

Abstract
The ParA family of proteins is involved in partition of a variety of plasmid and bacterial chromosomes. P1 ParA plays two roles in partition: it acts as a repressor of the par operon and has an undefined yet indispensable role in P1 plasmid localization. We constructed seven mutations in three putative ATP-binding motifs of ParA. Three classes of phenotypes resulted, each represented by mutations in more than one motif. Three mutations created ‘super-repressors’, in which repressor activity was much stronger than in wild-type ParA, while the remainder damaged repressor activity. All mutations eliminated partition activities, but two showed a plasmid stability defect that was worse than that of a null mutation. Four mutant ParAs, two super-repressors and two weak repressors, were analyzed biochemically, and all exhibited damaged ATPase activity. The super-repressors bound site-specifically to the par operator sequence, and this activity was strongly stimulated by ATP and ADP. These results support the proposal that ATP binding is essential but hydrolysis is inhibitory for ParA’s repressor activity and suggest that ATP hydrolysis is essential for plasmid localization.
Keywords: ATPase/bacterial chromosome segregation/mutagenesis/nucleotide switch/ParB
Introduction
Accurate distribution of daughter chromosomes at cell division is essential to ensure that each cell receives a complete copy of the genome. In bacteria, the process of chromosome partition involves the separation and positioning of daughter chromosomes in each cell cycle. The partition event is carried out with a high degree of precision: <0.03% of wild-type cells do not receive a copy of the chromosome at cell division in Escherichia coli (Hiraga et al., 1989). Low copy number plasmids are also stably inherited via active partition systems, and these systems are responsible for directing plasmids to distinct intracellular sites. The prophage of bacteriophage P1 exists as a low copy number plasmid and is located at the quarter and three-quarter positions of the rod-shaped E.coli cell for most of the cell cycle (Gordon et al., 1997). These positions become the mid-point of a newborn cell, but the plasmids duplicate and move to the quarter and three-quarter positions prior to the next cell division event. The P1 Par system consists of two proteins, ParA and ParB, which act on a centromere-like site called parS (Abeles et al., 1985). ParB, along with E.coli integration host factor (IHF), binds to parS to form a partition complex (Davis and Austin, 1988; Funnell, 1988b; Bouet et al., 2000). ParA, an ATPase (Davis et al., 1992), is required for the proper positioning of these partition complexes (Erdmann et al., 1999). Homologs of ParA and ParB are encoded by a variety of bacterial plasmids as well as by the chromosomes of a large number of bacterial species, although not by E.coli (for recent reviews see Gerdes et al., 2000; Hiraga, 2000). Several homologs have been shown to contribute to partition of the bacterial chromosomes (Ireton et al., 1994; Sharpe and Errington, 1996; Mohl and Gober, 1997).
The ParA-like family of proteins contain a specific version of the Walker A ATP-binding motif (Figure 1). Additional signature sequences, a Walker B motif, ‘motif 2’ and ‘motif 3’, are also conserved in members of this family (Motallebi-Veshareh et al., 1990; Koonin, 1993). Indeed, P1 ParA is an ATPase, and a lysine to glutamate change in the Walker A ATP-binding motif destroys this activity (Davis et al., 1996). While Walker A and B motifs are reliable indicators of ATP-binding activities, an important question concerns the role of motif 2 in nucleotide binding. A comparable sequence contributes to the ATP-binding sites of two distantly related ATPases whose structures with ADP have been solved. These are Azotobacter vinelandii nitrogenase iron protein (Fe protein) and E.coli ArsA (Georgiadis et al., 1992; Jang et al., 2000; Zhou et al., 2000). In this study, we probe ParA’s motif 2 and Walker B motifs, as well as Walker A, by mutagenesis and show that conserved residues are important for ATPase activity.
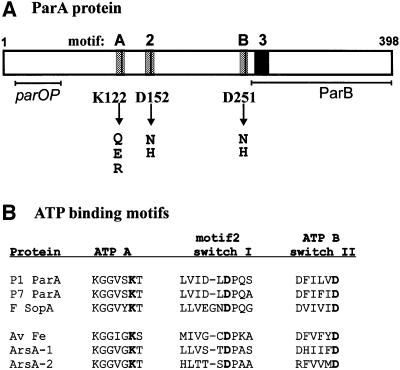
Fig. 1. P1 ParA protein and ATP-binding motifs. (A) Diagram of P1 ParA protein. The gray boxes represent the ATP-binding motifs (A = Walker or ATP A; B = Walker B or ATP B; 2 = motif 2). The residues changed in this study are indicated by vertical lines and arrows below the diagram. The black box represents the position of ‘motif 3’, another region of homology found in ParA-like proteins (Motallebi-Veshareh et al., 1990). The horizontal lines represent the regions required for species-specific interactions with DNA (parOP) and ParB, defined by domain-swapping experiments with the equivalent regions of P7 ParA (Radnedge et al., 1998). (B) Comparison of ATP-binding motifs found in three ParA-like proteins with demonstrated ATPase activities (P1 ParA, P7 ParA and F SopA; Davis et al., 1992; Watanabe et al., 1992) with those of A.vinelandii Fe protein and E.coli ArsA. ArsA consists of two homologous segments, each containing a nucleotide-binding site, joined by a linker sequence. The alignment is from Koonin (1993). The residues highlighted in bold are those changed in ParA (as in A). The protein accession numbers are P1 ParA, P07620; P7 ParA, S06099; F SopA, P08866; Fe protein, P00459; ArsA, A25937.
ParA plays at least two roles in P1 plasmid partition, a positioning one during the segregation reaction and a transcriptional one as the repressor of the par operon (Friedman and Austin, 1988; Erdmann et al., 1999). Adenine nucleotide binding is necessary for both these roles. In vivo, deletion of the Walker A motif eliminates all ParA function (Davis et al., 1996). In vitro, binding of ATP and ADP affect all ParA’s activities and its conformation, as measured by dimerization and circular dichroism (CD) (Davis et al., 1992; Davey and Funnell, 1994, 1997; Bouet and Funnell, 1999). ParA binds specifically to an inverted repeat sequence in the par promoter, parOP, and this activity is stimulated strongly by adenine nucleotides. Among ATP-dependent DNA-binding proteins, however, it has the unusual property that both ADP, the product of hydrolysis, and ATPγS, a non-hydrolyzable analog of ATP, are better cofactors than ATP (Davey and Funnell, 1994). The latter observation implied that the act of ATP hydrolysis was deleterious to ParA’s DNA-binding activity and thus to its repressor activity. ParA can also interact with the partition complex at parS, measured in a gel mobility shift assay, but this interaction requires ATP (Bouet and Funnell, 1999). These observations led us to propose that the ADP-bound (or non-hydrolyzing) form of ParA was the repressor form and the ATP-bound form was the partition form. In this way, the state of the bound nucleotide constituted a type of molecular switch.
ParB also influences ParA’s activities in vitro and in vivo. It acts as a corepressor, stimulating the repressor activity of ParA, but ParB has no repressor activity on its own (Friedman and Austin, 1988). In vitro, ParB stimulates ParA’s ATPase activity (Davis et al., 1992) and its DNA-binding activity towards parOP (Davey and Funnell, 1997). In the latter case, however, ParB could stimulate ParA’s DNA-binding activity only in the presence of ATP and only to a level seen with ParA (alone) in the presence of ADP. One possible explanation is that ParB stimulates ParA’s DNA-binding activity by shielding it from the negative effects of ATP hydrolysis.
In this study, we have functionally characterized the ATP-binding site in P1 ParA by a biological and biochemical examination of ParA proteins containing mutations in residues predicted to be important in ATP binding and hydrolysis. In particular, we examined the idea that the ADP form, or non-hydrolyzing ATP form is the preferred repressor form of ParA. Our results support the predictions that ParA’s repressor activity requires nucleotide binding but is inhibited by ATP hydrolysis, and that ParA requires ATP hydrolysis to perform its partition function.
Results
We previously have examined the roles of ATP binding and hydrolysis in ParA function in vitro using wild-type ParA and various nucleotides and nucleotide analogs (Davey and Funnell, 1994, 1997; Bouet and Funnell, 1999). To examine further these roles in vivo as well as in vitro, we created seven missense changes in three putative nucleotide-binding regions of parA (Figure 1). The invariant lysine residue in the Walker A, or P-loop, motif was changed to glutamate, glutamine and arginine residues. Two conserved aspartate residues, in the Walker B motif and in motif 2, were each changed to histidine and asparagine. One of these mutant proteins, P1 ParA K122E, has been described previously (Davis et al., 1996). The K122E mutation damaged partition and repression activities in vivo, and ParA’s ATPase and site-specific DNA-binding activities in vitro. The genes for these mutant ParA proteins were cloned into a variety of plasmid contexts so that we could measure their activities in vivo.
Three mutations created super-repressor ParA proteins
We first tested the effect of the mutations on ParA’s repressor function by swapping the mutations for wild-type parA sequence in the plasmid pBEF131, which contains the par operon with a lacZ gene fused early in-frame into the parB gene (Figure 2). Three of the mutant ParAs, K122Q, D251N and D251H, showed repressor activity that was as strong as that of wild-type ParA, measured as their effects on β-galactosidase expression in this plasmid context. ParA K122E, as has been shown previously, and ParA K122R, D152H and D152N were weaker repressors than wild-type ParA. They were, however, not entirely defective (compare β-galactosidase levels in their presence with that in the absence of ParA). This plasmid system provided a convenient estimate of repressor activity, but it was not ideal since the repressors were controlling their own expression as well as that of β-galactosidase. Indeed, immunoblots of cells containing these plasmids showed that the weak repressors were expressed at much higher levels than the strong repressors (data not shown). We therefore designed a new reporter system to correct this problem. First, lacZ was fused in-frame early in the parA gene. A parOP-parA::lacZ cassette was inserted into bacteriophage λ, which was integrated into the bacterial chromosome to provide a unit-copy reporter, and into the plasmid pST52 as a high copy number reporter system. The par genes were expressed from a modified β-lactamase promoter, blaP2 (modified to be stronger; Materials and methods), on a pBR322 derivative called pEF8. Immunoblots showed that intracellular concentrations of all mutant proteins were similar to each other and to that of wild-type ParA when their genes were expressed from blaP2 (data not shown).
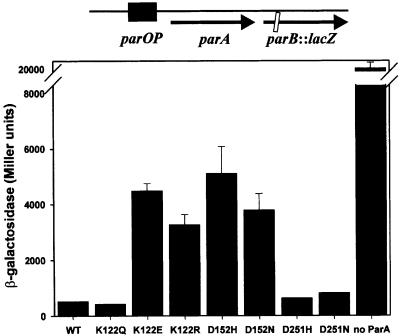
Fig. 2. Repressor activities of mutant ParA proteins under autogenous control. Results of a typical experiment measuring β-galactosidase levels of a parB::lacZ fusion in the presence or absence of wild-type and mutant ParAs. The diagram above the graph describes the arrangement of pBEF131 and its mutant derivatives. These plasmids were used to transform DH5Δlac, and β-galactosidase assays were performed as described by Miller (1992). The error bars represent the variation in duplicate readings in this experiment. WT, wild type.
Expression of wild-type parA alone from blaP2 was not sufficient to detect significant repressor activity in either reporter system (Figure 3). The weak repressors identified in the previous assay, ParA K122E, K122R, D152H and D152N, also showed no measurable repressor activity using the unit-copy reporter (Figure 3A). Three mutant ParAs, ParA K122Q, D251H and D251N, however, showed stronger repressor activity than that of wild-type ParA; they were ‘super-repressors’. When tested using the unit-copy reporter system, the repression was so strong that β-galactosidase levels were similar to those of DH5Δlac alone, and usually <5 Miller units (data not shown). To obtain numbers higher than these background levels, we measured repressor activity of ParA K122Q, D251H and D251N using the high copy reporter system (Figure 3B). These mutant ParAs reduced β-galactosidase expression to low but measurable levels, which were ∼40- to 50-fold lower than unrepressed levels.
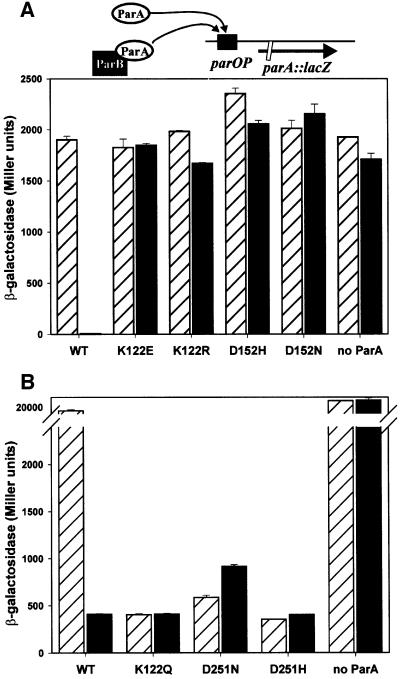
Fig. 3. Repressor activities of mutant ParA proteins with and without ParB. The effect of wild-type and mutant ParA proteins on the β-galactosidase levels of a parA::lacZ fusion was measured in the absence (hatched bars) or presence (filled bars) of ParB. The parA and parB genes were expressed from the blaP2 promoter, in trans to the parA::lacZ fusion (see diagram above the graphs). The version of ParA present is indicated below each set of bars, and ‘no ParA’ represents β-galactosidase expression in the absence of ParA. (A) The parOP-parA::lacZ cassette was in single copy (on bacteriophage λ, integrated into the chromosome of E.coli DH5Δlac at attB). (B) The parOP-parA::lacZ cassette was in multicopy on the plasmid pST52 in the strain DH5Δlac. The error bars represent the variation in duplicates in each experiment.
All mutant ParAs are insensitive to the corepressor ParB
ParB stimulates the repressor activity of ParA, although ParB has no regulatory effect on its own (Friedman and Austin, 1988). We asked whether the mutations introduced into ParA altered the sensitivity to ParB in repression. As stated above, expression of wild-type parA alone from blaP2 was insufficient to detect repression; however, when both parA and parB were expressed from blaP2, strong repression was observed (Figure 3). In contrast, the repressor activities of none of the mutant ParAs were affected by ParB. These results suggest that the mutants either do not interact with ParB or the interaction is not productive. Interestingly, the repressor activity of wild-type ParA in the presence of ParB was very similar to the repressor activities of the super-repressor ParAs, regardless of the presence of ParB.
All mutant ParA proteins are defective for partition
We next asked whether any of these mutant ParAs could support partition. The repressor activity of ParA is dispensable for plasmid stability in vivo as long as the par genes are expressed from a weak promoter (Davis et al., 1996). Therefore, to examine partition activity independently of repressor action, we placed the parA and parB genes under the control of the constitutive β-lactamase promoter (blaP1, as adapted by Su et al., 1992) in a pBR322 derivative called pEF5. We tested the ability of wild-type or mutant ParAs, in the presence of ParB, to stabilize a ΔparA miniP1 plasmid, pBEF250. In the absence of ParA, pBEF250 was not stably maintained in E.coli populations (Table I). pEF5 encoding wild-type ParA could stabilize this miniP1 plasmid, but none of the mutant parA genes could provide the partition activity of ParA (Table I).
Table I. Stability of miniP1 plasmids with mutant ParA proteins.
| Plasmid providing Par proteins (ParA allele) | Percentage retention of miniP1 (pBEF250) after overnight growth |
|---|---|
| pEF5.1 (wild-type) | 85 ± 4 |
| pEF5.2 (D251N) | 14 ± 2 |
| pEF5.3 (D251H) | 12 ± 2 |
| pEF5.4 (D152H) | nda |
| pEF5.5 (D152N) | 34 ± 2 |
| pEF5.6 (K122Q) | 15 ± 5 |
| pEF5.7 (K122E) | 22 ± 3 |
| pEF5.8 (K122R) | nda |
| pBEF251 (ΔparA) | 10 ± 5 |
| pBR322 (no parA or parB) | 28 ± 5 |
‘Retention’ represents the ratio of the frequency of cells with pBEF250 after overnight growth divided by the frequency of cells with pBEF250 at the start, expressed as a percentage. The data represent the average of 3–5 independent experiments, depending on the pEF5 derivative.
and, not determined by this assay. Colonies containing pBEF250 with these plasmids grew extremely slowly in the presence of ampicillin and choramphenicol (see Results).
Two of the weak repressor mutants, ParA K122R and ParA D152H, showed a partition defect that was worse than the lack of ParA. Colonies containing both pBEF250 and a pEF5 plasmid encoding either ParA K122R or ParA D152H grew extremely slowly on agar plates containing chloramphenicol and ampicillin, antibiotics that selected for both plasmids, and it was almost impossible to grow these cells in liquid culture. In comparison, colonies containing both pBEF250 and pBR322 grew normally (although pBEF250 was unstable; Table I). The ParA K122R or ParA D152H colonies, when sampled, contained pBEF250 in <10% of the cells, even though the colonies were grown in the presence of both antibiotics. The slow growth was due to an inability to maintain pBEF250; the colonies grew normally on media that contained only ampicillin (selection for the pEF5 derivative). This effect was dependent on the presence of ParB; if the pEF5 derivative contained no parB gene, the colonies grew as well as those containing pBR322 (although pBEF250 was still unstable; data not shown). This phenotype appears identical to the ‘ParPD’ (propagation defective) phenotype identified by Austin and colleagues, also caused by specific mutations in the parA gene (M314I and T158M; Youngren and Austin, 1997).
The seven mutations that we have created therefore divided into three distinct classes, based on their in vivo phenotypes (Table II). Class I were super-repressors, class II were weak repressors and partition defective, and class III were weak repressors and ParPD. Each class, however, was not restricted to changes at a single residue and, in fact, different changes at one residue (Lys122) yielded mutants of each class. We chose mutations at each of the three locations to study biochemically, and in particular we chose the mutations in the first two classes to examine further the role of ATP in the repressor and super-repressor activities of ParA.
Table II. Three classes of mutant ParAs.
| Class | ParAs | Repressor activity |
Partition |
|
|---|---|---|---|---|
| –ParB | +ParB | activity | ||
| Wild-type | + | ++ | + | |
| I | K122Q | |||
| D251H | ++ | ++ | – | |
| D251N | ||||
| II | K122E | –/+ | –/+ | – |
| D152N | ||||
| III | K122R | –/+ | –/+ | – (ParPD) |
| D152H | ||||
The in vivo properties of the parA alleles (Figure 3 and Table I) divided them into three categories, compared with wild-type parA+: class I are the super-repressors, class II are weak repressors and partition defective, and class III are weak repressors and ParPD. Proteins with the mutations shown in bold were purified for subsequent biochemical analyses.
Mutations in each of the three conserved motifs damage ATPase activity
We purified two of the super-repressor mutant proteins, ParA K122Q and ParA D251H. Our previous results suggested that the repressor activity of ParA, as measured in vitro as the ability to bind to parOP, required adenine nucleotides but was inhibited by the act of ATP hydrolysis (Davey and Funnell, 1994; Bouet and Funnell, 1999). ParA bound to ADP or ATPγS was a better site-specific DNA-binding protein than ParA bound to ATP. We predicted that a ParA protein that was unable to hydrolyze, but could bind ATP, might be a better repressor than wild-type ParA. For comparison, we also purified two weak repressors, ParA K122E and ParA D152N. The K122E and D152N mutations were chosen because they behaved most like loss-of-function mutations in vivo, and because ParA K122E has been examined previously (Davis et al., 1996). All four mutant proteins were expressed and purified using the same procedure as that used for wild-type ParA. The mutant proteins were soluble and behaved similarly during the purification process, which involved two ion-exchange chromatography steps. This behavior suggested that the mutant proteins were folded similarly to wild-type ParA. To confirm this, we probed their domain structure by partial proteolysis. All proteins produced a similar pattern of bands upon limited digestion with trypsin, albeit to slightly different extents depending on the ratio of ParA to trypsin (Figure 4). This result suggests that all show the same overall domain structure. ParA D152N was the most sensitive to trypsin but still produced the same cleavage products, suggesting it is folded properly but less stably than the other ParA proteins. It should also be noted that all ParAs were relatively resistant to trypsin, compared, for example, with ParB. At these ratios of trypsin to protein, ParB would be digested extensively (Surtees and Funnell, 1999; J.Surtees and B.Funnell, unpublished).
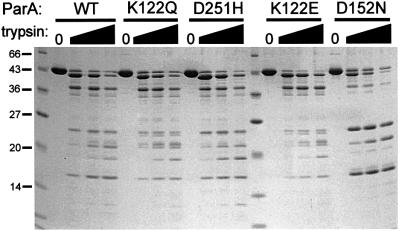
Fig. 4. Partial proteolysis of wild-type and mutant ParA proteins. ParA protein (2.5 µg per lane) was incubated with or without trypsin in 25 mM HEPES–KOH pH 7.5, 200 mM NaCl, 5 mM MgCl2, 5% (v/v) glycerol, 0.05 mM Na2EDTA, 0.5 mM dithiothreitol for 4 h at room temperature, and then analyzed by electrophoresis in a 15% SDS–polyacrylamide gel. In each set of four lanes, the ratio (w/w) of trypsin to ParA was 0, 1:200, 1:100 and 1:50. The numbers at the left show the size (in kDa) of the molecular size markers in the first lane.
We examined whether the mutations introduced into ParA had affected its ability to hydrolyze ATP and the ability of ParB to stimulate this activity. All four mutant proteins showed reduced ATPase activity, which was not stimulated by ParB (Table III). Therefore, these mutations have altered ParA’s interactions with ATP, consistent with a location in the ATP-binding pocket of ParA. The mutations could affect catalysis directly or indirectly by damaging ATP binding. We typically measured ATPase activity at 0.5 mM ATP, a concentration >10-fold higher than the Kd of wild-type ParA for ATP. At 2 mM ATP, however, none of the mutant ParAs showed higher levels of ATP hydrolysis (data not shown). Thus, at physiological concentrations of ATP, its hydrolysis by ParA has been reduced. These results indicate that ATPase activity is not essential for ParA’s repressor function, but suggest that ATP hydrolysis is required for its partition activities.
Table III. ATPase activity of mutant ParA proteins.
| ParA | ATP hydrolyzed (pmol) |
|
|---|---|---|
| –ParB | +ParB | |
| Wild-type | 45 | 278 |
| K122Q | 14 | 14 |
| K122E | 9 | 10 |
| D152N | 23 | 24 |
| D251H | 13 | 14 |
| None | 4 | 16 |
ParA (800 ng) was incubated in the absence or presence of ParB (1 µg) in 50 mM HEPES–KOH pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.5 mM [γ-32P]ATP, 10% glycerol, 1 µg BSA and 2 µg of sonicated salmon sperm DNA in a volume of 10 µl for 90 min at 37°C. Portions were spotted onto PEI-TLC plates, which were developed in 0.5 M LiCl in 1 M formic acid, dried, and exposed to a phosphor screen for quantification in a PhosphorImager. The low level of ATPase seen with ParB alone is most probably due to a minor contamination from a DNA-dependent ATPase.
The super-repressors bind to parOP and to ATP and ADP
We examined the site-specific DNA-binding activities of these ParA proteins using a DNase I footprinting assay. The P1 par promoter region (parOP) contains a large inverted repeat that is recognized by ParA and is located between the ribosome-binding site and the –10 promoter signal of the par operon (Hayes et al., 1994). Wild-type ParA protects >150 bp surrounding this inverted repeat sequence from DNase I digestion (Davis et al., 1992; Davey and Funnell, 1994). We first tested DNA-binding activity in the presence and absence of ADP, the preferred nucleotide cofactor for ParA’s site-specific DNA-binding activity (Davey and Funnell, 1994). Both super-repressors, ParA K122Q and ParA D251H, bound to parOP in the presence of ADP, but very weakly in its absence (Figure 5A and B). This behavior is similar to that of wild-type ParA. ParA D251H did not bind as strongly as wild-type ParA to parOP under typical conditions, but the activity of ParA D251H improved at lower salt concentrations (e.g. 25 mM NaCl). Since ParA D251H was a better repressor than wild-type ParA in vivo, it is possible that the intracellular environment is such that its DNA-binding activity is also better in vivo. In the presence of ADP, the pattern of protection from DNase I by ParA K122Q and D251H was indistinguishable from that of wild-type ParA (Figure 5A and B). All showed extensive protection of a large region surrounding the inverted repeat sequence in parOP. Therefore, the super-repressors must be able to bind ADP, even though ATP hydrolysis has been damaged.
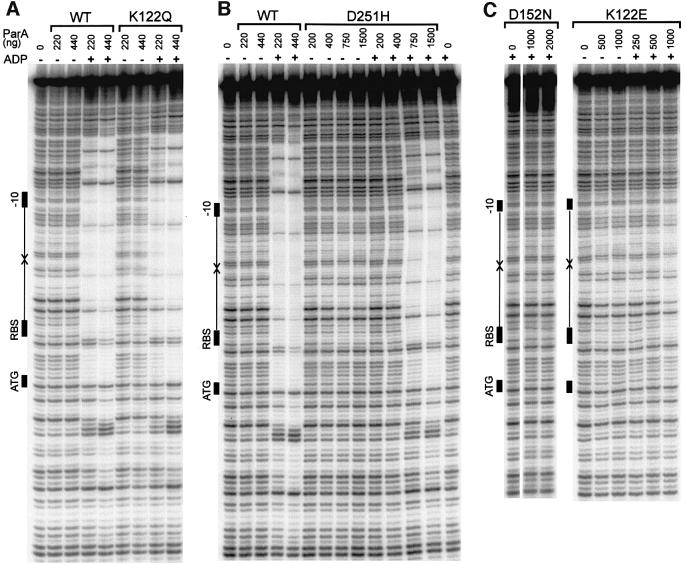
Fig. 5. Site-specific DNA-binding activities of wild-type and mutant ParA proteins. DNA binding to the parOP sequence was measured by DNase I protection assays (Materials and methods). The diagram at the left of each gel shows the elements of the promoter sequence parOP: the arrows represent the inverted repeats that are the recognition sequences for ParA, and the black rectangles show the positions of the –10 promoter signal, the ribosome-binding site (RBS) and the start codon for the parA gene (ATG). (A) DNA binding by wild-type ParA and ParA K122Q. The amount of ParA (in ng) is indicated above each lane. ADP, when present, was at 2 mM. (B) DNA binding by ParA D251H. The experiments were performed as in (A) except that the salt in the buffer was reduced to 25 mM NaCl. The amount of ParA (in ng) in each assay is indicated above each lane. ADP, when present, was at 5 mM. (C) DNA binding of ParA D152N and ParA K122E. The amount of ParA (in ng) in each assay is indicated above each lane. ADP, when present, was at 2 mM.
In contrast, the two weak repressors, ParA K122E and D152N, bound very poorly or not at all to parOP (Figure 5C). As has been shown previously, ParA K122E showed very little binding to parOP even in the presence of ADP, although there was some weak protection within the inverted repeat sequence at high protein concentrations in the presence of ADP. However, this pattern of protection was not the extensive footprint seen with wild-type or super-repressor ParAs. The other weak repressor, ParA D152N, did not bind to parOP (Figure 5C).
Both ATP and ADP stimulate the DNA-binding activity of wild-type ParA, although ADP is a better cofactor than ATP (Davey and Funnell, 1994). Formally, the super-repressor ParAs might bind ADP but not ATP. We compared the ability of ATP and ADP to serve as cofactors for ParA’s site-specific DNA-binding activity (Figure 6). DNA binding by both ParA K122Q and ParA D251H was stimulated by ATP and by ADP. ParA K122Q appeared to be able still to discriminate between ATP and ADP, since its binding activity in the presence of ADP was slightly better than that in the presence of ATP. On the other hand, ParA D251H showed little difference in its response to ATP and ADP. These results show that ParA K122Q and ParA D251H can bind ATP and ADP, and that both nucleotides can serve as cofactors for their DNA-binding activity.
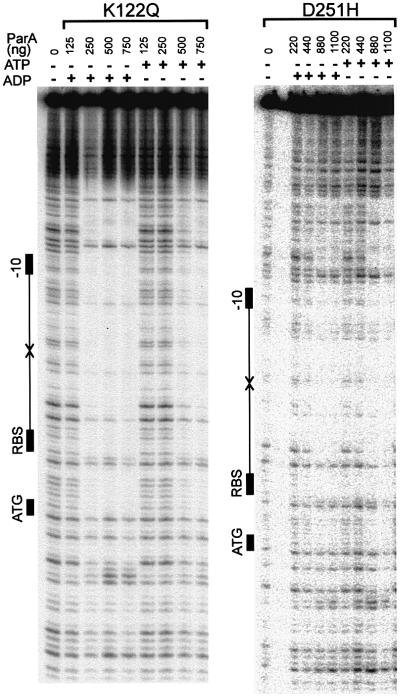
Fig. 6. Comparison of ATP and ADP as cofactors for DNA binding by ParA K122Q and D251H. DNA binding to the parOP sequence by ParA K122Q (left) and ParA D251H (right) was measured in the presence of 2 mM ATP or ADP. The symbols to the left of each gel are described in the legend to Figure 5.
We attempted to measure ATP binding directly to wild-type and mutant ParA proteins using a spin-gel filtration column assay (as in Klemm et al., 1997). While we could measure binding to ATP by wild-type protein, we could detect none to any of the mutant proteins including ParA K122Q and ParA D251H (with or without DNA; data not shown). Since the DNase I footprinting experiments clearly show that ParA K122Q and D251H do bind ADP and ATP (Figures 5 and 6), we conclude that ATP binding by the mutants has been altered so it did not survive the gel filtration step in this non-equilibrium assay. It is perhaps not surprising that we have altered the affinity for ATP and ADP with these mutations, and this assay (as well as others we have tried) was not sensitive enough to measure directly the ATP bound to ParA. However, our results show that at high but nevertheless physiological concentrations of nucleotide, the super-repressors do bind nucleotide and their DNA-binding activities are strongly stimulated by this bound nucleotide.
Finally, we examined whether the mutant ParAs, in particular the super-repressors, could interact with the partition complex consisting of ParB and IHF assembled at parS. Using wild-type protein, we can detect this interaction in a gel mobility shift assay performed in the presence of ATP and magnesium (Bouet and Funnell, 1999). Figure 7 shows that ParA K122Q was unable to interact with the partition complex in this assay, even at high protein levels. ParA D251H, K122E and D152N were also unable to interact with the partition complex in this assay (data not shown).
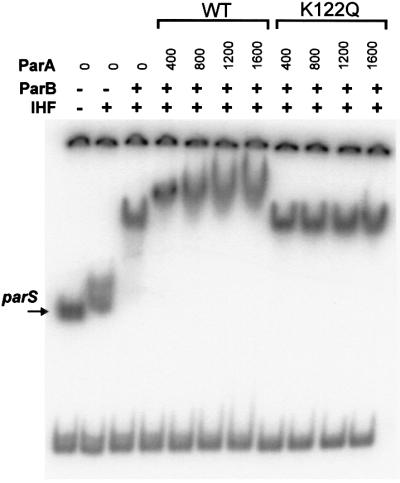
Fig. 7. Interaction of wild-type and mutant ParA with the partition complex. The ability of wild-type ParA and ParA K122Q to interact with the partition complex (ParB and IHF) at parS was tested in a gel mobility shift assay as described previously (Bouet and Funnell, 1999). Briefly, the binding reactions were performed in the presence of 2 mM ATP, and analyzed in a polyacrylamide gel containing 0.1 mM ATP and 10 mM MgCl2 in the gel and the running buffer. The substrate was a restriction digest of a plasmid (pBEF165) that produced three fragments including a 252 bp fragment containing parS. Where indicated, the reaction mixtures contained 80 ng of IHF and 400 ng of ParB.
Discussion
The ParA family of proteins share three motifs that are thought to contribute to their nucleotide-binding site(s) (Koonin, 1993). We have mutated three residues (one in each motif) that are conserved in all members of this family to probe the ATP-binding site of P1 ParA protein. Seven different changes altered the repressor and partition activities of ParA. Four were examined biochemically, ParA K122Q, K122E, D152N, and D251H, and these ParAs showed reduced or no ATPase activity. Our data support the proposal that these motifs interact with ATP and that the nature of this interaction determines ParA function. The phenotypes of the mutants divided into three distinct classes and, notably, different changes in the same residue produced mutants of different classes (Table II).
The first important conclusion from these studies is that the conserved lysine and aspartate residues in the Walker A, Walker B and motif 2 regions are important for ATPase activity. The observations that the purification properties and proteolytic digestion patterns of the mutant proteins were similar to those of wild-type ParA argue that no major structural changes have been effected by the mutations. Our results support the prediction that these motifs form part of the ATP-binding site of ParA.
The crystal structures of two distantly related ATPases with bound nucleotide have been determined. These are the A.vinelandii Fe protein encoded by the nifH gene (Georgiadis et al., 1992; Schindelin et al., 1997; Schlessman et al., 1998; Jang et al., 2000) and the E.coli ArsA component of the arsenite resistance pump (Zhou et al., 2000). Fe protein, in particular, has structural information for the nucleotide-free, MgADP and MgADP-AlF4 (thought to mimic a transition state in ATP hydrolysis) form (for reviews see Seefeldt and Dean, 1997; Gatti et al., 2000). Very recently, the structure of another related ATPase, the cell division protein MinD from Pyrococcus furiosus, showed that these residues are part of its ATP-binding site (Hayashi et al., 2001). The lysine and aspartate residues that are equivalent to those changed here in ParA are invariant (Figure 1), and are all very close to each other and to the terminal phosphate and the magnesium ion in the nucleotide-binding site. The lysine side chain interacts with oxygen on the β- and γ-phosphates of ADP and ATP, respectively. Both aspartates (equivalent to ParA D152 and D251) interact with Mg2+, either directly or through a water molecule. In Fe protein, the asparatate residues transduce signals from ATP binding and hydrolysis to other regions of the protein involved in electron transfer and in interaction with the MoFe protein, the requisite protein partner in nitrogenase catalysis. By analogy to the G-protein class of GTPases (the ATP-binding sites of Fe protein and MinD are structurally similar to the GTP-binding site of these proteins; Jang et al., 2000; Hayashi et al., 2001), the regions containing these conserved aspartate residues have been called the ‘switch I’ and ‘switch II’ motifs (motif 2 and Walker B, respectively). We suggest that this terminology be adopted also for the ParA class of ATPases for several reasons. First, ATP and ADP binding do induce different conformational changes in ParA, as measured directly by CD spectroscopy (Davey and Funnell, 1997). Secondly, the influence of ATP and ADP on ParA’s activities indicates that changes at the nucleotide-binding site are communicated to other regions of the protein. ATP and ADP binding have different effects on the interaction of ParA with parOP and with ParB, and ParB stimulates the ATPase activity of ParA (Davis et al., 1992; Davey and Funnell, 1994, 1997; Bouet and Funnell, 1999). The N-terminus of ParA is thought to interact with parOP, and the C-terminal 140 amino acids probably contain the region that interacts with ParB (Figure 1; Radnedge et al., 1998). Therefore, changes that occur at the nucleotide-binding site due to ATP binding and hydrolysis must signal changes in the interactions between ParA and DNA and between ParA and ParB via communication with these N- and C-terminal domains. The mutations that we have isolated here presumably all have altered or removed these signaling interactions, and they will be interesting to examine in more detail biochemically and structurally.
ATP and ADP promote the dimerization of ParA (Davey and Funnell, 1994). Fe protein is a dimer, and ArsA is essentially a dimer of two homologous domains that each contain a nucleotide-binding site (Figure 1). Their nucleotide-binding sites are situated close to the dimer interface (Schindelin et al., 1997; Schlessman et al., 1998; Jang et al., 2000; Zhou et al., 2000). In Fe protein, nucleotide binding does alter the contacts at the dimer interface, and we expect that this also occurs when ParA binds adenine nucleotides, resulting in a stronger dimerization interaction. An interesting distinction between these proteins and MinD is that the latter is monomeric.
The second important conclusion from these results is that disabling the ability to hydrolyze ATP while retaining the ability to bind nucleotide made ParA a better repressor protein. In vivo, four mutations damaged repressor activity, but three, K122Q, D251H and D251N, converted the protein to a super-repressor form (Figure 3). We have examined biochemically the distinction between the super-repressor class (ParA K122Q and D251H) and the weak repressor/partition class (ParA K122E and D152N). The super-repressors bound to parOP and to ADP and ATP. The weak repressors bound weakly or not at all to parOP. While we have not shown formally that the ability of the weak repressors to bind ATP has been damaged, we suggest that this is the case, and that it is the lack of nucleotide binding that is responsible for damaged ATPase and DNA-binding activities. Considering that the positively charged lysine side chain at position 122 should interact with an negatively charged phosphate in the ATP-binding site, the lysine to glutamate change should interfere strongly with ATP binding. We predict that the D152N change also eliminates nucleotide binding.
The behavior of the super-repressors correlates with our previous studies of wild-type ParA in the presence of ATP, ADP and ATP analogs (Davey and Funnell, 1994, 1997). Adenine nucleotides fulfill at least two functions with respect to ParA’s site-specific DNA-binding activity towards parOP. First, a bound adenine nucleoside di- or triphosphate is required to see significant DNA-binding activity towards parOP. We think this relates to the ability of these bound nucleotides to promote dimerization of ParA, so that the dimer form is the preferred DNA-binding form of ParA. The second function relates to the ability of ParA to catalyze ATP hydrolysis. ParA is an unusual ATP-binding protein, in that both the product of hydrolysis, ADP, and non-hydrolyzable analogs of ATP, such as ATPγS, are equivalent cofactors for DNA-binding activity and both are better than ATP. This observation suggested that some conformational change is promoted by the catalytic event itself (rather than the production of product, for example) that is deleterious to ParA’s DNA-binding activity towards parOP. In fact, CD of wild-type ParA showed that ADP and ATPγS promoted slightly different conformations from that promoted by ATP (Davey and Funnell, 1997). We propose that the super-repressors retain the first activity of ATP, the ability of bound nucleotide to promote DNA binding, but have lost the deleterious effect promoted by ATP hydrolysis and thus have become super-repressors.
From our previous in vitro studies, we hypothesized that ParB acts as a corepressor by eliminating the negative effects of ATP hydrolysis on ParA’s repressor activity. This proposal is supported by the observation that super-repressor activity is identical to the repressor activity of wild-type ParA in the presence of ParB (Figure 3). Since ATP hydrolysis has been damaged, the corepressor function of ParB is unnecessary. ParB may increase the time that ADP remains bound to wild-type ParA, for example, or it might uncouple the negative effect of ATP hydrolysis from the DNA-binding domain via protein– protein signals that alter protein conformation.
The third main conclusion from these experiments is that ATP hydrolysis is important for the partition activity of ParA. The super-repressors, which can still bind ATP, were unable to promote partition (Table I) even when partition was not dependent on repressor activity. These mutants also could not interact with the partition complex (ParB and IHF) assembled at parS (Figure 7, data not shown). These results are consistent with previous ones in which we showed that the ATP-bound form of wild-type ParA, but not the ADP-bound form, could interact with the partition complex (Bouet and Funnell, 1999). However, with wild-type ParA, we also saw an interaction using ATPγS, suggesting that ATP binding, but not hydrolysis, was required. The current results suggest that the latter is not the case and that ATP hydrolysis is necessary for this interaction, or that the conformation of wild-type protein with ATPγS is not identical to that of the hydrolysis-deficient mutants with ATP as far as its partition activity is concerned. We need to dissect the interaction of ParA with the partition complex further in order to understand the mechanistic role of ATP hydrolysis in partition.
The ParPD phenotype involves ATP binding and/or hydrolysis
The partition phenotype of two parA mutants, parAK122R and parAD152H, appeared worse than that of parA null mutants (Table I) and the same as that described previously as a ParPD phenotype caused by an M314I or T158M mutation in ParA (Youngren and Austin, 1997). Several observations suggest that ParPD ParAs cause P1 plasmids to segregate in clumps rather than as individual units, and that the clumps are promoted by ParB acting at parS. First, the ParPD phenotype and the interference we observed were dependent on parB. Secondly, very high expression of parB leads to the same problem (no growth of colonies containing miniP1) even in the presence of wild-type parA (Funnell, 1988a). Thirdly, immunofluorescence experiments suggest that P1 plasmids are grouped together by large concentrated foci of ParB (Erdmann et al., 1999). It has been suggested that ParPD ParAs may actively prevent the unpairing of plasmids mediated by ParB (or a disaggregation if more than two plasmids are grouped together), presumably by an inappropriate interaction between ParA and ParB (Youngren and Austin, 1997). Alternatively, ParPD ParAs may prevent ParB–parS complexes from dissociating from a host factor that tethers the plasmids to the cell. The observation here that some parPD mutations map to the ATP-binding site indicates that ATP is involved in the ParPD interactions of ParA.
ATP-binding sites of the ParA family of proteins
The sequence similarity among the ATP-binding motifs in the ParA-like family of partition (or putative partition) proteins strongly suggests that these proteins will have similarly structured ATP-binding sites. Mutations in the Walker A and B motifs of Bacillus subtilis Soj, a ParA homolog, alter behavior in vivo (Quisel et al., 1999). In this case, however, the analogous Walker A K16Q change behaved more like a loss-of-function mutation, and a G12V change was proposed to alter ATP hydrolysis but not binding. Although the biochemical properties of the mutant Soj proteins have not been determined, it was suggested that alterations in ATP binding and hydrolysis affect the intracellular state of Soj (Quisel et al., 1999). We think that the Walker A, switch I and switch II motifs will all contribute to the ATP-binding sites in this family of proteins, and that the signaling to other regions of the proteins may be conserved. Another region of conservation among ParA-like proteins, motif 3 (Figure 1), is essentially adjacent to the Walker B/switch II motif and is within the region involved in interactions with ParB. It represents an attractive candidate for an effector domain that is signaled by changes in the switch II motif due to ATP binding and/or hydrolysis.
Materials and methods
Escherichia coli strains, media and reagents
Plasmids were maintained in the E.coli K12 strain DH5 [F– endA1 hsdR17 (rK–mK+) supE44 thi-1 recA1 gyrA96 relA1] or DH5Δlac. All bacterial cells were grown in LB medium or on LB plates (Silhavy et al., 1984). When used, the concentrations of ampicillin and chloramphenicol were 100 and 25 µg/ml, respectively. Restriction enzymes and enzymes used for cloning were purchased from New England Biolabs. [α-32P]dATP was from DuPont-NEN, and ATP, ADP and bovine serum albumin (BSA) were from Sigma.
Site-directed mutagenesis and plasmid construction
The plasmids constructed and used in this study are summarized in Table IV. The site-directed mutations were introduced into the single-stranded form of pBEF230 as described (Kunkel et al., 1991) using the following oligonucleotides: CGGTGTGTCA(G/C)(A/C)AACTGTATCG (K122Q and K122E), CGGTGTGTCAAGAACTGTATCG (K122R), GGTTATTGACCTT(A/C)ATCCGCAATC, (D152N and D152H) and CTTTATCCTCGTT(A/C)ATAGTGGTCC (D251N and D251H). All mutations were between the unique XhoI and BglII sites in parA.
Table IV. Plasmids used in this study.
| Plasmid | Description |
|---|---|
| pBEF101 | parA, expressed from parOP in pBR322 (Funnell, 1988a) |
| pBEF116 | parA + parB, expressed from parOP (P1 HindIII–PvuI region in pBR322) |
| pBEF131 | parA + parB::lacZ, expressed from parOP [the 19th codon of parB was fused to lacZ in pMLB1034, a translational fusion vector (Silhavy et al., 1984)] |
| pBEF198 | parA, expressed from T7 promoter in pET17b (Davey and Funnell, 1994) |
| pBEF230 | par XhoI–PvuI fragment in pBlueScript SK+ |
| pBEF239b | parA::lacZ, expressed from parOP in pST52 |
| pBEF250b | miniP1 plasmid pLG44 (Funnell and Gagnier, 1995) deleted for parOP and parA (ΔHindIII–SacII) |
| pBEF251 | ΔparA version of pEF5 (ΔXho–SacII region of parA from pEF5) |
| pEF1 seriesa | parAs + parB::lacZ, expressed from parOP (from pBEF131) |
| pEF3 seriesa | parAs, expressed from T7 promoter (as pBEF198) |
| pEF5 seriesa | parAs + parB, expressed from blaP1 in pBR322 |
| pEF7 seriesa | parAs, expressed from blaP2 in pBR322 |
| pEF8 seriesa | parAs + parB, expressed from blaP2 in pBR322 |
aEach plasmid in these series is denoted as pEFx.y, ‘x’ represents the parent series and ‘y’ represents the parA allele (i.e. wild-type, mutant or null). y = 1 (wild-type parA), 2 (D251N), 3 (D251H), 4 (D152H), 5 (D152N), 6 (K122Q), 7 (K122E), 8 (K122R), 9 (ΔparA: ΔXhoI–BglII).
bpBEF239 and pBEF250 encode resistance to chloramphenicol; all others encode resistance to ampicillin.
For subsequent constructions, the XhoI–BglII fragment of each mutant gene was swapped with the corresponding fragment of wild-type parA into a variety of plasmid backgrounds. These parent plasmids were pBEF101, pBEF116, pBEF131 and pBEF198 (Table IV).
We constructed two versions of the β-lactamase promoter (blaP) using synthetic oligonucleotides (Figure 8). The first, blaP1, was adapted from Su et al. (1992), and contained the –35 and –10 promoter signals, a ribosome-binding site, an EcoRI site and 5′ overhangs that allowed cloning into BglII and NdeI sites. These oligonucleotides were cloned into the BglII–NdeI sites of pBEF198, which replaced the T7 promoter with the blaP promoter. The blaP1 promoter was used for partition (plasmid stability) assays. For repression assays, a stronger version called blaP2 was constructed that included a G to T substitution in the –10 signal, which increased its resemblance to the prokaryotic –10 consensus sequence (immunoblots showed that this blaP1 to blaP2 change increased protein expression by 10- to 20-fold). The EcoRI–XhoI fragments from these constructs were then used to replace the parOP promoter in pBEF101 and pBEF116 derivatives, resulting in pBR322 derivatives containing either parA or parA and parB under the control of blaP (Table IV).
Fig. 8. Sequence of oligonucleotides used to construct the β-lactamase promoters blaP1 and blaP2. The –35 and –10 promoter signals, the start of transcription (+1) and the ribosome-binding site (RBS) are indicated. The sequence of blaP1 was adapted from Su et al. (1992). In blaP2, the first base in the –10 region was changed from G to T (arrow), and 5 bp were inserted immediately after the transcription start site to create a SacI site that was used to distinguish easily between the two promoters.
parA null mutants were constructed by deleting the sequence between XhoI and BglII (pEF derivatives) or between XhoI and SacII (pBEF251) in parA. The deleted sequences were replaced by a 10 bp HindIII linker or an 8 bp PstI linker, respectively, to preserve the parA reading frame and avoid interfering with the expression of parB.
Construction of parOP-lacZ reporter systems
The PpuMI site of pMLB1034 (Silhavy et al., 1984) was changed to a HindIII site using synthetic linkers. The EcoRI–XhoI fragment (made blunt at the XhoI end) from pBEF116 was then cloned into the EcoRI–SmaI sites of the modified pMLB1034. This strategy created a HindIII fragment that contained parOP and lacZ fused in-frame to the first 158 bp of parA. This HindIII fragment was then inserted into λD69 DNA (Mizusawa and Ward, 1982). Phage packaging was performed in vitro (Promega) and the resulting phages were used to infect and lysogenize E.coli N99Δlac (F– galK2 ΔargF-lac). The lysogens were screened for lacZ activity by plating on agar plates containing X-gal. One lysogen was exposed to UV light to induce phage production and the phages were used to lysogenize DH5Δlac, resulting in the strain DH5Δlac(λEF1). This reporter strain thus contains a single copy of the reporter gene integrated into its chromosome.
Plasmid pBEF239 carries the parOP-parA::lacZ gene fusion (the HindIII fragment above) in the plasmid pST52, which is compatible with pBR322 derivatives (Som and Tomizawa, 1982).
Plasmid stability assays
A colony of E.coli DH5 cells containing pBEF250 and one of the pEF5 derivatives was picked from an LB agar plate containing ampicillin and chloramphenicol, diluted into LB with ampicillin, and grown overnight at 37°C. Samples were taken from the beginning and the end of the growth period and plated for single colonies on LB agar plates with ampicillin. Resulting colonies were transferred using toothpicks onto LB plates with chloramphenicol to monitor the presence of the miniP1 plasmid pBEF250.
Protein purification
ParB FrV was purified as described previously (Davey and Funnell, 1997). The mutant ParA proteins were purified using the protocol previously described for wild-type ParA (Davey and Funnell, 1994). ParA FrIV refers to ParA eluted from a MonoQ chromatography step, at which it was judged at least 98% pure by Coomassie blue staining of protein gels. Occasionally the protein was purified further by heparin affinity chromatography to remove contaminating ATPases found in some preparations. In this case, the FrIV protein was diluted to 100 mM KCl in 25 mM HEPES pH 7.5, 0.1 mM EDTA, 10% (v/v) glycerol, and then loaded on a 5 ml heparin–Sepharose column equilibrated with the same buffer. The ParA proteins (FrV) were eluted with a 100 mM to 1 M KCl linear gradient.
DNase I protection assays
DNase I protection assays were done as previously described (Davey and Funnell, 1994) with the following modifications. The reaction mixtures (25 µl) contained 10–50 fmol 32P-end-labeled par promoter fragment prepared from pMD9 (Davey and Funnell, 1994), 2 µg of sonicated salmon sperm DNA, 2 µg of BSA, 2 mM CaCl2, 50 mM Tris acetate or Tris–HCl pH 7.5, 50 mM NaCl, 10 mM MgCl2, and ParA as indicated in the figure legends.
Acknowledgments
Acknowledgements
We thank Zhen Zhang and Sal Forcucci for excellent technical assistance. We also thank Alan Davidson and Jun Liu for many helpful suggestions that improved the manuscript. This research was supported by a grant from the Canadian Institutes of Health Research to B.E.F.
References
- Abeles A.L., Friedman,S.A. and Austin,S.J. (1985) Partition of unit-copy miniplasmids to daughter cells. III. The DNA sequence and functional organization of the P1 partition region. J. Mol. Biol., 185, 261–272. [DOI] [PubMed] [Google Scholar]
- Bouet J.-Y. and Funnell,B.E. (1999) P1 ParA interacts with the P1 partition complex at parS and an ATP–ADP switch controls ParA activities. EMBO J., 18, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouet J.Y., Surtees,J.A. and Funnell,B.E. (2000) Stoichiometry of P1 plasmid partition complexes. J. Biol. Chem., 275, 8213–8219. [DOI] [PubMed] [Google Scholar]
- Davey M.J. and Funnell,B.E. (1994) The P1 plasmid partition protein ParA. A role for ATP in site-specific DNA binding. J. Biol. Chem., 269, 29908–29913. [PubMed] [Google Scholar]
- Davey M.J. and Funnell,B.E. (1997) Modulation of the P1 plasmid partition protein ParA by ATP, ADP and P1 ParB. J. Biol. Chem., 272, 15286–15292. [DOI] [PubMed] [Google Scholar]
- Davis M.A. and Austin,S.J. (1988) Recognition of the P1 plasmid centromere analog involves binding of the ParB protein and is modified by a specific host factor. EMBO J., 7, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.A., Martin,K.A. and Austin,S.J. (1992) Biochemical activities of the ParA partition protein of the P1 plasmid. Mol. Microbiol., 6, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Davis M.A., Radnedge,L., Martin,K.A., Hayes,F., Youngren,B. and Austin,S.J. (1996) The P1 ParA protein and its ATPase activity play a direct role in the segregation of plasmid copies to daughter cells. Mol. Microbiol., 21, 1029–1036. [DOI] [PubMed] [Google Scholar]
- Erdmann N., Petroff,T. and Funnell,B.E. (1999) Intracellular localization of P1 ParB protein depends on ParA and parS. Proc. Natl Acad. Sci. USA, 96, 14905–14910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S.A. and Austin,S.J. (1988) The P1 plasmid-partition system synthesizes two essential proteins from an autoregulated operon. Plasmid, 19, 103–112. [DOI] [PubMed] [Google Scholar]
- Funnell B.E. (1988a) Mini-P1 plasmid partitioning: excess ParB protein destabilizes plasmids containing the centromere parS. J. Bacteriol., 170, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funnell B.E. (1988b) Participation of Escherichia coli integration host factor in the P1 plasmid partition system. Proc. Natl Acad. Sci. USA, 85, 6657–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funnell B.E. and Gagnier,L. (1995) Partition of P1 plasmids in Escherichia coli mukB chromosomal partition mutants. J. Bacteriol., 177, 2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti D., Mitra,B. and Rosen,B.P. (2000) Escherichia coli soft metal ion-translocating ATPases. J. Biol. Chem., 275, 34009–34012. [DOI] [PubMed] [Google Scholar]
- Georgiadis M.M., Komiya,H., Chakrabarti,P., Woo,D., Kornuc,J.J. and Rees,D.C. (1992) Crystallographic structure of the nitrogenase iron protein for Azotobacter vinelandii. Science, 257, 1653–1659. [DOI] [PubMed] [Google Scholar]
- Gerdes K., Moller-Jensen,J. and Jensen,R.B. (2000) Plasmid and chromosome partitioning: surprises from phylogeny. Mol. Microbiol., 37, 455–466. [DOI] [PubMed] [Google Scholar]
- Gordon G.S., Sitnikov,D., Webb,C.D., Teleman,A., Straight,A., Losick,R., Murray,A.W. and Wright,A. (1997) Chromosome and low copy plasmid segregation in E.coli: visual evidence for distinct mechanisms. Cell, 90, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Hayashi I., Oyama,T. and Morikawa,K. (2001) Structural and functional studies of MinD ATPase: implications for the molecular recognition of the bacterial cell division apparatus. EMBO J., 20, 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes F., Radnedge,L., Davis,M.A. and Austin,S.J. (1994) The homologous operons for P1 and P7 plasmid partition are autoregulated from dissimilar operator sites. Mol. Microbiol., 11, 249–260. [DOI] [PubMed] [Google Scholar]
- Hiraga S. (2000) Dynamic localization of bacterial and plasmid chromosomes. Annu. Rev. Genet., 34, 21–59. [DOI] [PubMed] [Google Scholar]
- Hiraga S., Niki,H., Ogura,T., Ichinose,C., Mori,H., Ezaki,B. and Jaffe,A. (1989) Chromosome partitioning in Escherichia coli: novel mutants producing anucleate cells. J. Bacteriol., 171, 1496–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton K., Gunther,N.W. and Grossman,A.D. (1994) spoOJ is required for normal chromosome segregation as well as the initiation of sporulation in Bacillus subtilis. J. Bacteriol., 176, 5320–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S.B., Seefeldt,L.C. and Peters,J.W. (2000) Insights into nucleotide signal transduction in nitrogenase: protein with MgADP bound. Biochemistry, 39, 14745–14752. [DOI] [PubMed] [Google Scholar]
- Klemm R.D., Austin,R.J. and Bell,S.P. (1997) Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell, 88, 493–502. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. (1993) A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol., 229, 1165–1174. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A., Bebenek,K. and McClary,J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol., 204, 125–139. [DOI] [PubMed] [Google Scholar]
- Miller J.H. (1992) A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Mizusawa S. and Ward,D.F. (1982) A bacteriophage λ vector for cloning with BamHI and Sau3A. Gene, 20, 317–322. [DOI] [PubMed] [Google Scholar]
- Mohl D.A. and Gober,J.W. (1997) Cell cycle-dependent polar localization of chromosome partitioning proteins in Caulobacter crescentus. Cell, 88, 675–684. [DOI] [PubMed] [Google Scholar]
- Motallebi-Veshareh M., Rouch,D.A. and Thomas,C.M. (1990) A family of ATPases involved in active partitioning of diverse bacterial plasmids. Mol. Microbiol., 4, 1455–1463. [DOI] [PubMed] [Google Scholar]
- Quisel J.D., Lin,D.C.H. and Grossman,A.D. (1999) Control of development by altered localization of a transcription factor in B.subtilis. Mol. Cell, 4, 665–672. [DOI] [PubMed] [Google Scholar]
- Radnedge L., Youngren,B., Davis,M. and Austin,S. (1998) Probing the structure of complex macromolecular interactions by homolog specificity scanning: the P1 and P7 plasmid partition systems. EMBO J., 17, 6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin H., Kisker,C., Schlessman,J.L., Howard,J.B. and Rees,D.C. (1997) Structure of ADP-AlF4–-stabilized nitrogenase complex and its implications for signal transduction. Nature, 387, 370–376. [DOI] [PubMed] [Google Scholar]
- Schlessman J.L., Woo,D., JoshuaTor,L., Howard,J.B. and Rees,D.C. (1998) Conformational variability in structures of the nitrogenase iron proteins from Azotobacter vinelandii and Clostridium pasteurianum. J. Mol. Biol., 280, 669–685. [DOI] [PubMed] [Google Scholar]
- Seefeldt L.C. and Dean,D.R. (1997) Role of nucleotides in nitrogenase catalysis. Acc. Chem. Res., 30, 260–266. [Google Scholar]
- Sharpe M.E. and Errington,J. (1996) The Bacillus subtilis soj-spoOJ locus is required for a centromere-like function involved in prespore chromosome partitioning. Mol. Microbiol., 21, 501–509. [DOI] [PubMed] [Google Scholar]
- Silhavy T.J., Berman,M.L. and Enquist,L.W. (1984) Experiments with Gene Fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Som T. and Tomizawa,J. (1982) Origin of replication of Escherichia coli plasmid RSF1030. Mol. Gen. Genet., 187, 375–383. [DOI] [PubMed] [Google Scholar]
- Su G.F., Brahmbhatt,H.J., Wehland,J., Rohde,M. and Timmis,K.N. (1992) Construction of stable LamB–Shiga toxin B subunit hybrids: analysis of expression in Salmonella typhimurium aroA strains and stimulation of B subunit-specific mucosal and serum antibody responses. Infect. Immun., 60, 3345–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surtees J.A. and Funnell,B.E. (1999) P1 ParB domain structure includes two independent multimerization domains. J. Bacteriol., 181, 5898–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe E., Wachi,M., Yamasaki,M. and Nagai,K. (1992) ATPase activity of SopA, a protein essential for active partitioning of F-plasmid. Mol. Gen. Genet., 234, 346–352. [DOI] [PubMed] [Google Scholar]
- Youngren B. and Austin,S. (1997) Altered ParA partition proteins of plasmid P1 act via the partition site to block plasmid propagation. Mol. Microbiol., 25, 1023–1030. [DOI] [PubMed] [Google Scholar]
- Zhou T.Q., Radaev,S., Rosen,B.P. and Gatti,D.L. (2000) Structure of the ArsA ATPase: the catalytic subunit of a heavy metal resistance pump. EMBO J., 19, 4838–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]