

Abstract
Mutation of BRCA2 causes familial early onset breast and ovarian cancer. BRCA2 has been suggested to be important for the maintenance of genome integrity and to have a role in DNA repair by homology- directed double-strand break (DSB) repair. By studying the repair of a specific induced chromosomal DSB we show that loss of Brca2 leads to a substantial increase in error-prone repair by homology-directed single-strand annealing and a reduction in DSB repair by conservative gene conversion. These data demonstrate that loss of Brca2 causes misrepair of chromosomal DSBs occurring between repeated sequences by stimulating use of an error-prone homologous recombination pathway. Furthermore, loss of Brca2 causes a large increase in genome-wide error-prone repair of both spontaneous DNA damage and mitomycin C-induced DNA cross-links at the expense of error-free repair by sister chromatid recombination. This provides insight into the mechanisms that induce genome instability in tumour cells lacking BRCA2.
Keywords: BRCA2/DNA repair/homologous recombination/single-strand annealing/sister chromatid exchange
Introduction
Women who inherit loss-of-function mutations in either of the breast cancer susceptibility genes, BRCA1 and BRCA2, have a high risk of developing breast cancer (Rahman and Stratton, 1998). Since the wild-type allele is lost from tumours arising in heterozygous carriers, both BRCA1 and BRCA2 are thought to act as tumour suppressor genes. BRCA1 and BRCA2 encode unrelated nuclear proteins, which can both interact with Rad51 (Mizuta et al., 1997; Scully et al., 1997; Sharan et al., 1997; Chen et al., 1998), the eukaryotic equivalent of bacterial RecA. Rad51 catalyses strand exchange during homology-directed repair of DNA double-strand breaks (DSBs) by gene conversion. A direct interaction between BRCA2 and Rad51 has been demonstrated, and is mediated by a series of internal BRC repeats encoded by BRCA2 exon 11 (Chen et al., 1998), and an additional non-BRC domain located at the C-terminus of the protein (Mizuta et al., 1997; Sharan et al., 1997). These physical interactions, and the observation that BRCA1 and BRCA2 co-localize with Rad51 in ionizing radiation (IR)-induced nuclear foci (Chen et al., 1998), suggested a role for BRCA1 and BRCA2 in DNA repair by homologous recombination (HR). Subsequent studies, which have demonstrated that mouse and human cells deficient for wild-type BRCA1 or BRCA2 suffer from chromosome instability (Tirkkonen et al., 1997; Gretarsdottir et al., 1998; Patel et al., 1998; Tutt et al., 1999; Xu et al., 1999; Ban et al., 2001) and have a heightened sensitivity to DNA lesions that are repaired by HR (Patel et al., 1998; Shen et al., 1998; Scully et al., 1999; Yu et al., 2000; Wang et al., 2001a), have supported this contention.
Mammalian cells can repair DNA DSBs by both HR and by non-homologous end-joining (NHEJ) (Karran, 2000; Khanna and Jackson, 2001). NHEJ of DSBs is non-conservative and is often associated with deletions, insertions and translocations. HR accounts for 30–50% of endonuclease-induced DSB repair events in dividing mammalian cells and can occur by two main pathways: gene conversion and single-strand annealing (SSA) (Liang et al., 1998). During gene conversion, the DSB is processed to produce 3′ single-stranded tails, which recruit Rad51 and thereby seek out a homologous template on the sister chromatid or homologous chromosome from which to accurately resynthesize the sequence surrounding the DSB (Baumann and West, 1998). Use of the identical sister chromatid in gene conversion, as opposed to homologous chromosomes, maintains genome integrity and is the preferred repair template (Johnson and Jasin, 2000). Gene conversion can occur in the absence (here referred to as GC) or presence (CO) of a crossover or exchange event. Sister chromatid crossover (CO) events can be equal (error-free events termed sister chromatid exchanges, SCE) or unequal depending on the template used for repair. Wild-type cells suppress unequal sister chromatid CO or CO events between chromosomes (Richardson et al., 1998; Johnson and Jasin, 2000) because these can cause duplications, deletions or translocations (Lupski, 1998; Jasin, 2000). An alternative, Rad51- independent, HR repair pathway is SSA. This competes with the GC pathway for the common 3′ single-stranded repair intermediate (Ivanov et al., 1996; Kang and Symington, 2000; Lambert and Lopez, 2000). SSA aligns and anneals regions of homology on either side of a DSB, repairing it but deleting the intervening sequence, causing deletions between repetitive elements or chromosome translocations when DSBs occur on more than one chromosome (Richardson and Jasin, 2000). Vertebrate cells with large repetitive genomes must, therefore, tightly regulate homologous DNA repair pathways in order to avoid genome instability (Jasin, 2000). Recently, embryonic stem (ES) cells with disruptions in Brca2 have been shown to be compromised for repair of restriction enzyme-induced DSBs by GC (Moynahan et al., 2001). It remains unknown whether the disruption of repair by sister chromatid GC is associated with repair of damage by error-prone recombination pathways such as gene conversion with unequal CO or SSA. Here we ask whether disruption of Brca2 in ES cells is associated with an increased frequency of DNA repair using these pathways.
Common causes of spontaneous DSBs are arrested replication forks (Sasaki, 1980; Haber, 1999). Sister chromatid GC and equal sister chromatid CO events are thought to be an accurate mechanism responsible for their repair. SCE can be seen in untreated metaphase cells and following treatment with DNA-damaging agents, and are suggested to arise from the repair of arrested replication forks by equal sister chromatid CO (Sonoda et al., 1999). We therefore also examine the effect of disruption of Brca2 on the frequency of these events relative to other exchanges and aberrations that have arisen by error-prone repair. Our results suggest a mechanism for chromosome instability caused by loss of BRCA2.
Results
Strategy for assessment of the role of Brca2 in DSB repair in ES cells
We wished to create a cell line carrying a conditionally mutable allele of the Brca2 gene to test its role in DNA repair and HR. It is thought that null mutations for Brca2 result in early embryonic lethality probably due to cell cycle arrest mediated by checkpoint activation (Bertwistle and Ashworth, 1998). The choice of DSB repair pathway may be cell cycle regulated; therefore, to avoid the confounding effect of significant cell cycle perturbation, we created a cell line with two hypomorphic Brca2 alleles. We used our previously described ES cell line carrying a hypomorphic allele Brca2Tr2014, which results in the truncation of the Brca2 open reading frame at amino acid 2014 (Connor et al., 1997a). We altered the other allele so that the final Brca2 exon (exon 27) was flanked by loxP sites, which could be conditionally deleted by transient expression of Cre recombinase. Deletion of exon 27 has also been shown to produce a hypomorphic allele, homozygosity for which causes ionizing radiation sensitivity in mouse ES cells (Morimatsu et al., 1998). An analogous truncating mutation in BRCA2 is associated with cancer predisposition in humans (Hakansson et al., 1997). Simultaneously with the modification of exon 27, we introduced a HR repair substrate, DR1Bsd. This allows the repair of an I-SceI-mediated DSB in DR1Bsd to be compared before and after Cre-mediated deletion of Brca2 exon 27 in the same (isogenic) cell line. An additional feature of the construct is that the modified exon 27 allele carries an in-frame myc (9E10) epitope tag, allowing monitoring of the endogenous Brca2 protein. The construct and the modified Brca2 allele are shown in Figure 1A.
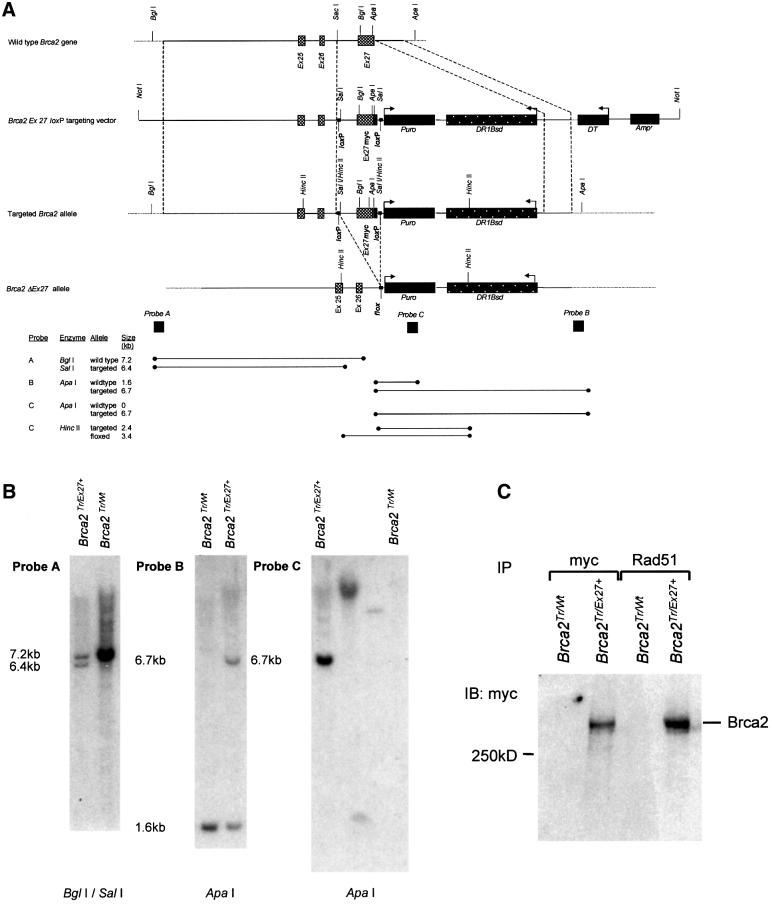
Fig. 1. Brca2 exon 27 ‘knock in’ strategy and analysis of clones. (A) Structure of the 3′ end of the mouse Brca2 locus and targeting vector containing HR repair substrate. The upper line represents the wild-type allele. The second and third lines represent the linearized targeting vector and the targeted allele, respectively. Exons 25, 26 and 27 are shown as grey boxes. The positions of relevant restriction enzyme sites are marked. 5′ and 3′ regions of homology between the targeting vector and the wild-type allele are enclosed between dashed lines flanking the ‘knock in’ region. This includes a mutated exon 27 with an in-frame 9E10 myc epitope tag shown as a black box. loxP sites flanking exon 27 myc are shown as small black squares. The lower line demonstrates the effect of Cre-mediated recombination between the labelled loxP sites to produce a Flox site. This deletes the intervening sequence including exon 27myc. Large black boxes represent the puromycin (Puro) selectable marker gene, the diphtheria toxin (DT) negative selection marker and the ampicillin (Amp) resistance gene. The HR repair substrate DR1Bsd is represented by a black speckled box. Regions of hybridization to the three probes (A, B and C) used in Southern blot analyses are indicated by large dark squares. The restriction enzymes and the probes used for Southern analysis of the targeted allele, and the positions and sizes of the fragments detected are shown at the bottom of the figure. (B) Southern blot analysis. The targeted allele was termed Brca2Ex27+ and the targeted cell line termed Brca2Tr/Ex27+. Southern blots of genomic DNA from parental cells Brca2Tr/Wt and a Brca2Tr/Ex27+-targeted clone subjected to restriction digestion and hybridization with the marked probe. Probe A is a flanking probe 5′ to the 5′ homology. Probe B is 3′ to the 3′ homology. Probe C is a fragment of Puro. In the left panel, BglI–SalI digestion shows the 7.2 kb wild-type fragment in Brca2Tr/Wt ES cells, and both the 7.2 kb wild-type fragment and the predicted 6.4 kb targeted fragment in Brca2Tr/Ex27+ cells. In the middle panel, ApaI digestion shows the 1.6 kb wild-type fragment and predicted 6.7 kb targeted fragment. These confirm correct integration into the Brca2 locus. The right panel shows ApaI digests probed with Puro, confirming the absence of additional random integrants in Brca2Tr/Ex27+ cells. Brca2Tr/Wt is a negative control. The two middle lanes are clones containing non-targeted random integrants. (C) Correct integration of the targeting construct into the wild-type allele of Brca2Tr/Wt ES cells was confirmed by confirmation of production of full-length myc-tagged Brca2 protein. Immunoblot analysis of whole-cell lysates of Brca2Tr/Wt and Brca2Tr/Ex27+ ES cells immunoprecipated (IP) with anti-myc and anti-Rad51 antibodies and immunoblotted (IB) with an anti-myc antibody.
Targeted modification of Brca2 in ES cells
We used targeted integration to obtain cell lines carrying a single copy of the DSB repair substrate at a defined chromosomal site. Following electroporation of Brca2Tr2014/Wt ES cells (Connor et al., 1997a) with the targeting construct, transformants were selected in puromycin and analysed by Southern blotting of genomic DNA (Figure 1B). In order to confirm integration into the wild-type allele, targeted clones and control parental Brca2Tr2014/Wt ES cells were lysed and the presence of a full-length myc-tagged Brca2 was confirmed by immunoprecipitation (IP) and immunoblotting (IB) using an anti-myc antibody (Figure 1C). The targeted allele was termed Brca2Ex27mycloxP (here Brca2Ex27+ for brevity) and the targeted cell line termed Brca2Tr2014/Ex27mycloxP (Brca2Tr/Ex27+). To confirm that addition of the six-amino-acid myc epitope to the C-terminus of Brca2 had not affected the ability of the protein to interact with Rad51, further reciprocal IP/IB experiments were performed using antibodies to Rad51 and to the myc epitope (Figure 1C and data not shown). Furthermore, co-localization of myc-tagged Brca2 and Rad51 in ionizing radiation-induced nuclear foci was confirmed by confocal immunofluorescent microscopy (data not shown). This established that the Brca2Ex27+ protein, in common with Brca2, was able to interact with Rad51.
Transient expression of Cre recombinase in ES cells causes recombination between loxP sites and deletion of intervening sequence, resulting in deletion of Brca2 exon 27 and part of the intron between exons 26 and 27 (Figure 1A). Thus, the truncation of Brca2 will remove the C-terminal Rad51 binding domain and the myc epitope tag. The Brca2Tr/Ex27+ cell line was transiently transfected with the expression vector pCAGGS driving expression of an EGFP–Cre recombinase fusion protein or with enhanced green fluorescent protein (EGFP) alone as control. Cells were analysed by fluorescence-activated cell sorter (FACS) and the green fluorescent protein (GFP)-positive population sorted to >99% purity and returned to culture. Genomic DNA extraction and Southern blot analysis revealed the expected deletion of exon 27 in 60–70% of the alleles (data not shown). Following exon 27 deletion, the floxed allele is termed Brca2ΔEx27. Brca2Tr/ΔEx27 and Brca2Tr/Ex27+ control clonal cell lines were derived as described in Materials and methods. The presence of a slightly smaller C-terminally truncated Brca2 with loss of the myc epitope tag was confirmed in Brca2Tr/ΔEx27 cell lines by IP using antibodies to the N-terminal region of Brca2, and to the myc epitope and IB using the Brca2 antibody. It was apparent that the deletion of exon 27 was associated with a reduction in the abundance of the truncated form of Brca2, Brca2ΔEx27 (Figure 2A).
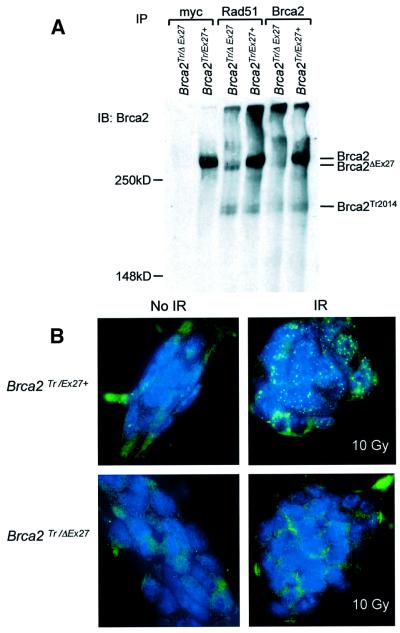
Fig. 2. Cre-mediated deletion of the C-terminus of Brca2 in ES cells. (A) Immunoblotting analysis of whole-cell extract of Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 clonal cell lines immunoprecipated (IP) with anti-myc, anti-Rad51 and N-terminal anti-Brca2 antibodies, and immunoblotted (IB) with the anti-Brca2 antibody. The positions of full-length Brca2, the exon 27-deleted (Brca2ΔEx27) and the exon 11-truncated (Brca2Tr2014) proteins are indicated. (B) Failure of normal induction of Rad51 foci in Brca2Tr/ΔEx27 ES cells. Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells mock irradiated (left upper and lower panels) or irradiated with 10 Gy (right upper and lower panels) were fixed and analysed by immunofluorescent microscopy. DNA is labelled with DAPI and appears blue. Rad51 was detected with anti-Rad51 antibody and a secondary FITC-conjugated antibody. The right upper panel shows induction of Rad51-containing nuclear foci in Brca2Tr/Ex27+ cells. The right lower panel shows failure of induction of Rad51 foci in Brca2Tr/ΔEx27 ES cells.
Brca2ΔEx27 and Brca2Tr2014 associate with Rad51 but inhibit X-ray-induced Rad51 nuclear focus formation
Despite deletion of the C-terminal Rad51 binding domain, Brca2ΔEx27 still contains the eight BRC repeats encoded by exon 11 of Brca2. Brca2Tr2014 is predicted to retain seven BRC repeats (Connor et al., 1997a). We wished to establish whether either Brca2Tr2014 or Brca2ΔEx27 associates with Rad51. IP with anti-Rad51 antibody and IB with antibody to the N-terminal region of Brca2 revealed that both Brca2Tr2014 and Brca2ΔEx27 associate with Rad51 (Figure 2A). The greater intensity of the Brca2ΔEx27 band when immunoprecipitated with anti-Rad51 antibody rather than anti-Brca2 may be due to a greater affinity of Brca2ΔEx27 for Rad51 than for the anti-Brca2 antibody. To ascertain whether Brca2ΔEx27 affected Rad51 nuclear focus formation, we irradiated Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells with 10 Gy of X-rays and analysed the formation of Rad51 nuclear foci 5 h later. We found that Brca2Tr/Ex27+ cells formed Rad51 foci after irradiation with 10 Gy, but this failed to induce Rad51 foci in Brca2Tr/ΔEx27 ES cells (Figure 2B). This shows that although Brca2ΔEx27 interacts with Rad51, the C-terminus of Brca2 is required for the formation or stabilization of IR-induced Rad51 foci.
The DR1Bsd recombination test substrate
To investigate the effect of these hypomorphic Brca2 mutations on the repair of DNA DSBs by homology-directed repair we have used a chromosomal DSB repair substrate that allows reporting of both gene conversion (GC and CO) and SSA homology-directed repair events at a defined chromosomal locus within our cell lines. This can be achieved both by antibiotic selection of colonies and by analysis of genomic DNA repair products. The DR1Bsd substrate contains a central zeocin selectable marker gene (Zeo) flanked by two differentially mutated blasticidin antibiotic resistance (Bsd) genes (Figure 3). S1Bsd is a full-length 693 bp Bsd gene that contains an in-frame insertion of the 18 bp recognition sequence of the restriction endonuclease I-SceI at a unique SalI site 279 bp into the coding sequence of Bsd. This insertion encodes two in-frame stop codons and renders Bsd non-functional. The 3′ repeat (5′ΔBsd) is a 659 bp promoterless fragment of Bsd inactivated by truncation of the 5′ 34 bp. Both repeats are in the same orientation and are therefore termed direct repeats.
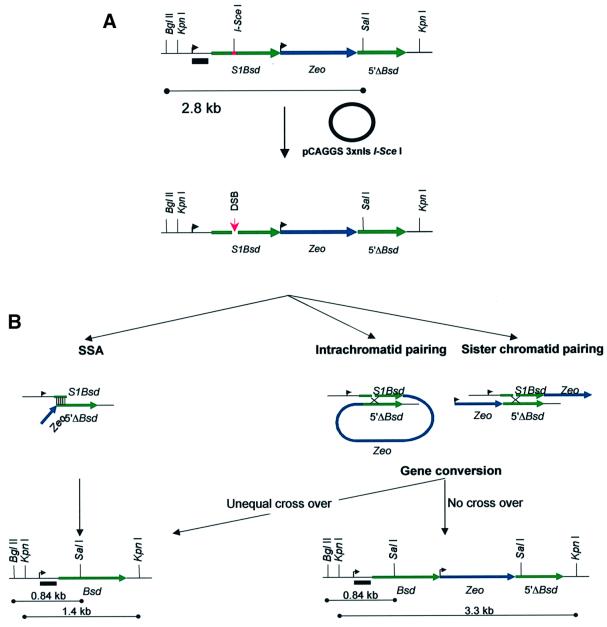
Fig. 3. HR repair substrate DR1Bsd. (A) The repair substrate is represented in a 5′ to 3′ orientation. Relevant restriction endonuclease recognition sites are marked. The 5′ mutated Bsd repeat is shown as a green line and is labelled S1Bsd. The upstream TK promoter sequence from pMCINeo is marked with an arrow. The site of mutation of the wild-type SalI site by insertion of the 18 bp recognition sequence of the I-SceI endonuclease is shown as a red bar. The central Zeo antibiotic selection marker is shown as a blue line with its upstream PGK promoter marked with an arrow. The downstream promoterless direct repeat 5′ΔBsd is marked as a green line. The position of the wild-type SalI site is marked. A black bar indicates the position of a TK promoter probe that can hybridize in all repair products (B, lower panels) equally. The effect of transient expression of I-SceI from the pCAGGS expression vector is illustrated. The upper line represents the undamaged repair substrate. The I-SceI expression vector is shown as a circle. The lower line demonstrates the site of induction of a DNA DSB at the I-SceI recognition sequence in S1Bsd. (B) Mechanisms by which wild-type Bsd may be created by HR repair of the I-SceI DSB in DR1Bsd. Repair by use of the SSA pathway is depicted in the left panels. This involves 5′–3′ resection of one strand on either side of the DSB, leaving a 3′ tail. When complementary Bsd sequences from S1Bsd and 5′ΔBsd on either side of the DSB are exposed, they can anneal. This is indicated by thin vertical lines. The single-stranded tails are resected by a nuclease, gaps are filled in and nicks ligated. This process deletes all sequence between S1Bsd and 5′ΔBsd, and thus results in the creation of wild-type Bsd and the deletion of Zeo. The repair product and the size of predicted restriction fragments are marked in the left lower panel. Repair of S1Bsd by use of the GC pathway (right panels) involves similar 5′–3′ resection to leave 3′ single-stranded tails. These invade and pair with homologous 5′ΔBsd sequence on either the same chromatid (central panel) or sister chromatid (right panel). The break may thus be repaired using wild-type sequence as the template. Regions of pairing are indicated with a cross and may be resolved either with or without a crossover (CO) event. If the substrate is repaired without CO, the repair product contains wild-type Bsd, Zeo and 5′ΔBsd. The repair product and the size of predicted restriction fragments are marked in the right lower panel. If an unequal CO event takes place, the central Zeo is removed. This product, referred to as the ‘Pop out’ repair product, is identical whether repair is by SSA or CO (left lower panel). Equal CO events recreate S1Bsd and are, therefore, not recovered.
DSB induction and repair events in DR1Bsd
Transient expression of the rare cutting endonuclease I-SceI linked to a triplicated nuclear localization signal (3 × nls I-SceI) is non-toxic in mouse ES cells and induces a DSB at a chromosomally integrated I-SceI site (Rouet et al., 1994; Moynahan et al., 2001), as in S1Bsd (Figure 3). Following repair by HR between S1Bsd and 5′ΔBsd, the disrupted SalI site of S1Bsd can be restored, recreating wild-type Bsd and consequently resulting in the resistance of ES cells to blasticidin (Figure 3). Repair of DR1Bsd by SSA leads to blasticidin resistance, but consequent deletion of Zeo renders cells sensitive to zeocin. HR by GC may occur using either 5′ΔBsd on the same chromatid as a donor (intra-chromatid GC) or 5′ΔBsd on the sister chromatid following DNA replication (sister chromatid GC). Clones derived by HR repair using GC will be resistant to both blasticidin and zeocin. Gene conversion may be associated with an unequal crossing over event, in which case the central Zeo gene is removed as an excised circle (intra-chromatid CO), or in sister chromatid CO Zeo is transferred to the donor sister chromatid. Therefore, I-SceI DSB repair of DR1Bsd by any of the above homology-directed mechanisms will induce blasticidin resistance in daughter cells. Whereas clones repaired by GC will be resistant to both blasticidin and zeocin, clones repaired by SSA or unequal CO will be resistant to blasticidin, but sensitive to zeocin.
The successful repair of DR1Bsd by HR mechanisms may also be detected and further characterized by Southern analysis of pooled blasticidin-resistant clones (Figure 3). Digestion of the HR repaired construct in blasticidin-resistant clones with BglII and SalI reveals a change in size of the fragment from 2.8 kb in the parental construct to 0.84 kb. Excision of the entire substrate with KpnI gives a 3.3 kb fragment if repair is by HR by GC or a 1.4 kb deletion product following repair by HR by SSA or CO. The outcomes of SSA and CO are identical at the DNA level in blasticidin-resistant cells. This repair product will be referred to here as the ‘Pop out’ recombination product.
Repair of the DSB by the NHEJ mechanism either involves precise re-ligation of the I-SceI overhangs, or more commonly, endonuclease processing of the broken ends. This is associated with small deletions and insertions. Under these circumstances, S1Bsd remains mutant and the clone is sensitive to blasticidin. Precise deletion of the 18 bp I-SceI site and accurate microannealing of the duplicated flanking SalI overhangs by NHEJ repair mechanisms might theoretically lead to restoration of wild-type Bsd. This event is reported to occur two orders of magnitude less frequently than HR between the repeats in similar constructs (Moynahan and Jasin, 1997; Lin et al., 1999). To test the requirement for a homologous repeat for recreation of wild-type Bsd in ES cells, we compared the frequency of blasticidin-resistant colonies induced by transient I-SceI expression in cell lines expressing full-length Brca2 containing either only S1Bsd or the entire repair substrate DR1Bsd. We found that the frequency of blasticidin resistance is 9 × 10–4 for S1Bsd alone compared with 5 × 10–2 for DR1Bsd (Figure 4A). Blasticidin resistance induced by NHEJ repair of S1Bsd is, therefore, a very rare event.
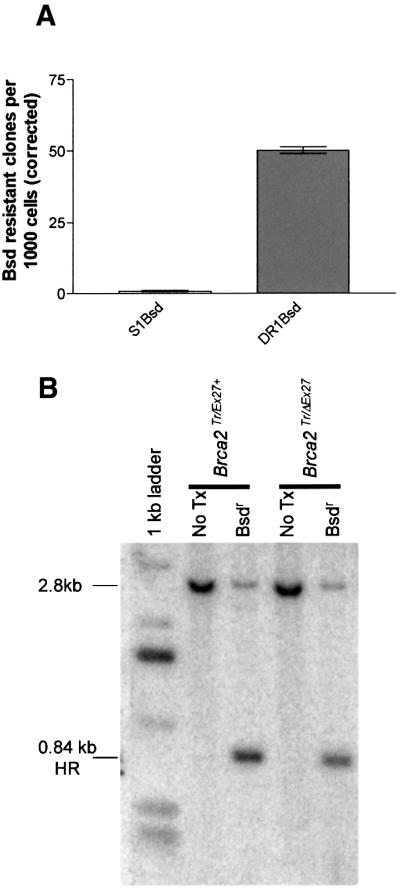
Fig. 4. Repair of DR1Bsd leads to blasticidin resistance by homology-directed repair. (A) Bar graph showing the number of blasticidin-resistant colonies per thousand cells plated (corrected for transfection and cloning efficiencies). The left column represents Brca2Tr/Wt cells containing S1Bsd alone and the right column Brca2Tr/Ex27+ cells that contain both the S1Bsd and the homologous donor repeat 5′ΔBsd in DR1Bsd. There is very little induction of blasticidin resistance in S1Bsd; therefore, the frequency of repair of a DSB in S1Bsd relative to wild-type Bsd by NHEJ is extremely low. Error bars represent ± 1 SEM. (B) A representative Southern blot of BglII–SalI-digested genomic DNA from Brca2Tr/Ex27+ or Brca2Tr/ΔEx27 ES cells before (marked No Tx) and after I-SceI-induced DSB repair and subsequent blasticidin selection (marked Bsdr). A TK promoter fragment (indicated in Figure 3) was used as a probe. A dominant 2.8 kb restriction fragment is seen in both cell lines before transfection of pCAGGS 3 × nls I-SceI and is from the unbroken DR1Bsd substrate. After DSB induction, repair and selection of blasticidin-resistant colonies, the predicted 0.84 kb HR fragment is dominant in all cell lines. This arises due to HR with 5′ΔBsd and transfer of the wild-type SalI-containing sequence from 5′ΔBsd to S1Bsd, to create Bsd.
Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells were transfected with pCAGGS 3 × nls I-SceI or with pCAGGS EGFP as control, replated and selected with blasticidin (see Materials and methods). DNA from pooled resistant colonies was digested with BglII and SalI, followed by Southern blotting. This confirmed that blasticidin-resistant colonies arise in both cell lines by HR with 5′ΔBsd (Figure 4B) rather than by NHEJ or another novel mechanism.
The effect of Brca2 exon 27 deletion on HR repair by gene conversion
As Rad51 binds the C-terminus of Brca2 and is known to have a key role in DSB repair by homologous strand invasion and gene conversion, we wished to test the effect of our Brca2ΔEx27 mutation on this process. Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells were transfected with pCAGGS 3 × nls I-SceI and selected with blasticidin as described in Materials and methods. In each of three experiments, using three Brca2Tr/Ex27+ and three independently derived Brca2Tr/ΔEx27 ES cell clones, resulting blasticidin-resistant clones were isolated, expanded and then double selected with blasticidin and zeocin. Parental Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cell clones were capable of continued growth in zeocin at the same concentration used for selection. Blasticidin-resistant clones surviving blasticidin + zeocin double selection represent repair events by gene conversion without crossing over. As the results are expressed as a proportion of blasticidin-resistant colonies, they are not affected by differences in transfection and cloning efficiency between cell lines or experiments. Of 71 viable Brca2Tr/Ex27+ blasticidin-resistant clones, 42 (59%) were also zeocin resistant, whereas of 72 viable Brca2Tr/ΔEx27 ES cell clones, only 11 (15%) were zeocin resistant (Figure 5A). There was, therefore, a 75% reduction in the proportion of HR repair due to GC. This demonstrates a role for Brca2 in HR repair of DNA DSBs by a conservative homology-directed GC mechanism. To validate this result, we subjected genomic DNA from thousands of pooled blasticidin-resistant Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cell clones from each of the three experiments to KpnI digestion and Southern blot analysis with a probe that hybridizes equally to both potential repair fragments. The relative intensity of the two repair fragments was determined on a phosphoimager and the proportion of HR repair due to GC calculated (Figure 5B). This confirmed a reduction in the proportion of HR due to GC.
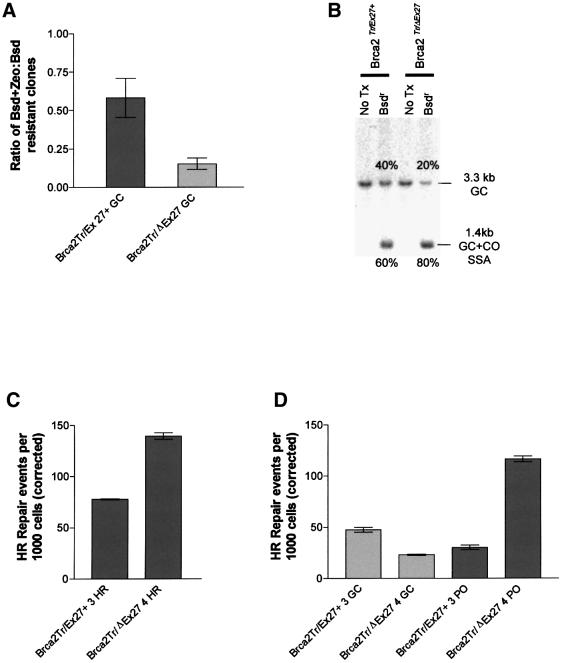
Fig. 5. Brca2 truncation affects choice of HR repair pathway. (A) The proportion of blasticidin-resistant clones that are also resistant to zeocin is plotted for Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells. The ratio expresses the proportion of all HR events due to GC without CO. The experiment was performed three times on three independent sets of Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cell clones. Error bars indicate ± 1 SEM. (B) A representative Southern blot analysis of KpnI-digested genomic DNA from Brca2Tr/Ex27+ or Brca2Tr/ΔEx27 ES cells before (marked No Tx) and after I-SceI-induced DSB repair and subsequent blasticidin selection (marked Bsdr). A TK promoter fragment was used as a probe (Figure 3). The untransfected control DNA has a single 3.3 kb fragment. After DSB induction, repair and selection of blasticidin-resistant colonies, the predicted 3.3 kb GC conservative HR product and the 1.4 kb SSA/CO ‘Pop out’ deletion product are both seen. The relative proportions of these products within each cell line were analysed by phosphoimager and are annotated adjacent to each fragment. DNA from three independent experiments was analysed. Brca2Tr/ΔEx27 ES cells show a reduced proportion of HR repair due to GC. (C) Absolute frequencies of overall HR repair events (GC, CO and SSA) are compared in Brca2Tr/ΔEx27 ES cells and compared with Brca2Tr/Ex27+ control. A successful HR repair event will produce a blasticidin-resistant daughter clone. The frequency of these events is expressed per 1000 cells, corrected for both the transfection and cloning efficiency, and represents the absolute frequency of HR repair of I-SceI-induced DSBs. Each experiment was performed in triplicate and repeated with at least three independently derived Brca2Tr/ΔEx27 ES cell clones and compared with Brca2Tr/Ex27+ control clones. A single representative experiment is shown. Error bars indicate ± 1 SEM. (D) Absolute frequency of HR GC repair events and ‘Pop out’ (CO and SSA) HR events are compared in Brca2Tr/ΔEx27 ES cells and compared with Brca2Tr/Ex27+ control. Whereas an HR GC repair event will produce a daughter clone doubly resistant to both blasticidin and zeocin, ‘Pop out’ HR will produce a clone resistant to blasticidin, but sensitive to zeocin. The frequency of the GC event was determined by double selection with blasticidin and zeocin after plating 2 × 105 ES cells transfected with pCAGGS 3 × nls I-SceI. Colony count was corrected for transfection efficiency and the cloning efficiency of parental Brca2Tr/ΔEx27 and Brca2Tr/Ex27+ ES cells in zeocin. The frequency of ‘Pop out’ HR events is calculated as the overall HR event frequency minus the HR GC event frequency. The absolute frequency of these events is shown per 1000 cells. Each experiment was performed in triplicate and repeated with at least three independently derived Brca2Tr/ΔEx27 ES cell clones and compared with Brca2Tr/Ex27+ control clones. A single representative experiment is shown. Error bars indicate ± 1 SEM.
The effect of Brca2 exon 27 deletion on the frequency and subclass of HR repair event
DR1Bsd can be repaired to give wild-type Bsd by any of the HR mechanisms shown in Figure 3. The reduction in the proportion of total HR repair due to GC in Brca2Tr/ΔEx27 ES cells may be due to a reduction in the absolute frequency of GC events, an increase in SSA/CO events or a combination of the two. Therefore, we wanted to quantify the effect of our hypomorphic Brca2ΔEx27 allele on the frequency of HR DSB repair overall, and GC and CO/SSA repair specifically. Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells were transfected with pCAGGS 3 × nlsI-SceI or with pCAGGS EGFP as control (see Materials and methods). The number of blasticidin-resistant colonies per plate was counted and was corrected for the cloning and transfection efficiency within each experiment. The proportion of HR due to GC was calculated from the ratio of blasticidin + zeocin-resistant: blasticidin-resistant colonies within each line (Table I). A statistically significant (p <0.0001) difference in the proportion of HR due to GC was found in all three experiments, the mean reduction being 70%. The absolute frequencies of subclasses of HR repair events for Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 lines are compared in Figure 5C and D and Table I. The absolute frequencies of all blasticidin-resistant HR events (GC, CO and SSA) and for the GC and ‘Pop out’ subclasses are expressed per 1000 cells. We find that the reduction in the proportion of HR due to GC in Brca2Tr/ΔEx27 ES cells is due to both a significant increase (234%) in the frequency of ‘Pop out’ (SSA/CO) deletion HR repair events and a significant decrease (42%) in the frequency of GC repair events. There is a significant increase (92%) in the frequency of HR events overall. This demonstrates that in the absence of wild-type Brca2 and the presence of a DSB in direct repetitive elements, ES cells shift their repair process away from error-free conservative intrachromosomal GC to non-conservative SSA or CO.
Table I. Brca2 truncation affects choice of HR repair pathway.
| DSB repair | Proportion of HR due to GC | HR events/103 cells | GC events/103 cells | PO events/103 cells |
|---|---|---|---|---|
| Brca2Tr/Ex27+ (1) | 0.43 | 50.31 | 21.500 | 28.8 |
| Brca2Tr/ΔEx27 (1) | 0.17 | 105.400 | 17.40 | 88 |
| p <0.0001 | p =0.0309 | p <0.0001 | ||
| 60% reduction | 110% stimulation HR | 19% inhibition GC | 206% stimulation PO | |
| Brca2 Tr/Ex27+ (3) | 0.61 | 77.79 | 47.500 | 30.2 |
| Brca2 Tr/ΔEx27 (4) | 0.17 | 139.420 | 23.01 | 116.4 |
| p <0.0001 | p =0.0005 | p <0.0001 | ||
| 72% reduction | 79% stimulation HR | 52% inhibition GC | 285% stimulation PO | |
| Brca2 Tr/Ex27+ (4) | 0.47 | 51.31 | 24.350 | 26.9 |
| Brca2 Tr/ΔEx27 (5) | 0.12 | 95.200 | 11.31 | 84.3 |
| p =0.0002 | p <0.0001 | 213% stimulation PO | ||
| 74% reduction | 86% | 54% inhibition GC | p <0.0001 | |
| Mean % change versus control | 70% reduction | 92% stimulation HR | 42% inhibition GC | 234% stimulation PO |
The first data column shows the proportion of HR repair events due to GC. p values are calculated using the χ2 test. The second, third and final data columns show the absolute frequency of overall HR, GC and ‘Pop out’ (PO) events respectively. p values were calculated using the unpaired Student’s t-test. Data are presented from three experiments on three independently derived Brca2Tr/ΔEx27 clones (1, 4 and 5) and control Brca2Tr/Ex27+ clones (1, 3 and 4).
Effect of Brca2ΔEX27 on both spontaneous and mitomycin C-induced SCE and chromosomal aberrations
We wished to extend our results by examining the role of Brca2 in the repair of DNA lesions more representative of global spontaneous DNA damage. The majority of spontaneous DSBs arising in cells are thought to occur at stalled replication forks, repair of which involves gene conversion from a sister chromatid (Sasaki, 1980; Haber, 1999). These events can be assayed by analysis of the frequency of spontaneous SCE. To examine the role of Brca2 in spontaneous HR between endogenous DNA sequences across the entire mouse genome we have analysed spontaneous SCE frequency by differential chromatid staining in independent sets of Brca2Tr/ΔEx27 and control Brca2Tr/Ex27+ ES cell clones. Loss of wild-type Brca2 in Brca2Tr/ΔEx27 ES cells is associated with a statistically significant reduction in spontaneous SCE (6.74 ± 0.27 SCE/metaphase) compared with Brca2Tr/Ex27+ ES cells (9.5 ± 0.43 SCE/metaphase; p < 0.0001; Figure 6C). This identifies a role for Brca2 in spontaneous recombination between sister chromatids.
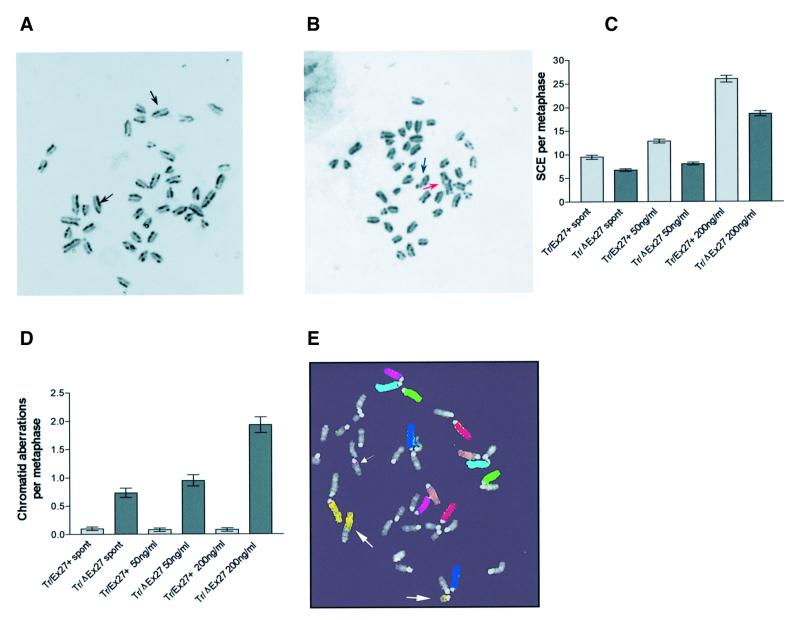
Fig. 6. Effect of Brca2 mutation on SCE and chromosomal aberration frequency. (A) Differential chromatid staining in mitomycin C-treated Brca2Tr/Ex27+ ES cell metaphases. Arrows indicate SCE events. (B) Differential chromatid staining in mitomycin C-treated Brca2Tr/ΔEx27 ES cell metaphases. The blue arrow indicates a chromatid break and the red a quadriradial chromatid exchange. Bar graphs show the numbers of SCE C) and chromatid aberrations (D) per metaphase in Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells untreated (spont) or treated with 50 or 200 ng/ml mitomycin C. Error bars = 1 SEM. Data are presented from two experiments using independently derived Brca2Tr/ΔEx27 clones and control Brca2Tr/Ex27+ clones. (E) A seven colour FISH image of a metaphase from an untreated Brca2Tr/ΔEx27 ES cell. The small arrow indicates a chromosome 4 insertion in a heterologous chromosome. The large arrows indicate a translocation and insertion between chromosome 5 and a heterologous chromosome.
Inter-strand DNA cross-links also cause replication fork arrest. Their repair involves co-operation of nucleotide excision repair and HR repair pathways to ‘un-hook’ the cross-link and repair the DSB by error-free gene conversion from a sister chromatid or by SSA, but not by NHEJ. (Van Houten et al., 1986; Faruqi et al., 1996; De Silva et al., 2000; Wang et al., 2001b). Mitomycin C inter-strand cross-links are thought to induce SCE by the gene conversion mechanism (Sonoda et al., 1999). To test whether the reduction in GC and increase in SSA demonstrated in Brca2Tr/ΔEx27 cells would lead to reduced induction of SCE and an increase in aberrant exchanges we used two different dose schedules of mitomycin C to induce inter-strand cross-links in independent sets of Brca2Tr/ΔEx27 and Brca2Tr/Ex27+ control ES cell clones.
We find that Brca2Tr/ΔEx27 cells have a significantly reduced induction of SCE when compared with Brca2Tr/Ex27+ cells at both mitomycin C doses: 18.6 ± 0.6 compared with 26 ± 0.7 SCE/metaphase (p <0.0001) at 200 ng/ml and 8.0 ± 0.3 compared with 12.9 ± 0.4 SCE/metaphase (p <0.0001) at 50 ng/ml (Figure 6A, B and C). The reduction in SCE frequency is very similar for spontaneous (29%) and mitomycin C-induced (29–38%) data sets. This suggests a similar defect in sister chromatid recombination in response to both spontaneous and mitomycin C-induced replication fork arrest. To compare the frequency of aberrant chromosomal alterations with SCE frequency, we measured chromatid aberrations in the same experiments. Brca2Tr/Ex27+ ES cells very rarely demonstrate spontaneous chromatid aberrations (0.1 ± 0.08/metaphase). In contrast, there is a 7.4-fold increase in spontaneous chromatid aberrations in Brca2Tr/ΔEx27 ES cells (0.74 ± 0.08/metaphase; p <0.0001; Figure 6D). In neither cell line did the presence of bromodeoxyuridine (BrdU) significantly influence the measurement of aberration frequency (data not shown). In addition to chromatid aberrations we used multicolour fluorescence in situ hybridization (FISH) to show the presence of chromosome-type aberrations in the same cells, including transmissible aberrations such as insertions (Figure 6E). Repair of mitomycin C inter-strand cross-links does not increase chromatid aberration frequency in Brca2Tr/Ex27+ ES cells at either 50 or 200 ng/ml (0.08 ± 0.08/metaphase). Compared with Brca2Tr/Ex27+ control cells, mitomycin C treatment of Brca2Tr/ΔEx27 ES cells induces a 12-fold increase in chromatid aberrations (0.95/metaphase) at 50 ng/ml and a 24-fold increase (1.93/metaphase) at 200 ng/ml (Figure 6A, B and D). These data indicate that Brca2 has a role in efficient HR repair of DNA cross-linking damage by equal sister chromatid crossover (SCE) and the suppression of non-conservative HR between dispersed homologous sequences.
Discussion
We have generated a conditionally mutable mouse ES cell model in order to examine the effect of a hypomorphic mutation in Brca2 (Brca2ΔEx27) on the repair of DNA by HR. The ES cell line Brca2Tr/Ex27+ contains a wild-type Brca2 protein tagged at the C-terminus with a myc epitope tag in addition to our previously described Brca2 truncation Brca2Tr2014 (Connor et al., 1997a). Upon transient expression of Cre recombinase, recombination occurs at loxP sites flanking exon 27, which truncates the wild-type Brca2 protein sequence by removing the extreme C-terminus. This region contains an interaction domain for Rad51 (Mizuta et al., 1997; Sharan et al., 1997) and a nuclear localization signal (Spain et al., 1999). The cell line Brca2Tr/ΔEx27 expresses two truncated forms of Brca2. Brca2Tr2014 retains seven BRC repeats. We show here that in common with a similar truncation in human BRCA2 (Marmorstein et al., 1998), this Brca2Tr2014 protein retains the ability to bind Rad51. As with other exon 11 truncations in both mouse and human BRCA2 (Yuan et al., 1999; Yu et al., 2000), it is associated with failure of IR-induced Rad51 focus formation. The truncated protein Brca2ΔEx27 has lost only the C-terminal Rad51 interaction domain, but despite binding Rad51 is also deficient in facilitation of induction of Rad51 foci by IR. Davies et al. (2001) have recently demonstrated a role for the Brca2 exon 11-encoded BRC repeats in binding Rad51 and abrogating the interaction of Rad51 and DNA. They propose a model where BRCA2 sequesters Rad51 in a form unable to bind DNA ready for relocalization to sites of DNA damage, at which point modification of the complex allows Rad51 to bind to the single-stranded DNA (ssDNA) tails of a DSB repair intermediate. Our results show that Brca2 exon 27 encodes a region essential for IR-induced Rad51 focus formation, suggesting that the C-terminus of Brca2 is required for assembly or stabilization of the Rad51 nucleoprotein filament.
Rad51 has been shown to have a role in homology-dependent strand invasion and gene conversion (GC and CO), but not in homology-directed repair by SSA (Baumann and West, 1998; Haber, 2000; Karran, 2000). We show that failure of normal formation of DSB-induced Rad51 foci in cells lacking the C-terminus of Brca2 is associated with defective DNA DSB repair using conservative HR repair by GC. Brca2Tr/ΔEx27 ES cells have a 4-fold (75%) reduction in the proportion of HR repair due to GC. A concurrent study has used ES cells homozygous for a Brca2 exon 27 deletion mutation with known sensitivity to IR (Morimatsu et al., 1998) and found a 5- to 6-fold reduction in non-crossover gene conversion when compared with a wild-type ES cell line (Moynahan et al., 2001). The DNA repair substrate used in that study specifically examined HR repair by the GC pathway, but did not examine HR by CO or by SSA.
Impaired Rad51 function in yeast and rodent cells causes both a decrease in GC and an increase in SSA (Ivanov et al., 1996; Kang and Symington, 2000; Lambert and Lopez, 2000; Osman et al., 2000). We addressed whether failure of the Brca2-dependent Rad51 nucleoprotein filament formation (Davies et al., 2001) is associated with an increase in use of the common 3′ ssDNA repair intermediate by Rad51-independent non-conservative SSA using a repair substrate (DR1Bsd) that can report homology-directed DNA DSB repair by GC, CO or SSA. By analysing the relative number of GC events and deletion recombination or ‘Pop out’ events we showed that the HR repair defect in Brca2Tr/ΔEx27 ES cells is specific to the GC HR repair pathway. The proportion of HR due to ‘Pop out’ deletion recombination (SSA or CO) in Brca2Tr/Ex27+ control cells is compatible with that reported with direct repeat constructs similar to DR1Bsd in wild-type mouse ES cells and in Chinese hamster ovary cells (Liang et al., 1998; Lambert et al., 1999; Dronkert et al., 2000). These ‘Pop out’ events are predominantly due to repair by SSA (Liang et al., 1998; Lambert et al., 1999; Dronkert et al., 2000). Although an identical product can be produced by gene conversion associated with a crossover, this is found to occur very rarely (Moynahan and Jasin, 1997; Richardson et al., 1998; Johnson and Jasin, 2000). We therefore believe that the DR1Bsd ‘Pop out’ deletion product arises predominantly by the SSA mechanism. The absolute frequency of HR events indicates that the reduction in the proportion of HR repair due to GC in Brca2Tr/ΔEx27 ES cells is due to both inhibition of GC and stimulation of SSA. The modest effect on GC is comparable to that found with a similar hypomorphic allele by Moynahan et al. (2001). This is less than the effect seen in Rad51-deficient models (Ivanov et al., 1996; Kang and Symington, 2000; Lambert and Lopez, 2000; Osman et al., 2000), perhaps due to the hypomorphic nature of the Brca2 alleles or the ability of some GC to take place in the absence of Brca2. The stimulation of SSA by loss of wild-type Brca2 explains the overall increase in HR repair seen in our model system and is similar to the effect of loss of Rad54 (Dronkert et al., 2000). This is consistent with the lack of effect of BRCA2 truncation on overall DSB rejoining (A.Tutt, unpublished observations) even in the absence of functional NHEJ (Wang et al., 2001a).
Having examined the role of Brca2 in repair of a site-specific DSB we extended our results to examine genome-wide repair of DNA by HR. Repair of arrested replication forks is thought to occur by HR using the error-free sister chromatid gene conversion mechanism (Sasaki, 1980; Haber, 1999). Using cytologically detectable SCE as a measure of these events, we found in untreated Brca2Tr/ΔEx27 ES cells that there was a significant reduction in SCE and a concomitant increase in chromatid and transmissible chromosome aberrations. These data indicate that Brca2 is involved in spontaneous HR by gene conversion from the sister chromatid, and demonstrate a role for Brca2 in the error-free repair of spontaneously occurring DNA damage.
HR by sister chromatid GC/CO is a dominant mechanism in the repair of inter-strand DNA cross-links, and mitomycin C is thought to induce SCE by this mechanism (Van Houten et al., 1986; Sonoda et al., 1999). Equal SCE is predicted to be an error-free mechanism of repair of inter-strand cross-links (Scully et al., 2000). HR repair of inter-strand cross-link-associated DSBs may also be achieved by SSA in mammalian cells (Faruqi et al., 1996). Repair by SSA will lead either to intrachromatid deletions of sequence between the homologous repeats or aberrant exchanges between chromatids causing chromatid aberrations. We show here that, in addition to DSB repair, Brca2 is also involved in repair of mitomycin C inter-strand cross-links by equal sister chromatid CO (SCE). Mitomycin C induces SCE, but aberrations are very infrequent in Brca2Tr/Ex27+ ES cells. Brca2Tr/ΔEx27 ES cells have a modest but statistically significant reduction in mitomycin C-induced SCE, consistent with the demonstrated reduction in DSB repair by GC. However, mitomycin C treatment of Brca2Tr/ΔEx27 ES cells induces frequent chromatid breaks and exchanges consistent with ectopic recombination between repeats by SSA. Others have attributed the induction of chromosome instability by mitomycin C in Brca2 mutant cells to the repair of inter-strand cross-links by NHEJ (Yu et al., 2000). Recent data indicate that while inter-strand cross-link repair requires functional HR pathways, the NHEJ pathway is not utilized in mammalian cells (De Silva et al., 2000). This, together with our finding of increased DNA repair by SSA in Brca2Tr/ΔEx27 ES cells, suggests that the mito mycin C-induced chromosome instability demonstrated in Brca2Tr/ΔEx27 ES cells, and in another hypomorphic Brca2 model (Yu et al., 2000), is due to repair of inter-strand cross-links by SSA rather than by NHEJ.
An increase in HR repair by SSA at the expense of GC, in the absence of Brca2, may have profound effects on genome stability. SSA aligns regions of homology as small as 29 bp (Sugawara et al., 2000) on both sides of a DSB and anneals them with the deletion of all intervening sequence, leading to intra-chromatid deletions. The presence of more than one DSB can lead to SSA between homologous repetitive elements on heterologous chromosomes, leading to translocation (Haber and Leung, 1996; Richardson and Jasin, 2000).
The recent analysis of the human genome sequence indicated that ∼50% of the sequence was made up of repetitive elements. Alu repeats are disproportionately represented in coding regions (Lander et al., 2001); recombination between Alu elements can lead to duplication or deletion of genes, or the formation of novel fusion genes (Lupski, 1998; Jasin, 2000; Pfeiffer et al., 2000). Both peri-centromeric and peri-telomeric regions of the human genome are richly endowed with highly homologous tandem duplications from elsewhere in the genome (Lander et al., 2001). These repeated sequences may also act as templates for error-prone recombination by SSA. It is of note that orthologues of Brca2 are not found in lower eukaryotes such as Saccharomyces cerevisiae, which have a substantially less repetitive genome than those of vertebrates.
In summary, these results demonstrate a role for Brca2 in the repair of DNA DSBs and DNA inter-strand cross-links by GC and SCE. Loss of wild-type Brca2 increases error-prone repair of these lesions, most likely by homology-directed SSA, suggesting a role for BRCA2 in regulation of the choice of conservative over non-conservative HR. Human tumours arise through a multi-step process of genetic change (Lengauer et al., 1998). The stimulation of error-prone DNA repair may accelerate this process (Loeb, 1991), allowing early acquisition of sufficient genetic changes necessary for early onset cancer predisposition in BRCA2 mutation carriers. Furthermore, the sensitivity of cells deficient in members of the GC pathway, including Brca2, to agents that induce DNA inter-strand cross-links (Takata et al., 2000; Yu et al., 2000) suggests that mitomycin C and platinum analogues may be of particular merit in the treatment of cancers in BRCA2 mutation carriers.
Materials and methods
DNA manipulations
Construction of Brca2 targeting vector. Mouse Brca2 sequences were isolated from a mouse 129/Sv genomic BAC library (Research Genetics) as described previously (Connor et al., 1997b). The Brca2 targeting vector was made as follows. A 1 kb 3′ homology fragment containing the Brca2 3′ UTR was generated by PCR from a BAC clone containing the 3′ Brca2 genomic fragment using the following primers: forward, 5′-GCAGATCTACTAGTAAGATGTGTACAGTT CCAGGC-3′; reverse, 5′-TGTTTGTAACTGGTGGCCTGAGAG-3′. This was cloned as an HpaI and BglII fragment into pKO SelectDT. A fragment containing bovine growth hormone poly(A) derived from pKO SelectPuro V810 was subcloned between ClaI and AscI sites in the intermediate pKO -3′ Brca2-DT vector. PGKPurobpA from pKO SelectPuro V810 was subcloned into the AscI site in the intermediate vector. The 5′ Brca2 long homology region was created by ligating a 6 kb NotI–SalI fragment, containing genomic sequences including 4.8 kb of the exon 24/25 intron, exons 25 and 26, the intervening intron and 0.45 kb of the exon 26/27 intron. An oligo linker containing a loxP site was inserted immediately 3′ to the 6 kb homology, and a second linker containing the sequence of the 9E10 myc epitope followed by a terminator and a second loxP site was inserted immediately 3′ to the first loxP site. The mouse 1.3 kb Brca2 genomic fragment containing exon 27 and the final 0.69 kb of the exon 26/27 intron was amplified by PCR from a BAC clone containing the 3′ Brca2 genomic fragment using the following primers: forward, 5′-CCGTGGGTCGACTCTGGAG GAAAGTAGATCAGACTCC-3′; reverse, 5′-ACCGGGGTCGACAG ACTCAACAGCTAATTTCTCACTGC-3′. This was digested with SalI and cloned, in-frame with the myc tag, into the unique SalI site between the loxP and myc–terminator–loxP linkers.
Construction of DR1Bsd. A silent mutation was created in Bsd at nucleotide position 3337 within pEF6/V5-His C (Invitrogen), generating a SalI site. Full-length BSD including SV40 poly(A) was amplified by PCR, and inserted between PstI and HindIII sites in pMC1Neo to replace Neo and create pMC1Bsd. A 22 bp linker containing the 18 bp I-SceI site and SalI overhangs was then inserted into the SalI site at position 279 in the coding sequence of Bsd within pMC1Bsd to create pMC1S1Bsd. 5′ΔBsd, lacking the 5′ 34 bp of Bsd, was created by PCR amplification of Bsd and SV40 poly(A) using an internal forward primer 5′-AAGAAGCTTGGATCCTCATTGAAAGAGCAACGG and reverse primer 5′-GCTCTAGCTAAAGCTTGACG, and cloned as a HindIII fragment downstream of S1Bsd in pMC1S1Bsd to give pMC1S1Bsd5′ΔBsd. Zeo was subcloned from pZeoSV (Invitrogen), replacing Neo in pPGKNeobpa. The PGK Zeo bpa cassette was subcloned by blunt ligation into the BamHI site between S1Bsd and 5′ΔBsd. This gave the final repair substrate plasmid pDR1Bsd. The substrate was released by digestion with NdeI and AseI, and ligated into a NdeI–AseI linker inserted in the unique Asp718 site 3′ of the Puro cassette within the Brca2 targeting construct. Sequencing revealed its insertion in a 3′ to 5′ orientation relative to the targeting construct.
Other plasmids. pS1Bsd Zeo was created by subcloning S1Bsd into pBluescriptKS. The PGK Zeo bpa cassette was then subcloned 3′ to S1Bsd in the polylinker.
pCAGGS EGFP-Cre was created by inserting a multiple cloning site between EcoRI sites in pCAGGS. A 1.9 kb XhoI–MluI EGFP-Cre fragment from pBs594 (Le et al., 1999) was subcloned into this multiple cloning site in pCAGGS. pCAGGS 3 × nls I-SceI was created by subcloning an 0.87 kb EcoRI–SalI fragment containing 3 × nls I-SceI from pPGK 3 × nls I-SceI (a gift from G.Donoho) into pCAGGS.
All cloning steps were verified by restriction digestion and ABI automated sequencing.
Targeting Brca2
Brca2Tr2014/Wt ES cells were electroporated with 20 µg of the NotI-linearized Brca2–DR1Bsd targeting construct, cultured for 24 h on gelatin-coated dishes and subsequently selected with puromycin (1 µg/ml) for 7 days. Puromycin-resistant colonies were isolated, expanded and frozen. Clones were screened for correct integration into the Brca2 locus by Southern blot analysis as depicted in Figure 1.
Cre-mediated deletion of Brca2 exon 27
Brca2Tr/Ex27+ ES cells were transiently transfected with pCAGGS EGFP-Cre or pCAGGS EGFP as control. Cells were analysed by FACS, and the GFP-positive population sorted to >99% purity and returned to culture. Genomic DNA was extracted from pooled clones, digested with HincII and subjected to Southern blot analysis, using a fragment of Puro as probe (probe C), to confirm deletion of sequence between loxP sites. Translational read-through into the intron is predicted to extend the open reading frame beyond exon 26 by only two amino acids. Brca2Tr/ΔEx27 and Brca2Tr/Ex27+ control clonal cell lines were derived by FACS sorting following transfection of pCAGGS EGFP-Cre or pCAGGS EGFP, respectively. Clones were isolated, expanded and frozen. Deletion of sequence between the loxP sites was confirmed by PCR using primers 5′ and 3′ to the loxP sites in the targeting construct: forward, 5′-TCCTTTGCTGGCTCCTGAGC-3′; reverse, 5′-TAGTGAGAC GTGCTACTTCC-3′.
Immunoprecipitation and immunoblotting
Whole-cell extracts were prepared by lysing ES cells in NETN buffer (20 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA pH 8.0, 10% glycerol, 0.5% NP-40) containing protease inhibitors. Immunoprecipitations were carried out in NETN buffer by pre-binding either 1 µl of rabbit polyclonal anti-Rad51 (gift from Stephen West), 10 µl of rat polyclonal antiserum raised against amino acids 20–246 of mouse Brca2 (S.Swift and A.Ashworth, unpublished), 5 µg of Jac6 rat monoclonal anti-myc raised against the 9E10 epitope or 2 µg of goat or rabbit polyclonal anti-9E10 myc (Santa Cruz) to 30 µl of protein A– or G–Sepharose beads (Sigma) for 1 h at 4°C, washing twice and incubating with 1–2 mg of whole-cell extract for 2 h at 4°C. Following washing, bound proteins were separated by 6% SDS–PAGE and detected by immunoblotting with either anti-mouse Brca2 N-terminal, Jac6 or polyclonal anti-myc primary antibodies, horseradish peroxidase-coupled secondary antibodies and an ECL detection system (Amersham).
Immunofluorescence
ES cells were grown on gelatin-coated glass cover slips in six-well plates. For irradiation experiments, plates were irradiated to a total dose of 5 or 10 Gy 250 kv X-rays, then cultured for 5 h at 37°C before fixation in 4% paraformaldehyde. Cells were permeabilized in 0.2% Triton X-100 in phosphate-buffered saline (PBS), blocked in 10% fetal calf serum (FCS) and then incubated in either 1:200 of Jac6 and 1:200 of anti-Rad51 or anti-Rad51 alone, washed in PBS and then incubated with fluorescein isothiocyanate (FITC)- or Texas Red-conjugated secondary antibodies. After further washes, coverslips were mounted in Mowiol (Calbiochem) and viewed on a Zeiss Axiomat epifluorescence microscope or Bio-Rad confocal microscope. Images were captured using SmartCapture 2 (Digital Scientific Ltd, Cambridge, UK). Experiments were performed at least three times using three independent sets of Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 clones.
Cell culture and transfection
ES cells were grown on gelatin-coated plates in Dulbecco’s modified Eagle’s medium supplemented with 15% FCS, l-glutamine, penicillin/streptomycin, β-mercaptoethanol (all Sigma), non-essential amino acids (Gibco) and ESGRO LIF (Chemicon). Antibiotic selection was performed in either 5 µg/ml blasticidin or 150–300 µg/ml zeocin, or both. Transfections were performed as follows: 2 × 105 ES cells per well of a six-well plate were transfected with 2 µg of supercoiled plasmid DNA using lipofectamine (Gibco) according to the manufacturer’s instructions.
I-SceI DSB repair assay
Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells were transfected with pCAGGS 3 × nls I-SceI or with pCAGGS EGFP as control. Twenty-four hours after transfection, cells were trypsinized, counted and replated at 2 × 105 cells/10 cm plate. Forty-eight hours after transfection, selection with blasticidin on triplicate plates or both blasticidin and zeocin in triplicate was commenced and continued daily for 7 days. Plates were fixed in methanol and stained with crystal violet. The total number of surviving colonies was counted on all plates. An independent analysis of the proportion of blasticidin-resistant colonies, which were also resistant to zeocin, was performed as follows. Blasticidin-resistant colonies (24 in each of three experiments) were isolated, expanded, replated in 24-well plates and then cultured in zeocin selection for 7 days. Plates were then fixed in methanol stained with crystal violet and each clone scored for viability. For analysis of genomic DNA repair products, blasticidin-resistant colonies were pooled, lysed, DNA extracted and subjected to restriction digestion and Southern blot analysis as described in Figures 4B and 5B.
Correction for cloning and transfection efficiencies
For analysis of transfection efficiency in each experiment, Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 ES cells were transfected with pCAGGS EGFP. Twenty-four hours after transfection, cells were subjected to FACS analysis. GFP-positive cells were expressed as a proportion of all living cells. A total of 2 × 105 cells were analysed per data point. In each experiment, the cloning efficiency of pCAGGS 3 × nls I-SceI transfected cells was determined by plating 1 × 104 cells per 10 cm plate in non-selective medium. To control for any differences in cloning efficiency in zeocin selection between Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 lines, cloning efficiency was also determined in zeocin. Colonies were fixed, stained and counted after 7 days, and the number expressed as a proportion of cells plated. Colony counts after blasticidin or blasticidin + zeocin selection were divided by a correction factor (transfection efficiency × cloning efficiency) to obtain the absolute frequency of total HR and GC events, respectively. The frequency of ‘Pop out’ deletion recombination is derived by subtracting the absolute frequency of blasticidin/zeocin-resistant colonies from that of blasticidin-resistant colonies. The frequency of ‘Pop out’ deletion recombination was also derived by Southern blot analysis as described in Figure 5B, followed by quantification using a phosphoimager. Experiments were performed at least three times using three independent sets of Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 clones. Analysis of the ability of NHEJ to recreate wild-type Bsd from S1Bsd alone was performed as follows. Brca2Tr2014/wt ES cells were electroporated with 20 µg of ScaI-linearized pS1Bsd Zeo and selected with zeocin for 7 days. Pooled zeocin-resistant clones were subjected to the I-SceI repair assay as described above.
FACS analysis and cell sorting
ES cells were trypsinized, washed in PBS and resuspended in 1–3 ml of PBS. Cells were analysed on either a Becton Dickenson FACS Calibur or FACS Vantage sorter using a 488 nm argon laser. GFP fluorescence was measured in the FL1 channel. The FLI GFP-positive gate was set to include <1% of untransfected control ES cells. For purification, cells expressing EGFP-Cre or EGFP control cells were double sorted to >90% GFP positive and then replated on gelatin for cloning.
Cytogenetic analyses
ES cells were cultured in medium containing 10 µM BrdU (Sigma) for two cell cycles (20 h) to differentially label sister chromatids. Colcemid (Karyomax; Gibco) was added for the final hour. For mitomycin C (Sigma) treatment, ES cells were incubated either with 200 ng/ml mitomycin C in the first hour of BrdU staining or 50 ng/ml for the final 6 h. Metaphase preparations were made using standard methods. Differential chromatid staining was achieved with the fluorescence plus Giemsa staining method (Perry and Wolff, 1974). More than 100 metaphases were analysed per clone for both SCE and chromatid aberrations. Experiments were performed using two independent sets of Brca2Tr/Ex27+ and Brca2Tr/ΔEx27 clones. Mouse RainbowFISH™ (Cambio, UK) specific paints for chromosomes 1–7 were employed to identify chromosome aberrations in ES cells. Procedures were as stated in the Mouse RainbowFISH protocol. Images were captured and processed using SmartCapture 2 (Digital Scientific Ltd).
Acknowledgments
Acknowledgements
We thank Stephen West for generously providing the anti-Rad51 antibody, Greg Donoho for pPGK 3 × nls I-SceI, M.Okabe for pCX EGFP (pCAGGS EGFP), and Ian Titley for FACS analysis and cell sorting. We thank Peter Kerr and other members of the Ashworth Laboratory for providing insightful comments. This work is supported by grants from the Cancer Research Campaign, the Medical Research Council, Breakthrough Breast Cancer and the Mary-Jean Mitchell Green Foundation.
References
- Ban S., Shinohara,T., Hirai,Y., Moritaku,Y., Cologne,J.B. and MacPhee,D.G. (2001) Chromosomal instability in BRCA1- or BRCA2-defective human cancer cells detected by spontaneous micronucleus assay. Mutat. Res., 474, 15–23. [DOI] [PubMed] [Google Scholar]
- Baumann P. and West,S.C. (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci., 23, 247–251. [DOI] [PubMed] [Google Scholar]
- Bertwistle D. and Ashworth,A. (1998) Functions of the BRCA1 and BRCA2 genes. Curr. Opin. Genet. Dev., 8, 14–20. [DOI] [PubMed] [Google Scholar]
- Chen P.L., Chen,C.F., Chen,Y., Xiao,J., Sharp,Z.D. and Lee,W.H. (1998) The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl Acad. Sci. USA, 95, 5287–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor F., Bertwistle,D., Mee,P.J., Ross,G.M., Swift,S., Grigorieva,E., Tybulewicz,V.L. and Ashworth,A. (1997a) Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nature Genet., 17, 423–430. [DOI] [PubMed] [Google Scholar]
- Connor F., Smith,A., Wooster,R., Stratton,M., Dixon,A., Campbell,E., Tait,T.M., Freeman,T. and Ashworth,A. (1997b) Cloning, chromosomal mapping and expression pattern of the mouse Brca2 gene. Hum. Mol. Genet., 6, 291–300. [DOI] [PubMed] [Google Scholar]
- Davies A.A., Masson,J.Y., McIlwraith,M.J., Stasiak,A.Z., Stasiak,A., Venkitaraman,A.R. and West,S.C. (2001) Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell, 7, 273–282. [DOI] [PubMed] [Google Scholar]
- De Silva I.U., McHugh,P.J., Clingen,P.H. and Hartley,J.A. (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol., 20, 7980–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert M.L., Beverloo,H.B., Johnson,R.D., Hoeijmakers,J.H., Jasin,M. and Kanaar,R. (2000) Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol., 20, 3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqi A.F., Seidman,M.M., Segal,D.J., Carroll,D. and Glazer,P.M. (1996) Recombination induced by triple-helix-targeted DNA damage in mammalian cells. Mol. Cell. Biol., 16, 6820–6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gretarsdottir S., Thorlacius,S., Valgardsdottir,R., Gudlaugsdottir,S., Sigurdsson,S., Steinarsdottir,M., Jonasson,J.G., Anamthawat-Jonsson,K. and Eyfjord,J.E. (1998) BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res., 58, 859–862. [PubMed] [Google Scholar]
- Haber J.E. (1999) DNA recombination: the replication connection. Trends Biochem. Sci., 24, 271–275. [DOI] [PubMed] [Google Scholar]
- Haber J.E. (2000) Recombination: a frank view of exchanges and vice versa. Curr. Opin. Cell Biol., 12, 286–292. [DOI] [PubMed] [Google Scholar]
- Haber J.E. and Leung,W.Y. (1996) Lack of chromosome territoriality in yeast: promiscuous rejoining of broken chromosome ends. Proc. Natl Acad. Sci. USA, 93, 13949–13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson S. et al. (1997) Moderate frequency of BRCA1 and BRCA2 germ-line mutations in Scandinavian familial breast cancer. Am. J. Hum. Genet., 60, 1068–1078. [PMC free article] [PubMed] [Google Scholar]
- Ivanov E.L., Sugawara,N., Fishman-Lobell,J. and Haber,J.E. (1996) Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics, 142, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M. (2000) Chromosome breaks and genomic instability. Cancer Invest., 18, 78–86. [DOI] [PubMed] [Google Scholar]
- Johnson R.D. and Jasin,M. (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J., 19, 3398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L.E. and Symington,L.S. (2000) Aberrant double-strand break repair in rad51 mutants of Saccharomyces cerevisiae. Mol. Cell. Biol., 20, 9162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P. (2000) DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev., 10, 144–50. [DOI] [PubMed] [Google Scholar]
- Khanna K.K. and Jackson,S.P. (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genet., 27, 247–254. [DOI] [PubMed] [Google Scholar]
- Lambert S. and Lopez,B.S. (2000) Characterization of mammalian RAD51 double strand break repair using non-lethal dominant-negative forms. EMBO J., 19, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S., Saintigny,Y., Delacote,F., Amiot,F., Chaput,B., Lecomte,M., Huck,S., Bertrand,P. and Lopez,B.S. (1999) Analysis of intrachromosomal homologous recombination in mammalian cell, using tandem repeat sequences. Mutat. Res., 433, 159–168. [DOI] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Le Y., Miller,J.L. and Sauer,B. (1999) GFPcre fusion vectors with enhanced expression. Anal. Biochem., 270, 334–336. [DOI] [PubMed] [Google Scholar]
- Lengauer C., Kinzler,K.W. and Vogelstein,B. (1998) Genetic instabilities in human cancers. Nature, 396, 643–649. [DOI] [PubMed] [Google Scholar]
- Liang F., Han,M., Romanienko,P.J. and Jasin,M. (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Lukacsovich,T. and Waldman,A.S. (1999) Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell. Biol., 19, 8353–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L.A. (1991) Mutator phenotype may be required for multistage carcinogenesis. Cancer Res., 51, 3075–3079. [PubMed] [Google Scholar]
- Lupski J.R. (1998) Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet., 14, 417–422. [DOI] [PubMed] [Google Scholar]
- Marmorstein L.Y., Ouchi,T. and Aaronson,S.A. (1998) The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl Acad. Sci. USA, 95, 13869–13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta R., LaSalle,J.M., Cheng,H.L., Shinohara,A., Ogawa,H., Copeland,N., Jenkins,N.A., Lalande,M. and Alt,F.W. (1997) RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc. Natl Acad. Sci. USA, 94, 6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimatsu M., Donoho,G. and Hasty,P. (1998) Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to γ-radiation and premature senescence. Cancer Res., 58, 3441–3447. [PubMed] [Google Scholar]
- Moynahan M.E. and Jasin,M. (1997) Loss of heterozygosity induced by a chromosomal double-strand break. Proc. Natl Acad. Sci. USA, 94, 8988–8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Pierce,A.J. and Jasin,M. (2001) BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell, 7, 263–272. [DOI] [PubMed] [Google Scholar]
- Osman F., Adriance,M. and McCready,S. (2000) The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet., 38, 113–125. [DOI] [PubMed] [Google Scholar]
- Patel K.J. et al. (1998) Involvement of Brca2 in DNA repair. Mol. Cell, 1, 347–357. [DOI] [PubMed] [Google Scholar]
- Perry P. and Wolff,S. (1974) New Giemsa method for the differential staining of sister chromatids. Nature, 251, 156–158. [DOI] [PubMed] [Google Scholar]
- Pfeiffer P., Goedecke,W. and Obe,G. (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis, 15, 289–302. [DOI] [PubMed] [Google Scholar]
- Rahman N. and Stratton,M.R. (1998) The genetics of breast cancer susceptibility. Annu. Rev. Genet., 32, 95–121. [DOI] [PubMed] [Google Scholar]
- Richardson C. and Jasin,M. (2000) Frequent chromosomal translocations induced by DNA double-strand breaks. Nature, 405, 697–700. [DOI] [PubMed] [Google Scholar]
- Richardson C., Moynahan,M.E. and Jasin,M. (1998) Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev., 12, 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P., Smih,F. and Jasin,M. (1994) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol., 14, 8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M.S. (1980) Chromosome aberration formation and sister chromatid exchange in relation to DNA repair in human cells. In Generoso,W.M. and De Serres,F.J. (eds), DNA Repair and Mutagenesis in Eukaryotes. Plenum Press, New York, NY, pp. 285–313. [DOI] [PubMed]
- Scully R., Chen,J., Plug,A., Xiao,Y., Weaver,D., Feunteun,J., Ashley,T. and Livingston,D.M. (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell, 88, 265–275. [DOI] [PubMed] [Google Scholar]
- Scully R., Ganesan,S., Vlasakova,K., Chen,J., Socolovsky,M. and Livingston,D.M. (1999) Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell, 4, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Scully R., Puget,N. and Vlasakova,K. (2000) DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene, 19, 6176–6183. [DOI] [PubMed] [Google Scholar]
- Sharan S.K. et al. (1997) Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature, 386, 804–810. [DOI] [PubMed] [Google Scholar]
- Shen S.X., Weaver,Z., Xu,X., Li,C., Weinstein,M., Chen,L., Guan,X.Y., Ried,T. and Deng,C.X. (1998) A targeted disruption of the murine Brca1 gene causes γ-irradiation hypersensitivity and genetic instability. Oncogene, 17, 3115–3124. [DOI] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,M.S., Morrison,C., Yamaguchi-Iwai,Y., Takata,M. and Takeda,S. (1999) Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol., 19, 5166–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain B.H., Larson,C.J., Shihabuddin,L.S., Gage,F.H. and Verma,I.M. (1999) Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc. Natl Acad. Sci. USA, 96, 13920–13925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N., Ira,G. and Haber,J.E. (2000) DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell. Biol., 20, 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M. et al. (2000) The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol., 20, 6476–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirkkonen M. et al. (1997) Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res., 57, 1222–1227. [PubMed] [Google Scholar]
- Tutt A., Gabriel,A., Bertwistle,D., Connor,F., Paterson,H., Peacock,J., Ross,G. and Ashworth,A. (1999) Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol., 9, 1107–1110. [DOI] [PubMed] [Google Scholar]
- Van Houten B., Gamper,H., Holbrook,S.R., Hearst,J.E. and Sancar,A. (1986) Action mechanism of ABC excision nuclease on a DNA substrate containing a psoralen crosslink at a defined position. Proc. Natl Acad. Sci. USA, 83, 8077–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Zeng,Z.C., Bui,T.A., DiBiase,S.J., Qin,W., Xia,F., Powell,S.N. and Iliakis,G. (2001a) Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA1 or BRCA2. Cancer Res., 61, 270–277. [PubMed] [Google Scholar]
- Wang X., Peterson,C.A., Zheng,H., Nairn,R.S., Legerski,R.J. and Li,L. (2001b) Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol. Cell. Biol., 21, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Weaver,Z., Linke,S.P., Li,C., Gotay,J., Wang,X.W., Harris,C.C., Ried,T. and Deng,C.X. (1999) Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell, 3, 389–395. [DOI] [PubMed] [Google Scholar]
- Yu V.P., Koehler,M., Steinlein,C., Schmid,M., Hanakahi,L.A., van Gool,A.J., West,S.C. and Venkitaraman,A.R. (2000) Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev., 14, 1400–1406. [PMC free article] [PubMed] [Google Scholar]
- Yuan S.S., Lee,S.Y., Chen,G., Song,M., Tomlinson,G.E. and Lee,E.Y. (1999) BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res., 59, 3547–3551. [PubMed] [Google Scholar]