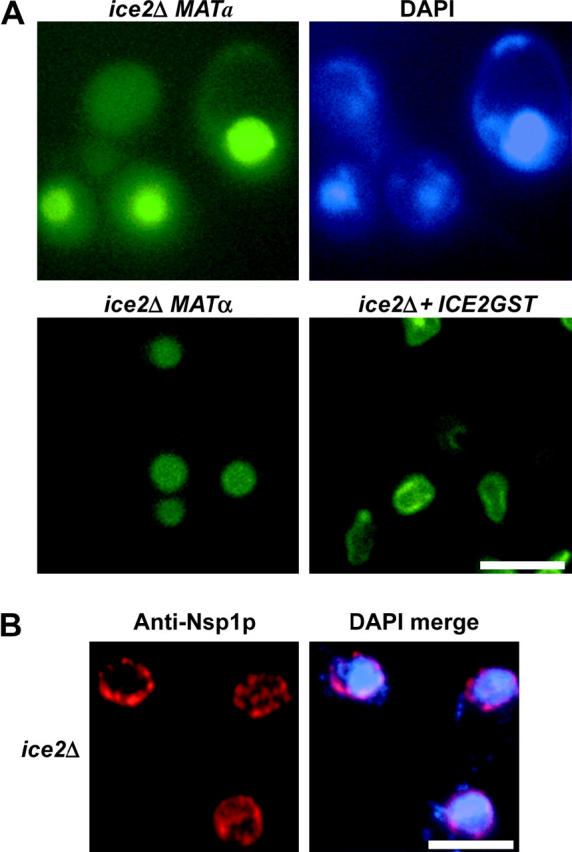
Ice2p is important for the INM location of Trm1-II-GFP but not for the nucleoporin, Nsp1p. (A) Location of Trm1-II-GFP. Trm1-II-GFP is nucleoplasmic in two independent YIL090WΔ/ice2Δ strains and ICE2-GST restores location of Trm1-II-GFP to the INM in the MATa YIL090WΔ/ice2Δ strain. (B) IF for Nsp1p. Nuclear pores are not affected by ice2Δ. Left, IF for Nsp1p showing punctate distribution; right, merge of Nsp1p and DNA locations in the respective strains shows that Nsp1p surrounds the nucleoplasm. Bar, 5 μm.
Trm1-II-GFP could poorly associate with the INM in ice2Δ cells because of a general defect in the nuclear membrane organization or because of a defect in tethering a subset of proteins to the INM. To distinguish between these possibilities, we assessed whether ice2Δ affects the subnuclear distribution of Nsp1p. No effect was detected (Figure 6B). The data support a role for Ice2p in targeting/tethering a subset of proteins to the INM.
DISCUSSION
The yeast deletion collections provide powerful tools to screen systematically for roles of all unessential proteins affecting any process of interest. Here we employed the collections in a cell biology study to understand mechanisms by which nuclear proteins achieve their appropriate subnuclear distribution, focusing on the INM. This easy screen could be applied to any cellular location for which a known resident protein exists. A priori one might have expected that proteins involved in organization of important structures, such as the INM, would be essential, prohibiting gene discovery using strains missing only unessential genes. However, we succeeded in identifying ORFs representing two different processes, using the unessential yeast collections. A collection in which each essential gene is replaced with a repressible version of the ORF is under construction (Mnaimneh et al. 2004). When this collection is completed the role of every yeast gene in targeting to any subcellular location can be learned using a similar strategy.
We show that Trm1-II-GFP is not associated with the INM in strains lacking NatC activity. Even though our data provide the first evidence that NatC-dependent N-acetylation is involved in subnuclear organization, a different NAT has been implicated in nuclear structure. Sir3p N-acetylation by NatA is necessary for mating-type silencing and gene silencing is mediated by chromatin sequestration at the INM (Wang et al. 2004). N-acetylation by NatC has previously been implicated in other types of protein targeting. For example, NatC-dependent killer Gag protein N-acetylation is essential for directing Gag into viral particles, presumably at the plasma membrane (Tercero et al. 1993; Ribas and Wickner 1998). NatC-dependent N-acetylation is also required for locating Arl3p to the Golgi in yeast (Behnia et al. 2004; Setty et al. 2004) and has been implicated in chloroplast targeting in Arabidopsis (Pesaresi et al. 2003). It will be interesting to learn if other yeast proteins require N-acetyl modification to associate with the INM.
How does N-acetylation affect INM location? Our data indicate that N-acetylation is necessary but may not be sufficient for Trm1p-II to locate to the INM. This is consistent with Behnia et al. (2004), who reported that the NatC-dependent N-acetylation is necessary for targeting Arl3p to the Golgi, but N-terminal alterations that allow N-acetylation of Arl3p by NatA or NatB motifs do not result in its Golgi distribution. The data are also consistent with the report that not all mutant Sir3p termini that are modified by NatA function appropriately (Wang et al. 2004). We must consider three explanations for why ectopic NatC motifs do not restore the INM location of Trm1-II-GFPΔL2. The first is that the altered proteins are not acetylated even though they have ectopic Nat motifs. Second, appropriate subnuclear distribution could require both N-acetylation that is achieved with the ectopic sequence and a Trm1p-specific N-terminal sequence or structure disrupted by the ectopic sequences. Third, the N-acetyl moiety might not interact directly with the INM, but rather it could regulate protein folding and exposure of other cis-located targeting motifs and the proteins with ectopic sequences may not achieve the appropriate structure. The latter possibility is consistent with the previous observation that a β-galactosidase fusion containing the N-terminal one-third of Trm1p-II fails to associate with the INM (Li et al. 1989).
Our genome-wide screen also uncovered a role for YIL090W/Ice2p in targeting/tethering the reporter protein to the INM. Interestingly, YIL090WΔ was previously implicated in nuclear structure/function because it was uncovered in a genome-wide study as being synthetically lethal with kar3Δ (Tong et al. 2004) and Kar3p is required for nuclear fusion during mating (reviewed in Rose 1996). However, YIL090W was also recently uncovered as Ice2p, important for the structure of the cortical ER (de Martin et al. 2005). YIL090W/Ice2p is an integral membrane protein with multiple transmembrane domains and GFP-tagged versions reside in the ER, rather than in the INM (Huh et al. 2003; de Martin et al. 2005). Although C-terminal tags can interfere with nucleoplasmic anchoring, allowing INM proteins to move to the contiguous ER (Holmer and Worman 2001), we show here that a YIL090W/Ice2p with a C-terminal GST tag complements the YIL090WΔ/ice2Δ, effectively eliminating the possibility that the ER location of the tagged versions is incorrect. We also eliminated the possibility that YIL090WΔ/ice2Δ disrupts nuclear membrane structure because nuclear pores appear to be unaffected by deletion of this gene (Figure 6B). We are considering two alternative explanations for why Trm1-II-GFP is partially released from the INM in ice2Δ cells. First, it is possible that Ice2p resides in both the ER and INM and thereby serves as a tether for ER and INM proteins in each location. More likely, Ice2p is an ER component that indirectly affects Trm1p-II subnuclear distribution perhaps by directing authentic tether(s) to the INM. It will be interesting to learn whether Ice2p plays an important role in targeting/tethering other proteins to the INM.
Acknowledgments
We thank G. Peng for construction of TRM1-GFP and insightful interactions and E. Phizicky for the yeast collection containing GST-ORF plasmids. Special thanks are given to M. Guilford, B. Stanley, and A. Stanley, Proteomics/Mass Spectroscopy Core Facility of the Section of Research Resources, Penn State College of Medicine, for conducting the MS analyses and protein sequencing. We thank members of the A.K.H. laboratory, especially K. Stauffer, for valuable scientific interactions. We thank N. C. Martin for scientific input and comments on the manuscript. This work was supported by a Pennsylvania State University Four Diamonds grant, by grants to A.K.H. from the National Science Foundation and the National Institutes of Health, and by a predoctoral fellowship from the American Heart Association to A.M.
References
- Behnia, R., J. R. Whyte and S. Munrop, 2004. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell. Biol. 6: 405–413. [DOI] [PubMed] [Google Scholar]
- Casolari, J. M., C. R. Brown, S. Komili, J. West, H. Hieronymus et al., 2004. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117: 427–439. [DOI] [PubMed] [Google Scholar]
- Chu, A., R. Rassadi and U. Stochaj, 1998. Velcro in the nuclear envelope: LBR and LAPs. FEBS Lett. 441: 165–169. [DOI] [PubMed] [Google Scholar]
- Cronshaw, J. M., A. N. Krutchinsky, W. Zhang, B. T. Chait and M. J. Matunis, 2002. Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158: 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martin, P. E., Y. Du., P. Novick and S. Ferro-Novick, 2005. Ice2p is important for the distribution and structure of the cortical ER network in Saccharomyces cerevisiae. J. Cell Sci. 118: 65–77. [DOI] [PubMed] [Google Scholar]
- Ellis, S. R., M. J. Morales, J. M. Li, A. K. Hopper and N. C. Martin, 1986. Amino-terminal extension generated from upstream AUG codon is not required for mitochondrial import of yeast tRNA methyltransferase. J. Biol. Chem. 261: 9703–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etcheverry, T., D. Colby and C. Guthrie, 1979. A precursor to a minor species of yeast tRNASer contains an intervening sequence. Cell 18: 11–26. [DOI] [PubMed] [Google Scholar]
- Gruenbaum, Y., R. D. Goldman, R. Meyuhas, E. Mills, A. Margalit et al., 2003. The nuclear lamina and its functions in the nucleus cytol. Nat. Rev. Mol. Cell. Biol. 226: 1–62. [DOI] [PubMed] [Google Scholar]
- Holmer, L., and H. J. Worman, 2001. Inner nuclear membrane proteins: functions and targeting. Mol. Life Sci. 58: 1741–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper, A. K., A. H. Furukawa, H. D. Pham and N. C. Martin, 1982. Defects in modification of cytoplasmic and mitochondrial transfer RNAs are caused by single nuclear mutations. Cell 28: 543–550. [DOI] [PubMed] [Google Scholar]
- Hughes, T. R., C. J. Roberts, H. Dai, A. R. Jones, M. R. Meyer et al., 2000. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat. Genet. 25: 333–337. [DOI] [PubMed] [Google Scholar]
- Huh, W. K., J. V. Falvo, L. C. Gerke, S. Carroll, R. W. Howson et al., 2003. Global analysis of protein localization in budding yeast. Nature 425: 686–691. [DOI] [PubMed] [Google Scholar]
- Ito, H., Y. Fukuda, K. Murata and A. Kimura, 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153: 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond, A. I., and J. E. Sleeman, 2003. Nuclear substructure and dynamics. Curr. Biol. 13: R825–R828. [DOI] [PubMed] [Google Scholar]
- Li, J.-M., A. K. Hopper and N. C. Martin, 1989. N2,N2-dimethylguanosine-specific tRNA methyltransferase contains both nuclear and mitochondrial targeting signals in Saccharomyces cerevisiae. J. Cell Biol. 109: 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnaimneh, S., A. P. Davierwala, J. Haynes, J. Moffat, W. T. Peng et al., 2004. Exploration of essential gene functions via titratable promoter alleles. Cell 118: 31–44. [DOI] [PubMed] [Google Scholar]
- Martzen, M. R., S. M. Mccraith, S. L. Spinelli, F. M. Torres, S. Fields et al., 1999. A biochemical genomics approach for identifying genes by the activity of their products. Science 286: 1153–1155. [DOI] [PubMed] [Google Scholar]
- Pesaresi, P., N. A. Gardner, S. Masiero, A. Dietzmann, L. Eichacker et al., 2003. Cytoplasmic N-terminal protein acetylation is required for efficient photosynthesis in Arabidopsis. Plant Cell 15: 1817–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky, E. M., M. R. Martzen, S. M. Mccraith, S. L. Spinelli, F. Xing et al., 2002. Biochemical genomics approach to map activities to genes. Methods Enzymol. 50: 546–559. [DOI] [PubMed] [Google Scholar]
- Polevoda, B., and F. Sherman, 2003. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325: 595–622. [DOI] [PubMed] [Google Scholar]
- Ribas, J. C., and R. B. Wickner, 1998. The Gag domain of the Gag-Pol fusion protein directs incorporation into the L-A double-stranded RNA viral particles in Saccharomyces cerevisiae. J. Biol. Chem. 273: 9306–9311. [DOI] [PubMed] [Google Scholar]
- Rose, A. M., P. B. M. Joyce, A. K. Hopper and N. C. Martin, 1992. Separate information required for nuclear and subnuclear localization: additional complexity in localizing an enzyme shared by mitochondria and nuclei. Mol. Cell. Biol. 12: 5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, A. M., H. G. Belford, W. C. Shen, C. L. Greer, A. K. Hopper et al., 1995. Location of N2,N2-dimethylguanosine-specific tRNA methyltransferase. Biochimie 77: 45–53. [DOI] [PubMed] [Google Scholar]
- Rose, M. D., 1996. Nuclear fusion in the yeast Saccharomyces cerevisiae. Annu. Rev. Cell. Dev. Biol. 12: 663–695. [DOI] [PubMed] [Google Scholar]
- Rout, M. P., J. D. Aitchison, A. Suprapto, K. Hjertaas, Y. Zhao et al., 2000. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148: 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty, S. R., T. I. Strochlic, A. H. Tong, C. Boone and C. G. Burd, 2004. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat. Cell. Biol. 6: 414–419. [DOI] [PubMed] [Google Scholar]
- Sikorski, R. S., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei, A., F. Hediger, F. R. Neumann and S. M. Gasser, 2004. The function of nuclear architecture: a genetic approach. Annu. Rev. Genet. 38: 305–345. [DOI] [PubMed] [Google Scholar]
- Tercero, J. C., J. D. Dinman and R. B. Wickner, 1993. Yeast MAK3 N-acetyltransferase recognizes the N-terminal four amino acids of the major coat protein (gag) of the L-A double-stranded RNA virus. J. Bacteriol. 175: 3192–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolerico, L. H., A. L. Benko, J. P. Aris, D. R. Stanford, N. C. Martin et al., 1999. Saccharomyces cerevisiae Mod5p-II contains sequences antagonistic for nuclear and cytosolic locations. Genetics 151: 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, A. H., G. Lesage, G. D. Bader, H. Ding, H. Xu et al., 2004. Global mapping of the yeast genetic interaction network. Science 303: 808–813. [DOI] [PubMed] [Google Scholar]
- Wang, X., J. J. Connelly, C. L. Wang and R. Sternglanz, 2004. Importance of the Sir3 N terminus and its acetylation for yeast transcriptional silencing. Genetics 168: 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson et al., 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]