

Abstract
The genetic code is defined by the specific aminoacylations of tRNAs by aminoacyl-tRNA synthetases. Although the synthetases are widely conserved through evolution, aminoacylation of a given tRNA is often system specific—a synthetase from one source will not acylate its cognate tRNA from another. This system specificity is due commonly to variations in the sequence of a critical tRNA identity element. In bacteria and the cytoplasm of eukaryotes, an acceptor stem G3:U70 base pair marks a tRNA for aminoacylation with alanine. In contrast, Drosophila melanogaster (Dm) mitochondrial (mt) tRNAAla has a G2:U71 but not a G3:U70 pair. Here we show that this translocated G:U and the adjacent G3:C70 are major determinants for recognition by Dm mt alanyl-tRNA synthetase (AlaRS). Additionally, G:U at the 3:70 position serves as an anti-determinant for Dm mt AlaRS. Consequently, the mitochondrial enzyme cannot charge cytoplasmic tRNAAla. All insect mitochondrial AlaRSs appear to have split apart recognition of mitochondrial from cytoplasmic tRNAAla by translocation of G:U. This split may be essential for preventing introduction of ambiguous states into the genetic code.
Keywords: aminoacyl-tRNA synthetases/genetic code evolution/mitochondrial tRNA recognition/tRNA recognition
Introduction
The genetic code is established through specific aminoacylation of tRNAs by aminoacyl-tRNA synthetases. The aminoacylation reaction proceeds in two steps, the first of which produces an aminoacyl adenylate through the condensation of the cognate amino acid with ATP and release of pyrophosphate. In the second step, the aminoacyl group is transferred to the 3′ end of the tRNA, producing aminoacyl-tRNA. Through this, the nucleotide triplet of the code is physically connected (through the tRNA) to the cognate amino acid. The specificity of the aminoacylation reaction is achieved through direct recognition of the cognate tRNA by the specific synthetase. In some cases, the recognition depends on the anticodon, while in others the relationship between the amino acid and the nucleotide triplet codon is indirect. In these cases, and even in those where synthetase contact is made with the anticodon, the acceptor stem contains determinants necessary for specific aminoacylation. These RNA sequences/structures in the acceptor stem (near the amino acid attachment site) can be considered an operational RNA code for specific aminoacylation (Schimmel et al., 1993). This RNA code may have pre-dated the genetic code and may have given rise to the genetic code (de Duve, 1988; Möller and Janssen, 1990; Di Giulio, 1992; Rodin et al., 1996; Musier-Forsyth and Schimmel, 1999).
Variations in the operational RNA code imbedded in acceptor stems can lead to ‘system’-specific recognition of tRNAs, where a synthetase of one system does not aminoacylate its cognate tRNA from another system. One way this can be accomplished is through sequence permutation, in which two homologous synthetases from separate systems use the same specific locations in the acceptor stem as sites of recognition, but target different bases at these particular sites. An example is species-specific recognition, where a particular species’ synthetase does not aminoacylate its cognate tRNA from a different species, despite the anticodons being the same. Glycyl-tRNA synthetases from Escherichia coli and humans do not to cross-aminoacylate their respective tRNAs (Shiba et al., 1994a), because the critical U73 nucleotide in E.coli tRNAGly is changed to an A in eukaryotic cytoplasmic tRNAGly (Hipps et al., 1995). Similarly, isoleucyl-tRNA synthetase from E.coli does not aminoacylate mammalian tRNAIle (Shiba et al., 1994b). In a well-studied case, tyrosyl-tRNA synthetases from prokaryotes and eukaryotic cytoplasm are unable to cross-aminoacylate their respective tRNAs (Chow and RajBhandary, 1993; Quinn et al., 1995). In both systems, the 1:72 base pair of the acceptor stem is a critical determinant for aminoacylation, but the G1:C72 pair in prokaryotes is replaced by C1:G72 in eukaryotes. A simple G:C to C:G transversion is sufficient to switch species-specific recognition of substrates that are charged with tyrosine (Quinn et al., 1995; Wakasugi et al., 1998). A switch in species-specific aminoacylation in this system was also achieved by Wakasugi et al. (1998) through a simple peptide transplant from one synthetase to another. This experiment demonstrated the role of this peptide in determining the specificity of recognition of the 1:72 base pair.
System-specific aminoacylation also exists within the eukaryotic cell, where two separate compartments (the mitochondria and cytoplasm) utilize separate translation machineries but essentially the same genetic code. In many eukaryotes, the nuclear genome encodes separate genes for the same enzyme activity in each compart ment. The mitochondrial enzyme is translated in the cytoplasm and transported to the mitochondria where it aminoacylates tRNAs encoded by the mitochondrial genome. These mitochondrial tRNAs are often unique in sequence and secondary structure, and differ significantly from cytoplasmic tRNAs (Martin, 1995; Sprinzl et al., 1998).
One particular example of animal mitochondrial tRNAs differing significantly from their cytoplasmic counterparts exists in the alanine system. Recognition of tRNAAla by E.coli alanyl-tRNA synthetase (AlaRS) depends largely on a single G3:U70 base pair in the acceptor stem (Hou and Schimmel, 1988; McClain and Foss, 1988). Introduction of this base pair into other tRNAs confers alanine acceptance upon them (Hou and Schimmel, 1988; McClain and Foss, 1988; Francklyn and Schimmel, 1989). The unpaired exocyclic 2-amino group of G3 is the major atomic determinant for aminoacylation with alanine (Musier-Forsyth et al., 1991, 1995). This G3:U70 pair, as well as base pairs 1:72 and 2:71 and the N73 discriminator base, are conserved in all known tRNAAla sequences from prokaryotes, archaea, eukaryote cytoplasm and chloroplasts. G3:U70 has been shown explicitly to be essential for alanine acceptance in E.coli, the yeast Saccharomyces cerevisiae, the insect Bombyx mori cytoplasmic tRNAAla, Caenorhabditis elegans mitochondrial tRNAAla, and human cytoplasmic tRNAAla (Hou and Schimmel, 1988, 1989; McClain and Foss, 1988; Ripmaster et al., 1995; Shiba et al., 1995; Chihade et al., 1998).
Interestingly, the Drosophila melanogaster mitochondrial (Dm mt) tRNAAla (like other animal mitochondrial alanine tRNAs) does not contain the G3:U70 base pair. In Dm mt tRNAAla, a G:U pair is found at position 2:71 (Figure 1). The lack of a canonical G3:U70 in Dm mt tRNAAla, as well as the unusual G2:U71 base pair, suggested the possibility that G:U had been translocated as a critical identity element from 3:70 to 2:71. If this is the case, then it would be the first example where the same base pair is recognized by homologous synthetases at different locations in the acceptor stem.
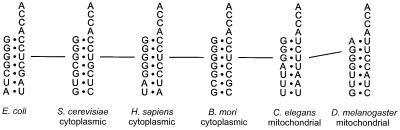
Fig. 1. Comparison of acceptor stems for alanyl-tRNAs. Lines connect the G:U base pair in each case.
To investigate this possibility, we sought to clone, express and purify Dm mt AlaRS. Having isolated the enzyme, we then focused on acceptor stem recognition and the role of the G2:U71 base pair. After verifying that Dm mt AlaRS was active, we constructed a series of substrates that tested the significance of the translocated G:U pair. In addition, we tested the role of sequence context for manifestation of the functional effects of the G:U pair.
Results
Identification of a gene encoding Dm mt AlaRS
The unusual location of the G:U pair in the acceptor stem of Dm mt tRNAAla raises the possibility that a translocated G:U is the critical element for recognition by AlaRS. The first step towards investigating this possibility was to identify a gene encoding Dm mt AlaRS. To identify gene candidates encoding a Dm mt AlaRS, a search was performed of the expressed sequence tag (EST) database of NCBI for clones with similarity in sequence to a human mt AlaRS gene fragment. Two distinct D.melanogaster gene candidates were identified (Chihade et al., 2000). In prokaryotes, distinct aminoacyl-tRNA synthetases exist for each amino acid. Because in eukaryotes two nuclear-encoded enzymes typically are found for each amino acid, one that is cytoplasmic and one mitochondrial, the two gene candidates found in the D.melanogaster genome most likely corresponded to the cytoplasmic and mitochondrial forms of AlaRS.
Sequencing of each of these clones (LD11251 and LD07142, Berkeley Drosophila Genome Project) revealed the presence of poly(A) tails, suggesting that the genes encode proteins that extend to the C-terminus. LD11251 encoded a polypeptide of 1012 amino acids with a predicted molecular mass of 113.1 kDa, and LD07142 encoded a polypeptide of 966 amino acids with a predicted molecular mass of 107.7 kDa.
Sequence alignments of the 1012 amino acid AlaRS encoded by LD11251 with known sequences of AlaRSs revealed an N-terminal extension that was not present in the 966 amino acid sequence predicted from clone LD07142. The N-terminal sequence of the protein encoded by LD11251 was predicted (by the program PSORTII; Nakai and Kanehisa, 1992) to be a suitable mitochondrial pre-sequence with a cleavage site between residues 11 and 12. Additionally, PSORTII predicted a probability of 21.7% for the protein encoded by clone LD11251 to be localized to the mitochondria, while a 0% probability was predicted for the protein encoded by clone LD07142 to be localized to the mitochondria. In addition, clone LD07142 encodes a four amino acid insert (between N58 and Y59 of the E.coli enzyme) immediately prior to motif 2 (a conserved sequence found in class II tRNA synthetases) of the active site domain that is typical of eukaryotic cytoplasmic AlaRSs (Chihade et al., 1998). Clone LD11251 does not encode this insert, suggesting that it is more prokaryote-like (as are most mitochondrial enzymes).
Lastly, previous phylogenetic analysis of clones LD11251 and LD07142 (Chihade et al., 2000) showed that the protein encoded by clone LD11251 groups most closely with the C.elegans mitochondrial AlaRS and is further from the cytoplasmic enzymes. Conversely, LD07142 encodes a protein that groups strongly with the cytoplasmic AlaRSs from several organisms. Thus, this analysis is also consistent with the conclusion that LD11251 encodes mitochondrial AlaRS from D.melanogaster.
Expression and purification of Dm mt AlaRS in Spodoptera frugiperda
Using methods described in Materials and methods, recombinant C-terminal His-tagged Dm mt AlaRS from Spodoptera frugiperda (Sf9) cells was purified to >90% homogeneity (Figure 2). The size of the purified protein (110 kDa) is within experimental error of the predicted molecular weight.
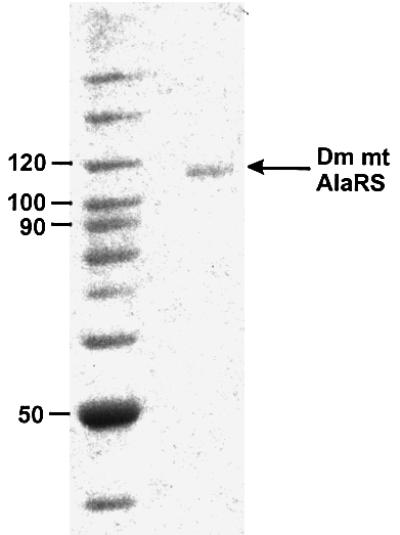
Fig. 2. SDS–PAGE analysis of purified Dm mt AlaRS. Left lane, molecular weight markers; right lane, 5 pmol Dm mt AlaRS. Samples were analyzed on a 8% (w/v) polyacrylamide gel and the proteins were visualized by staining with Coomassie blue.
Determination of Dm mt AlaRS dependence on the acceptor stem of alanine tRNA for substrate recognition
Early work established that bacterial AlaRS makes no contact with the anticodon, and that base substitutions into the anticodon do not affect aminoacylation (Hou and Schimmel, 1988; Park and Schimmel, 1988). Francklyn and Schimmel (1989) showed that RNA minihelices composed of the acceptor–TψC–stem–loop and microhelices based on the 7 bp acceptor stem are robust substrates for aminoacylation with alanine. These substrates maintain the essential substrate characteristics of full tRNAAla. The two orders of magnitude decrease in kcat/Km in the minihelix and microhelix substrates relative to full-length tRNA is due to an increased Km, suggesting that nucleotides outside of the acceptor stem contribute only binding energy to the aminoacylation reaction (Francklyn et al., 1992). To determine if the acceptor stem has a major role in establishing identity for tRNAAla in the Dm mt system, with no contribution from the anticodon, Dm mt synthetic tRNAAlas were made by in vitro transcription and tested with the Dm mt AlaRS. A wild-type Dm mt tRNAAla transcript and a transcript where two of the three anticodon nucleotides are swapped (G35C and C36G) were prepared. Wild-type and G35C,C36G tRNAAla were good substrates and essentially equivalent for the Dm mt AlaRS (see below). Thus, the anticodon of Dm mt tRNAAla does not contribute to recognition by Dm mt AlaRS. For this reason and because small chemically synthesized microhelices allow easy permutations of sequences that are localized to the acceptor stem, we chose to do further studies with chemically synthesized microhelices based on the 7 bp acceptor stem of Dm mt tRNAAla as substrates for Dm mt AlaRS.
Translocated acceptor stem G:U base pair is a major identity element for Dm mt AlaRS
To ensure that none of the sequence permutations of the microhelices destroyed the ability of the oligonucleotides to form the desired hairpin structure necessary for aminoacylation assays, melting temperatures of these substrates were determined. All Tms were above the temperature (37°C) at which aminoacylation assays were carried out (data not shown). We first verified that the ‘wild-type’ sequence of Dm mt microhelixAla was charged with alanine. Then, to investigate the significance of the G2:U71 base pair, we made Dm mt microhelixAla substrates with variations at the 2:71 position. Remark ably, a substrate containing a G2:C71 substitution was inactive for aminoacylation (Figure 3A). This result suggested that a G:U pair at 2:71 significantly determines recognition by the mitochondrial synthetase. This led us to test whether recognition could be induced by placement of a G3:U70 base pair into the microhelix that contained a G2:C71 substitution. However, this substitution did not allow recovery of aminoacylation (Figure 3A).
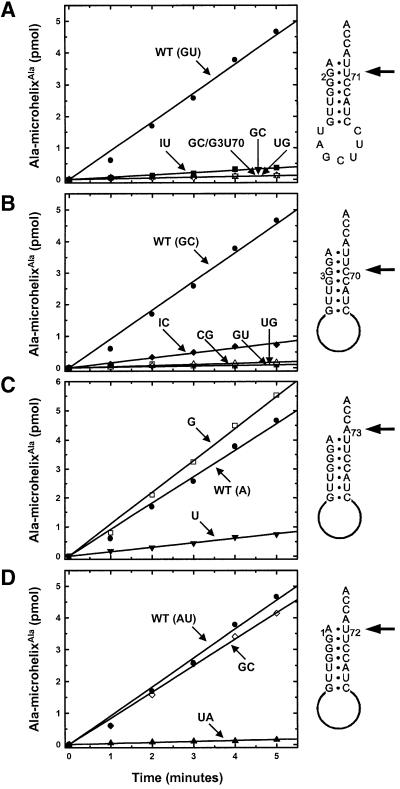
Fig. 3. Aminoacylation by Dm mt AlaRS at pH 7.5, 37°C of microhelices based on the acceptor stem of Dm mt tRNAAla. (A) Aminoacylation of microhelices with variations at the 2:71 position. (B) Aminoacylation of microhelices with variations at the 3:70 position. (C) Aminoacylation of microhelices with variations at the N73 position. (D) Aminoacylation of microhelices with variations at the 1:72 position. Positions of variation are highlighted with arrows pointing to the appropriate locations on the microhelices.
The G:U wobble pair presents a free, exocyclic amino group on the dyad axis of the RNA minor groove. (In a Watson–Crick G:C pair, this amino group is hydrogen-bonded and therefore not free.) This exocyclic 2-amino group is critical for G:U recognition at the 3:70 position by the conventional AlaRSs. With this in mind, we investigated whether the exocyclic 2-amino group of G2 is the major atomic determinant for the Dm mt enzyme. This possibility was tested by substituting inosine for guanosine at the 2-position. The I2:U71 Dm mt microhelixAla lost >90% of its activity relative to the wild-type substrate (Figure 3A). This loss in activity is qualitatively similar to the loss seen when the equivalent substitution of I3 for G3 is made in the E.coli system (Musier-Forsyth et al., 1991).
To determine whether a 2-amino group presented to the minor groove dyad axis by the opposite RNA strand is sufficient for recognition, we made a U2:G71 substitution into Dm mt microhelixAla. This substrate (with a G:U transversion) lost >99% of its activity relative to the wild-type substrate. Thus, like the conventional E.coli enzyme (Musier-Forsyth et al., 1995), Dm mt AlaRS is sensitive to the direction in which the free amino group is projected into the minor groove.
Collectively, the data presented in Figure 3 show that G2:U71 is a critical determinant for aminoacylation by Dm mt AlaRS. These results are thus consistent with the idea that a major determinant for the recognition by Dm mt AlaRS has been translocated in the acceptor stem from the 3:70 to the 2:71 position.
A Dm mt tRNAAla transcript containing a G2:C71 base pair, as well as one that contained a G2:C71, G3:U70 double mutation, was synthesized. Although the G2:C71 transcript was charged by Dm mt AlaRS, the activity was significantly compromised compared with wild-type transcript (Figure 4). The G2:C71, G3:U70 transcript was not charged. Thus, results with full-length tRNAAla largely parallel those seen with the microhelices.
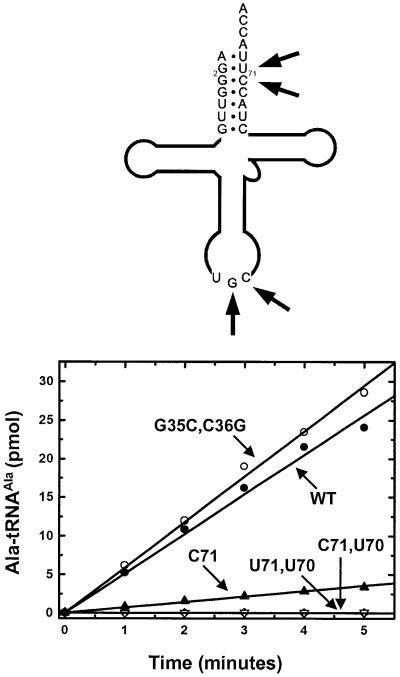
Fig. 4. Aminoacylation by Dm mt AlaRS at pH 7.5, 37°C of tRNAAla transcripts. The transcript substrate is shown above with positions of variation highlighted with arrows.
Introduction of the G2:U71 base pair into a non-alanine tRNA acceptor stem confers alanine acceptance upon the substrate
To demonstrate the importance of the G2:U71 base pair for recognition by the Dm mt AlaRS, we introduced this base pair into a non-alanine tRNA acceptor stem to see if this was enough to confer alanine acceptance upon this substrate. We chose to make a microhelix based upon the acceptor stem of Dm mt tRNATrp. This microhelix differs from the wild-type Dm mt microhelixAla at four of the seven base pairs of the acceptor stem, including the 2:71 position. As expected, this substrate is not aminoacylated by the Dm mt AlaRS (Figure 5). Interestingly, this substrate contains a G:C at the 3:70 position. We then substituted a single G into the 2-position of microhelixTrp, thus producing a substrate with a G2:U71 base pair. This microhelix was now a substrate for the Dm mt AlaRS, being charged at a rate that is within 28% of that of Dm mt wild-type microhelixAla. This result shows that a G2:U71 base pair is sufficient to confer alanine acceptance upon at least one ‘non-alanine’ acceptor stem and is consistent with the conclusion that G2:U71 is a critical determinant for aminoacylation by Dm mt AlaRS.
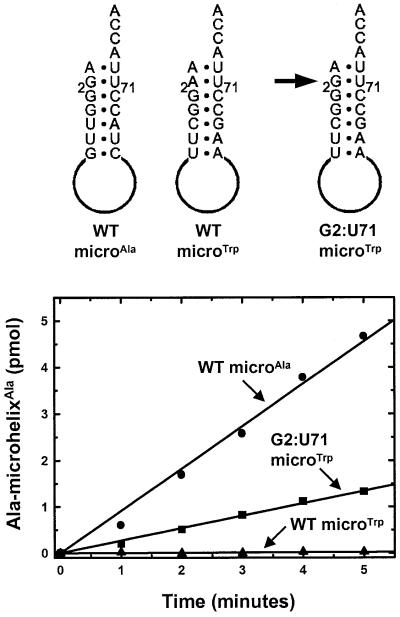
Fig. 5. Conference of alanine acceptance at pH 7.5, 37°C through introduction of a G2:U71 base pair into a microhelix based on the acceptor stem of Dm mt tRNATrp. The microhelix substrates are shown above.
Importance of the 3:70 position for aminoacylation by AlaRS
To determine the role, if any, of the 3:70 base pair in the Dm mt system, several microhelices with specific replacements of the G3:C70 pair were synthesized and tested. Four specific substitutions were made, including G:U, U:G, C:G and I:C. Strikingly, the G3:U70-substituted microhelix was inactive, even though it contained the G2:U71 pair that is important for the Dm mt enzyme (Figure 3B). These results, together with the finding that a G2:C71, G3:U70-containing microhelix and a G2:C71, G3:U70-containing Dm mt tRNAAla were each inactive for charging by the mitochondrial enzyme (see above), supports the idea that G3:U70 cannot replace the requirement for the translocated G2:U71 in this mitochondrial system.
The U3:G70 substitution was also inactive for charging (Figure 3B). Therefore, the G:U and U:G substitutions at the 3:70 position operationally block recognition of the G2:U71 base pair. This result suggests a strong selective pressure against placing a G:U (or U:G) pair at the 3:70 position in this mitochondrial system. [This idea was supported further through examination of a tRNAAla transcript that contains a G3:U70 base pair in the presence of G2:U71. This transcript, like the aforementioned microhelix, was completely inactive for aminoacylation (Figure 4).] Some activity was seen with the C3:G70 and I3:C70 substitutions of Dm mt microhelixAla (Figure 3B). Thus, while G:U is negatively selected at the 3:70 position, other substitutions are at least partly tolerated at 3:70.
Recognition of the N73 discriminator base of Dm mt tRNAAla is based on a purine system
Both Dm mt tRNAAla and E.coli tRNAAla have an A73 discriminator base. Escherichia coli AlaRS (Fischer et al., 1999) has a strong preference for a purine at N73. This preference for purine is thought to reflect a need to maintain the local conformation and stability of the RNA helix. However, because the 6-keto oxygen of guanosine appears to be a negative atomic determinant (Fischer et al., 1999), the advantages of the strong stacking characteristics of G are offset, making A73-containing substrates more active than those with G73. To determine if these preferences exist in the Dm mt system, two substrates, one with a G73 and the other with a U73 substitution, were tested. Significantly, G73 Dm mt microhelixAla was aminoacylated at a rate comparable with that of the wild-type substrate (Figure 3C). The U73-containing substrate was aminoacylated at a rate just 17% of that of the wild-type substrate. This result shows that, while the Dm mt enzyme lacks the A versus G preference of the E.coli enzyme, the two systems preserve the same preference for purine versus pyrimidine at N73.
Dm mt AlaRS tolerates variation in the 1:72 position of tRNAAla
In contrast to most E.coli tRNAs (including tRNAAla), which contain a G1:C72 base pair, the Dm mt tRNAAla has an A1:U72 pair. Interestingly, Beuning et al. (1997) reported that, in the E.coli system, a major groove carbonyl group presented by a U at position 72 is a blocking element for aminoacylation. Thus, recognition of 1:72 by Dm mt AlaRS is fundamentally different from that of E.coli AlaRS. To determine the importance of the 1:72 position for aminoacylation, we synthesized a microhelix containing G1:C72. This substitution results in substantially different atomic group presentations in both major and minor grooves. Nonetheless, the G1:C72 substrate was aminoacylated at a rate comparable to that of the wild-type substrate (Figure 3D).
An NMR structure of E.coli microhelixAla (Ramos and Varani, 1997) revealed substantial base stacking between the A at position 73 and the G of the opposite strand at position 1. This interaction may help achieve the proper orientation of the discriminator base during amino acid transfer (Fischer et al., 1999; Nagan et al., 2000). To investigate whether the stacking of the discriminator base and the nucleotide at position 1 is important in Dm mt tRNAAla, we made a transversion of the A1:U72 base pair. This produced a U1:A72 substrate that now has a pyrimidine at position 1. This pyrimidine should not stack as favorably with the discriminator base and, as might be expected, the U1:A72 substitution significantly compromised aminoacylation by the Dm mt AlaRS (Figure 3D). Thus, mitochondrial AlaRS appears not to depend significantly on specific elements of the 1:72 base pair, as long as helix stability is preserved through base stacking between nucleotides at positions 73 and 1.
Converting the Dm mt microhelixAla to a substrate for E.coli AlaRS
While the Dm mt microhelixAla is charged efficiently by the Dm mt AlaRS, this microhelix is not a substrate for the conventional E.coli AlaRS (Figure 6A). To understand how translocation of G:U is affected by sequence context, we sought to determine minimal changes in the Dm mt microhelixAla that allow for recognition by the E.coli enzyme. The acceptor stems for E.coli tRNAAla and Dm mt tRNAAla differ at all seven base pairs. As described above, we made a G2:C71, G3:U71 Dm mt microhelixAla that provided two of the three base pairs present in the first three positions of the E.coli tRNAAla sequence. This substrate was not aminoacylated by either Dm mt AlaRS or E.coli AlaRS (Figure 6B).
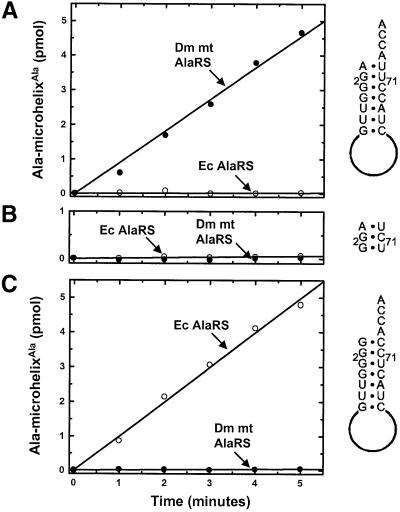
Fig. 6. Aminoacylation by Dm mt AlaRS and E.coli AlaRS at pH 7.5, 37°C of wild-type and substituted microhelices based on the acceptor stem of Dm mt tRNAAla. (A) Aminoacylation of wild-type microhelixAla. (B) Aminoacylation of G2:C71, G3:U70 microhelixAla. (C) Aminoacylation of G1:C72, G2:C71, G3:U70 microhelixAla.
The failure of E.coli AlaRS to aminoacylate this substrate was probably due to the blocking A1:U72 base pair (Beuning et al., 1997). Thus, we changed A1:U72 to G:C to give a G1:C72, G2:C71, G3:U70 Dm mt micro helixAla substrate. This substrate was not charged by the Dm mt AlaRS, but was charged by E.coli AlaRS (Figure 6C). Thus, while the acceptor stem of E.coli tRNAAla and of Dm mt tRNAAla differ at seven base pairs, only differences at the first three positions are sensed by the E.coli enzyme. That changes at the first three base pairs are required to convert Dm mt microhelixAla to a substrate for the E.coli enzyme shows that the functional effects of translocation of the critical G:U determinant require limited, but distinct sequence contexts.
Discussion
Our results show that the acceptor stem G2:U71, G3:C70 combination is essential for aminoacylation with alanine, in a specific example (Dm mt AlaRS) from the phylum Arthropoda, class Insecta. In a database search, this combination of base pairs was not found in any organism outside of Arthropoda. Although it is found in the two organisms in the class Branchiopoda for which there are sequences of mitochondrial tRNAAla, the G2:U71, G3:C70 combination is not conserved throughout all Arthropoda. Considering just the class Insecta, we examined sequences of mitochondrial tRNAAlas for conservation of the translocated identity element G2:U71 and the neighboring G3:C70 pair. The G2:U71, G3:C70 combination was found in all 11 organisms of the class Insecta for which tRNAAla sequences are available (Figure 7). Although we were only able to evaluate a small subset of the mitochondrial tRNAAla sequences from Insecta, it is a random sampling and can be considered representative of the class as a whole. Strikingly, bacterial and eukaryotic cytoplasmic tRNAAlass have the exactly inverted sequence G2:C71, G3:U70.
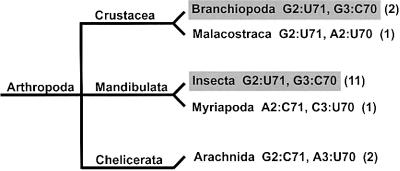
Fig. 7. Base pair combinations present at the 2:71, 3:70 position of mitochondrial tRNAAlas from Arthropods. Classes of organisms whose tRNAAlas contain the G2:U71, G3:C70 combination are highlighted in gray. The number of organisms in existing databases from each class is shown in parentheses. Only classes containing known mitochondrial tRNAAla sequences are shown.
The conservation of these two base pairs throughout Insecta is remarkable, especially considering that Insecta contains the largest number of species of any class (Margulis and Schwartz, 1998). Moreover, the wide distribution and conservation of G2:U71, G3:C70 shows that, among Insecta, these variations from the canonical alanine system have been fixed. Thus, they are not simply random but appear to have a specific function. A plausible explanation is that the modes of recognition of cytoplasmic and mitochondrial tRNAAlas were under evolutionary pressure to be distinct.
In the typical eukaryote, separate tRNA synthetases for the cytoplasm and the mitochondria are nuclear-encoded. The mitochondrial enzyme is synthesized in the cytoplasm and then imported. Thus, the mitochondrial and cytoplasmic proteins transiently co-exist in the same cytoplasmic compartment. If both enzymes were to recognize the same acceptor stem, one of the proteins could remain stable while mutations occurred in the other, gradually introducing changes to the genetic code (Ribas de Pouplana and Schimmel, 2001). [Mutational changes in synthetases recently have been demonstrated as capable of introducing ambiguity into the code in vivo (Doring et al., 2001).] However, if the two enzymes recognize distinct elements in the tRNA acceptor stems, then this possibility is eliminated. An alternative would be for one gene to encode both cytoplasmic and mitochondrial enzymes. Examples of this circumstance are seen in Arabidopsis thaliana and S.cerevisiae (Mireau et al., 1996; Dujon et al., 1997).
Chihade et al. (2000) showed that cytoplasmic AlaRSs of extant mitochondriate eukaryotes are mitochondrial in nature, suggesting that the original nuclear alaS was replaced by the mitochondrial counterpart. This situation undoubtedly contributes to AlaRS being one of the most conserved of all synthetases. Because eukaryotic AlaRSs are of mitochondrial origins, the alanine system may present a particular problem to the eukaryotic cell. Replacement of the nuclear gene by the mitochondrial counterpart would create a system in which the two enzymes are closely similar and that could lead to cross-acylation of substrates. Thus, a particularly strong pressure to have distinct mechanisms for recognition of mitochondrial and cytoplasmic tRNAAlas may exist. Consistent with this interpretation is that G:U at the 3:70 position blocks aminoacylation by Dm mt AlaRS (Figures 3B and 5). As a consequence, the mitochondrial enzyme is unable to aminoacylate substrates that carry the major recognition element of the cytoplasmic tRNA.
Translocation of a critical base pair is a novel way to achieve system-specific aminoacylation. Although there are examples of nucleotide variations at specific locations that cause species-specific aminoacylation by homologous enzymes (Sampson et al., 1989; Chow and RajBhandary, 1993; Shiba et al., 1994a,b; Hipps et al., 1995; Quinn et al., 1995; Wakasugi et al., 1998), this is the first known example where the same identity element is recognized at different positions in the sequence. The Dm mt enzyme shares a large amount of sequence similarity (21–34%) to AlaRSs from a variety of organisms. Thus, many features of the enzyme are conserved despite the variations in the RNA substrate. However, the Dm mt enzyme contains an insertion of 28 amino acids in the domain of the protein thought to contact the acceptor stem (Figure 8). Perhaps it is this insertion that allows the Dm mt synthetase to recognize an identity element that has been translocated. This insertion is only present in this insect mitochondrial AlaRS, is not found in the insect cytoplasmic AlaRSs for which sequences are available and is not present in other sequenced mitochondrial AlaRSs that have a cognate tRNA with a conventional G3:U70 base pair.
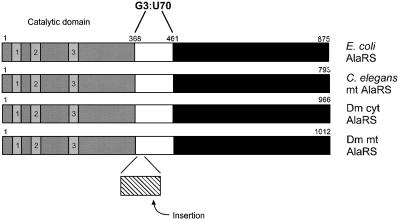
Fig. 8. Insertion of a 28 amino acid segment into Dm mt AlaRS. AlaRSs from E.coli, C.elegans mitochondria, D.melanogaster cytoplasm and D.melanogaster mitochondria are compared. The active site-containing domain of AlaRS with the three characteristic sequence motifs (1–3) of class II enzymes is shaded. The domain of E.coli AlaRS thought to contact the G3:U70 base pair (E.coli numbering shown) and homologous domains are white, while the insertion present in Dm mt AlaRS is shown with cross hatches. The C-terminal portions of the proteins are in black.
Materials and methods
Sequencing of a gene encoding Dm mt AlaRS
An EST encoding a putative Dm mt AlaRS was identified through a BLAST search with a human mt AlaRS gene fragment (Shiba et al., 1995) of the EST database of NCBI. The EST containing ‘clone’ LD11251 (Berkeley Drosophila Genome Project, Berkeley, CA) was sequenced and found to encode a protein of 1012 amino acids (Chihade et al., 2000).
Subcloning of Dm mt AlaRS
The Dm mt AlaRS gene was cloned into the E.coli expression vector pET-21b (Novagen, Madison, WI) for initial manipulation. Using the sequence of LD11251, primers were designed to amplify the 5′ end of the AlaRS gene minus the coding sequence for the 10 N-terminal amino acids. The ‘forward’ primer contained an NdeI site (DmmtNdeF1.1, 5′-GCAGCTGCAGCATATGCTGACAGC-3′) and the ‘reverse’ primer contained an EcoRI site (DmmtEcoRR1.2, 5′-CGGTGCAGGGAATTCACGGCCCTG-3′). PCR was performed with LD11251 as template and the Pfu proofreading polymerase (Stratagene, La Jolla, CA). The amplified product was digested with NdeI and EcoRI and ligated into pET-21b digested with the same enzymes to give plasmid pMAL014. A 2458 bp central piece of the Dm mt AlaRS gene was cut from LD11251 using NruI and SalI and ligated into pMAL014 digested with the same enzymes to give plasmid pMAL016. The 3′ end of LD11251 was amplified by PCR using a forward primer containing a SalI site (DmmtF2.1, 5′-CCAAGGGCCAAATCC-3′) and a reverse primer containing a NotI site (DmmtNotR2.2, 5′-GGAGGGTAATCACAACTGCGGCCGCTAGATAAAGC-3′). The amplified product was digested with SalI and NotI and ligated into plasmid pMAL016 digested with the same enzymes to give plasmid pMAL018. This plasmid contains the Dm mt AlaRS gene lacking the coding sequence for the 10 N-terminal amino acids but also encodes a C-terminal His6 tag derived from vector pET-21b. The Dm mt AlaRS gene was then cloned into the baculovirus expression vector pFastBac1 (Life Technologies, Inc., Rockville, MD) in two steps. The 5′ end of LD11251 was PCR-amplified using a forward primer containing a BamHI site (DmmtBamF3.1, 5′-GTTTTAATTGGGATCCGGCATGTACAAC-3′) and a reverse primer containing a NotI site, a sequence encoding a His6 tag, a stop codon and an XbaI site (Dmmt6HisR4.1, 5′-CCGCTCTAGACTAGTGATGGTGATGGTGATGTGCGGCCGCGCAGCCAAAGGGCAGTATTCGCGAGGCGG-3′). The amplified product was digested with BamHI and XbaI and was ligated into pFastBac1 digested with the same enzymes to give plasmid pMAL020. The 3′ end of the gene was obtained from plasmid pMAL018. For this purpose, pMAL018 was digested with NruI and NotI and ligated into pMAL020 digested with the same enzymes to give plasmid pMAL023. Thus, plasmid pMAL023 is based on pFastBac1 and contains the full-length Dm mt AlaRS gene with a sequence encoding a C-terminal His6 tag.
Expression and purification of histidine-tagged Dm mt AlaRS in the baculovirus expression system
Preparation of recombinant bacmid DNA was performed according to the Bac-to-Bac Baculovirus Expression System Instruction Manual (Life Technologies, Inc.). Bacmid DNA was isolated and used to transfect Sf9 insect cells using CellFECTIN reagent (Life Technologies, Inc.). Recombinant baculovirus was isolated and used to infect 106 Sf9 insect cells. Cells were grown for 3 days and harvested by centrifugation. Harvested cells were lysed by sonication in 10 ml of lysis buffer (20 mM Na2HPO4 pH 7.4, 250 mM NaCl, 10% glycerol, 20 mM β-mercaptoethanol, 30 mM imidazole). The lysate was centrifuged for 25 min at 16 000 g at 4°C. Ni-NTA resin (1.5 ml, Qiagen, Santa Clarita, CA) was added to the supernatant, the mix was rotated for 1 h at 4°C, applied to a 0.8 × 4 cm Poly-Prep column (Biorad, Hercules, CA), and the supernatant drained. The resin was then washed with 150 ml of lysis buffer and the bound protein was eluted with 15 ml of elution buffer (20 mM Na2HPO4 pH 7.4, 250 mM NaCl, 10% glycerol, 20 mM β-mercaptoethanol, 250 mM imidazole). Fractions (0.5 ml) were analyzed for purity and concentration by SDS–PAGE. Fractions containing protein of the expected molecular weight were dialyzed into 25 mM Tris pH 7.4, 250 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol.
Purification of an N-terminal fragment of E.coli AlaRS
Plasmid pQE_459–6H (Ribas de Pouplana and Schimmel, 1997) was used for the expression of the N-terminal 459 amino acids of E.coli AlaRS, a fragment that maintains wild-type levels of activity and substrate specificity with RNA microhelix substrates (Buechter and Schimmel, 1993). The protein was purified from E.coli TG1 cells as previously described (Ribas de Pouplana and Schimmel, 1997).
RNA substrate preparation
Dm mt tRNAAla was transcribed in vitro from a linearized plasmid DNA template and processed using a cis-acting hammerhead ribozyme (Fechter et al., 1998; Steer and Schimmel, 1999). A ‘BamHI–PstI ribozyme-tRNA’ gene under the control of a T7 RNA polymerase promoter was constructed by annealing together four DNA oligonucleotides and ligating them into pUC18 cut with BamHI and PstI. A BstNI site at the 3′ end of the gene allowed generation of linear template that provided the correct 3′-CCA terminus in the tRNA by in vitro runoff transcription. The transcription reaction mixtures contained 0.1 µg/µl BstNI-linearized plasmid in 40 mM Tris–HCl pH 7.9, 21 mM MgCl2, 2 mM spermidine, 10 mM dithiothreitol (DTT), ATP, CTP, GTP, UTP (4 mM each), 0.08 U/µl RNase inhibitor, 2 µg/µl yeast pyrophosphatase and T7 polymerase. The transcription reaction was stopped by phenol extraction and the transcript was ethanol precipitated. The transcript was cleaved in cis by the hammerhead ribozyme at 60°C in 4 mM Tris–HCl pH 8.0, 0.2 mM EDTA and 30 mM MgCl2. The tRNA transcript was purified by denaturing gel electrophoresis (12% polyacrylamide, 8 M urea), gel extraction and ethanol precipitation.
Microhelix RNA substrates were chemically synthesized using the phosphoramidite method (Expedite 8909 synthesizer, PE Biosystems, Foster City, CA). Synthesis products were purified by HPLC on a Dionex, DNAPac™ PA-100 column using a linear gradient from 0.28 to 0.66 M NH4Cl over 28 min. Thermal melting curves for microhelices were constructed from absorbance data obtained using a Cary 3E UV–visible spectrophotometer with a temperature controller. Melts were performed in 50 mM HEPES pH 7.5, 20 mM KCl and 10 mM MgCl2. Absorbances at 260 nM were measured at intervals of 1°C over a temperature range of 15–90°C.
Aminoacylation assays
Aminoacylation assays were performed at 37°C in a reaction mixture containing 50 mM HEPES pH 7.5, 0.1 mg/ml bovine serum albumin (BSA), 20 mM KCl, 10 mM MgCl2, 20 mM β-mercaptoethanol, 2 mM ATP, 5 µM alanine, 5 µM [3-3H]alanine, 50 µM RNA microhelix or 5 µM transcript, and 200 nM enzyme. Prior to assay, RNAs were annealed by heating to 90°C and slowly cooling in the absence of magnesium to 55–60°C, at which time MgCl2 was added (2 mM final concentration) with further cooling to room temperature. Reactions were initiated by the addition of reaction mix to the enzyme and RNA. Aliquots were removed at appropriate time intervals, spotted onto trichloroacetic acid-soaked filter pads, washed and measured by scintillation counting (Schreier and Schimmel, 1972).
Acknowledgments
Acknowledgements
We are grateful to Dr Heimo Strohmaier for help with the baculovirus expression system. We also thank Dr Lluis Ribas de Pouplana for plasmid pQE_459-6H, for many helpful discussions and for critically reviewing the manuscript, Professor Shana Kelly for technical help and suggestions, and Dr Karla Ewalt and Professor Karin Musier-Forsyth for helpful advice. This work was supported by grant number GM15539 from the National Institute of Health and by a fellowship from the National Foundation for Cancer Research. M.A.L. is a National Science Foundation Graduate Student Fellow. J.W.C. was a NIH postdoctoral fellow, 1996–1999.
References
- Beuning P.J., Gullotta,M. and Musier-Forsyth,K. (1997) Atomic group ‘mutagenesis’ reveals major groove fine interactions of a tRNA synthetase with an RNA helix. J. Am. Chem. Soc., 119, 8397–8402. [Google Scholar]
- Buechter D.D. and Schimmel,P. (1993) Dissection of a class II tRNA synthetase: determinants for minihelix recognition are tightly associated with domain for amino acid activation. Biochemistry, 32, 5267–5272. [DOI] [PubMed] [Google Scholar]
- Chihade J.W., Hayashibara,K., Shiba,K. and Schimmel,P. (1998) Strong selective pressure to use G:U to mark an RNA acceptor stem for alanine. Biochemistry, 37, 9193–9202. [DOI] [PubMed] [Google Scholar]
- Chihade J.W., Brown,J.R., Schimmel,P.R. and Ribas De Pouplana,L. (2000) Origin of mitochondria in relation to evolutionary history of eukaryotic alanyl-tRNA synthetase. Proc. Natl Acad. Sci. USA, 97, 12153–12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.M. and RajBhandary,U.L. (1993) Saccharomyces cerevisiae cytoplasmic tyrosyl-tRNA synthetase gene. Isolation by comple mentation of a mutant Escherichia coli suppressor tRNA defective in aminoacylation and sequence analysis. J. Biol. Chem., 268, 12855–12863. [PubMed] [Google Scholar]
- de Duve C. (1988) The second genetic code. Nature, 333, 117–118. [DOI] [PubMed] [Google Scholar]
- Di Giulio M. (1992) On the origin of the transfer RNA molecule. J. Theor. Biol., 159, 199–214. [DOI] [PubMed] [Google Scholar]
- Doring V., Mootz,H.D., Nangle,L.A., Hendrickson,T.L., de Crecy-Lagard,V., Schimmel,P. and Marliere,P. (2001) Enlarging the amino acid set of Escherichia coli by infiltration of the valine coding pathway. Science, 292, 501–504. [DOI] [PubMed] [Google Scholar]
- Dujon B. et al. (1997) The nucleotide sequence of Saccharomyces cerevisiae chromosome XV. Nature, 387, 98–102. [PubMed] [Google Scholar]
- Fechter P., Rudinger,J., Giege,R. and Theobald-Dietrich,A. (1998) Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity [published erratum appears in FEBS Lett., 1998, 441, 342]. FEBS Lett., 436, 99–103. [DOI] [PubMed] [Google Scholar]
- Fischer A.E., Beuning,P.J. and Musier-Forsyth,K. (1999) Identification of discriminator base atomic groups that modulate the alanine aminoacylation reaction. J. Biol. Chem., 274, 37093–37096. [DOI] [PubMed] [Google Scholar]
- Francklyn C. and Schimmel,P. (1989) Aminoacylation of RNA minihelices with alanine. Nature, 337, 478–481. [DOI] [PubMed] [Google Scholar]
- Francklyn C., Musier-Forsyth,K. and Schimmel,P. (1992) Small RNA helices as substrates for aminoacylation and their relationship to charging of transfer RNAs. Eur. J. Biochem., 206, 315–321. [DOI] [PubMed] [Google Scholar]
- Hipps D., Shiba,K., Henderson,B. and Schimmel,P. (1995) Operational RNA code for amino acids: species-specific aminoacylation of minihelices switched by a single nucleotide. Proc. Natl Acad. Sci. USA, 92, 5550–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.M. and Schimmel,P. (1988) A simple structural feature is a major determinant of the identity of a transfer RNA. Nature, 333, 140–145. [DOI] [PubMed] [Google Scholar]
- Hou Y.M. and Schimmel,P. (1989) Evidence that a major determinant for the identity of a transfer RNA is conserved in evolution. Biochemistry, 28, 6800–6804. [DOI] [PubMed] [Google Scholar]
- Margulis L. and Schwartz,K.V. (1998) Five Kingdoms: An Illustrated Guide to the Phyla of Life on Earth. W.H.Freeman and Co., New York, NY.
- Martin N. (1995) Organellar tRNAs: biosynthesis and function. In Söll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. American Society for Microbiology, Washington, DC, pp. 127–140.
- McClain W.H. and Foss,K. (1988) Changing the acceptor identity of a transfer RNA by altering nucleotides in a ‘variable pocket’. Science, 241, 1804–1807. [DOI] [PubMed] [Google Scholar]
- Mireau H., Lancelin,D. and Small,I.D. (1996) The same Arabidopsis gene encodes both cytosolic and mitochondrial alanyl-tRNA synthetases. Plant Cell, 8, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller W. and Janssen,G.M. (1990) Transfer RNAs for primordial amino acids contain remnants of a primitive code at position 3 to 5. Biochimie, 72, 361–368. [DOI] [PubMed] [Google Scholar]
- Musier-Forsyth K. and Schimmel,P. (1999) Atomic determinants for aminoacylation of RNA minihelices and relationship to genetic code. Acc. Chem. Res., 32, 368–375. [Google Scholar]
- Musier-Forsyth K., Usman,N., Scaringe,S., Doudna,J., Green,R. and Schimmel,P. (1991) Specificity for aminoacylation of an RNA helix: an unpaired, exocyclic amino group in the minor groove. Science, 253, 784–786. [DOI] [PubMed] [Google Scholar]
- Musier-Forsyth K., Shi,J.-P., Henderson,B., Bald,R., Furste,J.P., Erdmann,V.A. and Schimmel,P. (1995) Base-analog-induced aminoacylation of an RNA helix by a tRNA synthetase. J. Am. Chem. Soc., 117, 7253–7254. [Google Scholar]
- Nagan M.C., Beuning,P., Musier-Forsyth,K. and Cramer,C.J. (2000) Importance of discriminator base stacking interactions: molecular dynamics analysis of A73 microhelix(Ala) variants. Nucleic Acids Res., 28, 2527–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K. and Kanehisa,M. (1992) A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics, 14, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J. and Schimmel,P. (1988) Evidence for interaction of an aminoacyl transfer RNA synthetase with a region important for the identity of its cognate transfer RNA. J. Biol. Chem., 263, 16527–16530. [PubMed] [Google Scholar]
- Quinn C.L., Tao,N. and Schimmel,P. (1995) Species-specific microhelix aminoacylation by a eukaryote pathogen tRNA synthetase dependent on a single base pair. Biochemistry, 34, 12489–12495. [DOI] [PubMed] [Google Scholar]
- Ramos A. and Varani,G. (1997) Structure of the acceptor stem of Escherichia coli tRNA Ala: role of the G3.U70 base pair in synthetase recognition. Nucleic Acids Res., 25, 2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas de Pouplana L. and Schimmel,P. (1997) Reconstruction of quaternary structures of class II tRNA synthetases by rational mutagenensis of a conserved domain. Biochemistry, 36, 15041–15048. [DOI] [PubMed] [Google Scholar]
- Ribas de Pouplana L. and Schimmel,P. (2001) Operational RNA code for amino acids in relation to genetic code in evolution. J. Biol. Chem., 276, 6881–6884. [DOI] [PubMed] [Google Scholar]
- Ripmaster T.L., Shiba,K. and Schimmel,P. (1995) Wide cross-species aminoacyl-tRNA synthetase replacement in vivo: yeast cytoplasmic alanine enzyme replaced by human polymyositis serum antigen. Proc. Natl Acad. Sci. USA, 92, 4932–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin S., Rodin,A. and Ohno,S. (1996) The presence of codon– anticodon pairs in the acceptor stem of tRNAs. Proc. Natl Acad. Sci. USA, 93, 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson J.R., DiRenzo,A.B., Behlen,L.S. and Uhlenbeck,O.D. (1989) Nucleotides in yeast tRNAPhe required for the specific recognition by its cognate synthetase. Science, 243, 1363–1366. [DOI] [PubMed] [Google Scholar]
- Schimmel P., Giege,R., Moras,D. and Yokoyama,S. (1993) An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl Acad. Sci. USA, 90, 8763–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier A.A. and Schimmel,P.R. (1972) Transfer ribonucleic acid synthetase catalyzed deacylation of aminoacyl transfer ribonucleic acid in the absence of adenosine monophosphate and pyrophosphate. Biochemistry, 11, 1582–1589. [DOI] [PubMed] [Google Scholar]
- Shiba K., Schimmel,P., Motegi,H. and Noda,T. (1994a) Human glycyl-tRNA synthetase: wide divergence of primary structure from bacterial counterpart and species-specific aminoacylation. J. Biol. Chem., 269, 30049–30055. [PubMed] [Google Scholar]
- Shiba K., Suzuki,N., Shigesada,K., Namba,Y., Schimmel,P. and Noda,T. (1994b) Human cytoplasmic isoleucyl-tRNA synthetase: selective divergence of the anticodon-binding domain and acquisition of a new structural unit. Proc. Natl Acad. Sci. USA, 91, 7435–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K., Ripmaster,T., Suzuki,N., Nichols,R., Plotz,P., Noda,T. and Schimmel,P. (1995) Human alanyl-tRNA synthetase: conservation in evolution of catalytic core and microhelix recogntion. Biochemistry, 34, 10340–10349. [DOI] [PubMed] [Google Scholar]
- Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steer B.A. and Schimmel,P. (1999) Major anticodon-binding region missing from an archaebacterial tRNA synthetase. J. Biol. Chem., 274, 35601–35606. [DOI] [PubMed] [Google Scholar]
- Wakasugi K., Quinn,C.L., Tao,N. and Schimmel,P. (1998) Genetic code in evolution: switching species-specific aminoacylation with a peptide transplant. EMBO J., 17, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]