

Abstract
We studied the function of a Trypanosoma brucei topoisomerase II using RNA interference (RNAi). Expression of a topoisomerase II double-stranded RNA as a stem–loop caused specific degradation of mRNA followed by loss of protein. After 6 days of RNAi, the parasites’ growth rate declined and the cells subsequently died. The most striking phenotype upon induction of RNAi was the loss of kinetoplast DNA (kDNA), the cell’s catenated mitochondrial DNA network. The loss of kDNA was preceded by gradual shrinkage of the network and accumulation of gapped free minicircle replication intermediates. These facts, together with the localization of the enzyme in two antipodal sites flanking the kDNA, show that a function of this topoisomerase II is to attach free minicircles to the network periphery following their replication.
Keywords: kinetoplast DNA/minicircle/RNAi/topoisomerase II
Introduction
Trypanosoma brucei is a protozoan parasite that causes sleeping sickness in Africa. Trypanosomes have attracted considerable attention from the research community not only because they cause disease but also because they have an unusual biology. Among their most amazing features is a unique mitochondrial DNA, termed kinetoplast DNA (kDNA). kDNA is a network consisting of thousands of DNA circles topologically interlocked in a planar array (for a review see Shapiro and Englund, 1995). In vivo the network is condensed into a disk-shaped structure within the mitochondrial matrix near the flagellar basal body. There are two types of DNA circles catenated in a network, a few dozen maxicircles (each 23 kb) and several thousand minicircles (each 1 kb). The maxicircles are homologs of mitochondrial DNAs in other eukaryotes, encoding rRNAs and mitochondrial proteins that function mainly in energy transduction (for a review see Simpson, 1987). Maxicircle transcripts are edited by addition or deletion of uridine residues at specific sites, and minicircles encode guide RNAs that control editing specificity (for a review see Estevez and Simpson, 1999).
Knowledge about the mechanism of kDNA replication has come mainly from studies of Crithidia fasciculata, an insect parasite related to the trypanosomes. Crithidia fasciculata is an ideal subject for biochemical studies, and much of our research, and that in other laboratories, has involved analysis of the function and intracellular localization of kDNA replication proteins. These studies, together with investigations of the structure and dynamics of kDNA replication intermediates, have led to a fairly detailed model for kDNA replication (Shapiro and Englund, 1995; Abu-Elneel et al., 2001; Drew and Englund, 2001).
In the current model for C.fasciculata kDNA replication, covalently closed minicircles are released vectorially from the kDNA disk (towards the flagellar basal body) by a topoisomerase II (topo II) (Englund, 1979; Drew and Englund, 2001). The free minicircles then undergo replication, ultimately forming singly and multiply gapped progeny (Kitchin et al., 1984, 1985; Birkenmeyer and Ray, 1986; Birkenmeyer et al., 1987). The partially or fully replicated minicircles migrate to two antipodal sites that flank the kDNA disk, and in these sites they are thought to undergo primer removal (Engel and Ray, 1999) and repair of some gaps in the multiply gapped species (Shapiro and Englund, 1995). Then, in another topo II-mediated reaction, the gapped progeny minicircles are attached to the periphery of the network. At this stage, the partially replicated network contains two zones. Covalently closed minicircles, not yet replicated, are situated in the central zone of the network, and gapped minicircles, having completed replication, are localized in the peripheral zone (Ferguson et al., 1992; Pérez-Morga and Englund, 1993a; Guilbride and Englund, 1998). As replication proceeds, the central zone shrinks and the peripheral zone enlarges until, at the end of replication, the minicircles have doubled in copy number and all contain gaps. The minicircle gaps are then repaired, and the network splits in two (Pérez-Morga and Englund, 1993b). The latter reaction is not understood, but it probably involves a topo II-mediated selective unlinking of minicircles along the scission line of the network. See Shapiro and Englund (1995) and Morris et al. (2001) for reviews on kDNA replication.
There have been few investigations on kDNA replication in T.brucei, but studies on a closely related parasite, Trypanosoma equiperdum, indicate that trypanosome kDNA replication closely resembles that of C.fasciculata (Ntambi et al., 1986; Ryan and Englund, 1989a,b). Nevertheless, there is one major mechanistic difference involving the attachment of free progeny minicircles to the network. In both cases, minicircles are thought to attach to the network at opposite sides of the kDNA disk. In C.fasciculata, minicircles become distributed uniformly around the periphery, apparently because of a relative movement of the network disk and the sites of minicircle attachment (Simpson and Simpson, 1976; Pérez-Morga and Englund, 1993a). In T.brucei, on the other hand, progeny minicircles are found only at opposite sites of the network, apparently because there is no relative movement (Hoeijmakers and Weijers, 1980; Ferguson et al., 1994).
Although studies on C.fasciculata continue to be fruitful, there are now compelling reasons to focus attention on kDNA replication in T.brucei. One reason is the rapidly progressing genome project (El-Sayed et al., 2000) that allows identification of many candidate genes for kDNA replication proteins. Another is the growing arsenal of genetic techniques available in T.brucei (e.g. Wirtz and Clayton, 1995; Wirtz et al., 1999). One such technique is RNA interference (RNAi), a method in which expression of a double-stranded RNA (dsRNA) in trypanosomes causes selective degradation of the cognate mRNA (Ngo et al., 1998; Bastin et al., 2000; Bringaud et al., 2000; LaCount et al., 2000; Shi et al., 2000; Wang et al., 2000). Our laboratory recently developed two RNAi vectors that, after integration into a T.brucei chromosome, express a specific dsRNA upon induction with tetracycline (Wang et al., 2000). RNAi also functions in other organisms including Caenorhabditis elegans (Fire et al., 1998) and Drosophila melanogaster (Kennerdell and Carthew, 1998).
In this study, we use RNAi to evaluate the function of a T.brucei topo II. This topo II gene was cloned in 1990 (Strauss and Wang, 1990), but its protein product has never been studied or localized intracellularly. However, its 66% identity with the C.fasciculata mitochondrial topo II (Melendy and Ray, 1989) suggested that it could be mitochondrial. Although the C.fasciculata enzyme localizes in the two antipodal sites flanking the kDNA disk (Melendy et al., 1988), there is no genetic evidence regarding its function. In preliminary studies, we reported that induction of topo II-specific dsRNA in T.brucei caused inhibition of parasite growth as well as dyskinetoplasty, the loss of the kDNA network (Wang et al., 2000). Here we describe detailed studies on the loss of kDNA. We found that the kDNA gradually shrinks in size during RNAi, and that the major reason for progressive kDNA loss is the inefficient attachment of newly replicated minicircles to the network.
Results
RNAi causes selective degradation of topo II mRNA and loss of topo II protein
We used a construct for expression of a stem–loop RNA in which the stem contains a 584 bp sequence derived from the topo II-coding region (Figure 1A). After linearizing this construct with EcoRV, we transfected it into T.brucei 29-13, a transgenic cell line that expresses the tetracycline repressor and T7 RNA polymerase (Wirtz et al., 1999). We used phleomycin to select cells in which the construct had integrated into the non-transcribed rDNA spacer. After obtaining a clonal cell line by limiting dilution, we induced synthesis of the stem–loop RNA from a procyclin promoter by adding tetracycline to the culture. As reported earlier (Wang et al., 2000), the cells grew normally for the first 6 days. They then gradually stopped growing, and eventually they died (Figure 1B).
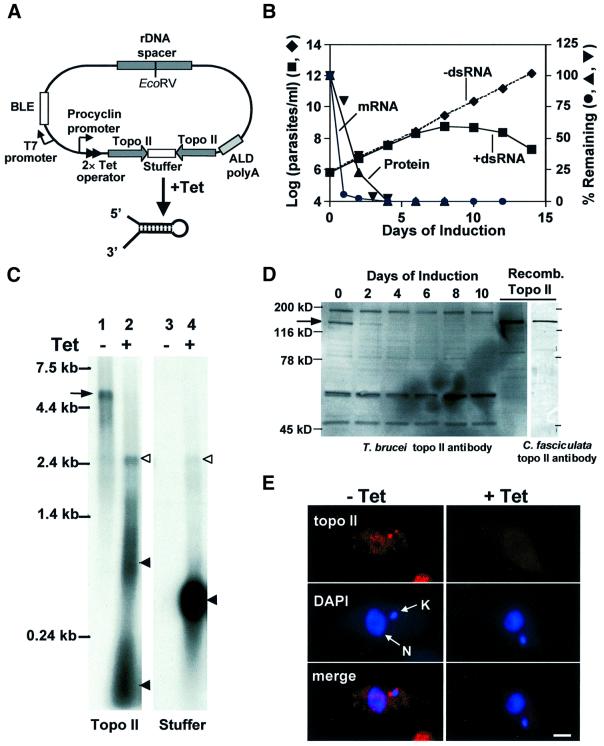
Fig. 1. dsRNA expression causes degradation of topo II mRNA and loss of topo II protein. (A) Stem–loop construct for topo II RNAi (Wang et al., 2000): see Materials and methods for more details. BLE, phleomycin resistance gene; ALD polyA, aldolase poly(A) addition sequence; Tet, tetracycline. The construct is not drawn to scale. (B) Effect of topo II RNAi on cell growth. RNAi was uninduced (filled diamonds) or induced with 1 µg/ml tetracycline (filled squares). The graph also shows topo II mRNA levels (filled circles, evaluated by a northern blot as in C) and topo II protein levels (filled upright triangles and filled inverted triangles, evaluated by western blots in two independent experiments including that in D). (C) Northern blot of total RNA of trypanosomes transfected with the topo II stem–loop construct. Lane 1, RNA from uninduced cells, with topo II probe. Lane 2, RNA from cells induced with tetracycline for 24 h, topo II probe. Lane 3, RNA from uninduced cells, stuffer probe. Lane 4, RNA from induced cells, stuffer probe. As a control, this blot was also probed for tubulin mRNA, and there was no difference between uninduced and induced cells (not shown). Arrow, topo II mRNA; open arrowheads, dsRNA containing topo II and stuffer sequences; filled arrowheads, fragments of stem–loop dsRNA. (D) Western blot of whole-cell lysates (1 × 106 cell equivalents/lane) from uninduced (day 0) and tetracycline-induced cells (days 2–10) using T.brucei antibody. Recombinant T.brucei topo II (40 ng) was loaded on the same gel as a control. The arrow indicates topo II protein. Right-hand gel: recombinant topo II recognized by C.fasciculata topo II antibody. (E) Immunofluorescence of uninduced cells (left panels) and cells induced with tetracycline for 6 days (right panels) using C.fasciculata topo II antibody. kDNA (K) and nuclear DNA (N) are stained with DAPI. Scale bar, 2 µm.
We analyzed topo II mRNA from cells before and 24 h after tetracycline induction of RNAi on a northern blot. Tetracycline induction resulted in massive expression of stem–loop dsRNA, as detected by the topo II probe (Figure 1C, lane 2) and the stuffer probe (lane 4). There is a faint band, at ∼2.4 kb, containing both topo II sequences and stuffer sequences (open arrowhead, lanes 2 and 4), which probably represents full-length stem–loop product. However, most of the stem–loop RNA is degraded, forming smears at ∼1 kb and <0.2 kb (detected by the topo II probe, lane 2, filled arrowheads) or ∼0.5 kb (detected by the stuffer probe, lane 4, solid arrowhead). The 5.4 kb topo II mRNA (marked by an arrow, lane 1) was almost completely degraded in the sample treated with tetracycline (lane 2). This degradation is specific, as there is no loss of tubulin mRNA (not shown). In an evaluation of the kinetics of mRNA degradation, we found that there was barely detectable topo II mRNA between day 1 and day 12 (Figure 1B, filled circles).
Using a western blot with antibody against recombinant T.brucei topo II, we measured the amount of topo II protein in parasites at different times after induction (Figure 1D). The topo II (∼130 kDa, in good agreement with its predicted size; Strauss and Wang, 1990) is indicated by an arrow (see right-hand lanes for detection of recombinant enzyme). Two days after RNAi induction, the cells lost ∼75% of their topo II protein, and there was virtually no protein detected by 3–4 days after induction. Several proteins that bind non-specifically to the topo II antibody did not change in level following the tetracycline induction (Figure 1D). Figure 1B shows the kinetics of loss of topo II protein based on two independent experiments (upright triangles and inverted triangles).
As mentioned above, the T.brucei topo II is homologous to a well characterized topo II from C.fasciculata (Pasion et al., 1992) that is located in the mitochondrial matrix at two antipodal sites flanking the kDNA disk (Melendy et al., 1988). However, the T.brucei enzyme has never been localized intracellularly. Using antibody against C.fasciculata topo II (this antibody binds T.brucei topo II, as shown in the last lane of Figure 1D), we found the same antipodal localization for the T.brucei enzyme (Figure 1E, left panels). Six days after RNAi induction, the topo II was undetectable in those sites (Figure 1E, right panels).
RNAi of topo II causes dyskinetoplasty
The most remarkable phenotype caused by topo II RNAi, easily visualized by 4′,6-diamidino-2-phenylindole (DAPI) staining, was a loss of kDNA (dyskinetoplasty). Figure 2A (left panel) shows a DAPI-stained image of cells prior to tetracycline induction. As expected, each cell contains a nucleus (N) and a much smaller kinetoplast (K). In contrast, 9 days after tetracycline induction, cells have clearly stained nuclei but either have no detectable kinetoplast or, in some cases, have a small kinetoplast remnant (right panel).
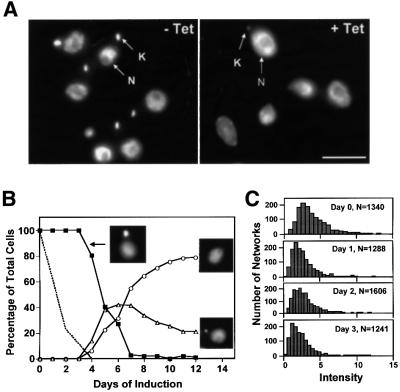
Fig. 2. Cells expressing topo II dsRNA became dyskinetoplastic. (A) Cells were fixed and treated with DAPI to stain the nucleus (N) and kinetoplast (K). The left panel shows uninduced cells, and the right panel shows cells 9 days after induction with tetracycline. Scale bar, 5 µm. (B) Time course of appearance of dyskinetoplasty. Cells were fixed, stained with DAPI and photographed as in (A). More than 150 cells at each time point were scored by eye according to the size of kinetoplast [normal appearing kDNA (filled squares), abnormally small kDNA (open triangles) and no kDNA (open circles); examples are shown in inset images]. The dashed line shows the loss of topo II protein (same data as Figure 1B). (C) The distribution of DAPI staining intensity of kDNA from uninduced (day 0) and tetracycline-induced cells (days 1, 2 and 3). The staining intensity was analyzed automatically using a IPLab software script. The typical kinetoplast present after day 4 was too small to be identified reliably with the parameters used for days 0–3. N, number of cells analyzed. Intensity values are arbitrary.
In a study of the kinetics of the disappearance of the kinetoplast, we found that the cells lost this structure gradually. As visualized by fluorescence microscopy of DAPI-stained cells, all had a normal appearing kinetoplast during the first 3 days after induction of RNAi. Then, starting at day 4, we detected cells with an abnormally small kinetoplast and even a few that appeared dyskinetoplastic (Figure 2B). By day 12, ∼80% of the cells were dyskinetoplastic and 20% had a small kinetoplast. To analyze the kDNA size more precisely during the first 3 days of RNAi, we used an automated method developed by M.E.Drew in our laboratory (Drew and Englund, 2001). At each time point, we photographed >1000 individual cells with a CCD camera and then measured the DAPI staining intensity of each kDNA network using IPLab software. We plotted the number of networks as a function of staining intensity (Figure 2C). The histograms showed a small and gradual decrease in DAPI staining intensity during the first 3 days, indicating that the effect on kDNA begins soon after RNAi induction. Using an analogous automated method, we observed a similar result when measuring the size of the kDNA networks in the same DAPI-stained cells (data not shown).
Analysis of kDNA network size by sucrose gradient centrifugation and electron microscopy (EM)
To study further the loss of kDNA, we centrifuged total DNA samples through sucrose gradients and detected the kDNA in each fraction by dot-blotting using a minicircle probe (Figure 3). In uninduced cells (day 0), most of the kDNA sedimented as intact networks (peak indicated by vertical arrow). The small amount of radioactivity at the top of the gradients probably represented free minicircle replication intermediates (Englund, 1979). The hybridizing material at the bottom of the tubes, especially at days 3 and 5, may represent aggregated networks. As a function of time after RNAi induction, there was a gradual reduction in size of networks and, between days 1 and 3, an accumulation of radioactivity near the top of the gradient. By day 5, there was a striking decline in total kDNA detected by the minicircle probe. These results, together with the DAPI staining intensity measurements (Figure 2C), indicate that kDNA networks decrease in size gradually. Eventually there is a nearly complete loss of kDNA detected by the minicircle probe.
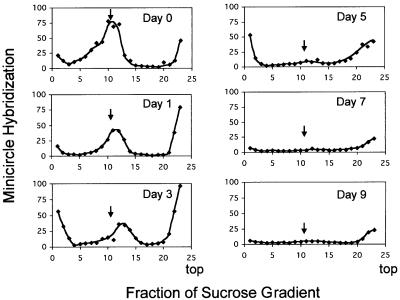
Fig. 3. Analysis of loss of kDNA by sucrose gradient centrifugation. Total DNA from cells without (day 0) or with induction of RNAi by tetracycline (days 1–9) was purified and nuclear DNA was sheared mildly. Samples (4 × 107 cell equivalents) were then centrifuged through 5–20% sucrose gradients. Fractions (5% of total) were dot-blotted and hybridized to a 32P-labeled minicircle probe and quantified by phosphorimaging. Minicircle hybridization is expressed in arbitrary units. The bimodal distribution at day 0 probably indicates unit-size networks (marked by an arrow in all panels) and larger networks that are undergoing replication. Radioactivity at the top of gradients at day 0 is probably free minicircle replication intermediates. At later times, the material at the top of the gradient probably includes network fragments. The radioactivity at the bottom of the gradients in some panels is probably due to aggregates of kDNA, possibly formed when networks sediment into the walls of the tubes.
We obtained data consistent with these conclusions using EM of isolated networks (Figure 4). Figure 4A shows half of a normal kDNA network, a planar structure ∼6 µm in diameter (Ferguson et al, 1994). After 5 days of RNAi, we found heterogeneously sized and irregular network fragments averaging ∼3 µm in diameter (Figure 4B). After 9 days, we found even smaller network fragments (Figure 4C).
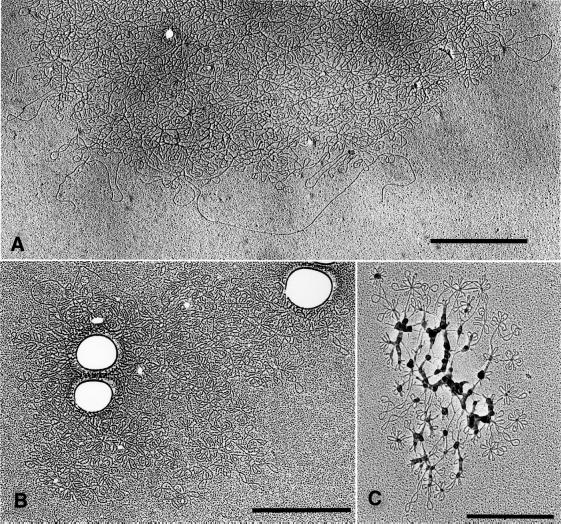
Fig. 4. EM of kDNA networks after RNAi. (A) EM showing half of a kDNA network from an uninduced cell. (B) A network fragment from a cell induced for 5 days. (C) A fragment from a cell induced for 9 days (we do not know the reason for the blobs associated with the DNA). Scale bar, 1 µm.
Both minicircles and maxicircles are lost following RNAi
To measure the kinetics of loss of minicircles and maxicircles, we induced a culture with tetracycline and then purified total DNA at different times. We digested the DNA from each sample with HindIII and XbaI and analyzed it by Southern hybridization (5 × 105 cell equivalents/lane) (Figure 5). These enzymes cut maxicircles into six fragments, and we detected the 1.4 kb XbaI fragment with a specific probe (Figure 5A). The hybridization of this maxicircle band was measured by phosphorimaging and the results were plotted in Figure 5D. Because T.brucei minicircles are highly heterogeneous in sequence (Steinert and Van Assel, 1980), these enzymes produce a variety of products, and we used a full-length cloned minicircle probe to detect them (Figure 5B). The predominant products are singly cleaved minicircles (1 kb), and there are also fragments from multiply cleaved minicircles (<1 kb) and a faint ladder of multimeric catenanes. We measured minicircle products between 0.5 and 1 kb by phosphorimaging, and the results are also plotted in Figure 5D. Figure 5C shows a control experiment demonstrating that there was no significant loss of nuclear DNA during the 9 days, as detected by probing the same blot for THT1, the gene for a trypanosome hexose transporter (Bringaud and Baltz, 1993). As shown in Figure 5D, there was little loss of either minicircle or maxicircle DNA during the first 2 days after induction of RNAi. Subsequently there was a parallel decline of both sequences, and loss was nearly complete by day 9.
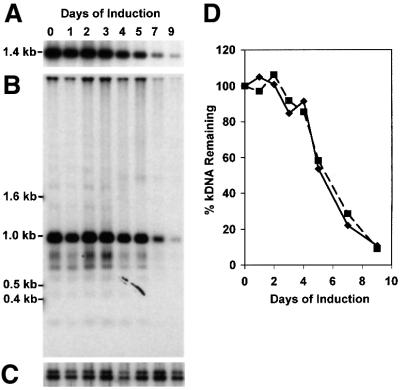
Fig. 5. Loss of minicircles and maxicircles. (A) Loss of maxicircle sequences. Total DNA (from 5 × 105 cells) was digested overnight by HindIII–XbaI, fractionated on a 1% agarose gel and transferred to a nylon membrane. The membrane was hybridized to the maxicircle probe (a random-primed PCR product that recognizes the 1.4 kb XbaI fragment). Only the 1.4 kb region is shown because >90% of the maxicircle signal was detected in this fragment. (B) Loss of minicircle sequences. The blot was stripped and rehybridized to a minicircle probe (a T.equiperdum minicircle fragment; Ntambi and Englund, 1985). Full-length linearized minicircles are 1 kb, and smaller fragments are multiply cleaved species. The ladder above 1 kb derives from undigested minicircle catenanes. (C) Levels of nuclear DNA during topo II RNAi. The same blot was reprobed for THT1, a nuclear gene encoding the trypanosome hexose transporter. (D) Kinetics of minicircle (filled squares) and maxicircle (filled diamonds) loss after induction of RNAi. Maxicircles were measured by phosphorimaging of the 1.4 kb fragment (A) and minicircles were measured by phosphorimaging of the fragments between 0.5 and 1 kb (B). To correct for unequal loading of the lanes, the amount of minicircle and maxicircle DNA was normalized to nuclear DNA (C).
Effect of RNAi on free minicircle replication intermediates
kDNA minicircles are released from the network for replication, and the free minicircle intermediates have been well characterized in C.fasciculata (Kitchin et al., 1984, 1985; Birkenmeyer et al., 1985; Birkenmeyer and Ray, 1986) and T.equiperdum (a close relative of T.brucei) (Ryan et al., 1988; Ryan and Englund, 1989b). To assess changes in the T.brucei free minicircle population as a consequence of RNAi, we extracted total DNA at different times after RNAi induction (Figure 6A). We fractionated the DNA by agarose gel electrophoresis in the presence of ethidium bromide, and detected free minicircles by Southern hybridization. Lane M shows minicircle markers (form I, covalently closed; form II, gapped; form III, linearized). Lane 0 shows minicircles from uninduced cells, and subsequent lanes show those from tetracycline-induced cells after the indicated number of days. After 1 day of RNAi, there was relatively little change in the level or composition of free minicircle replication intermediates (as indicated by phosphorimaging, Figure 6B), but by the second day there was a 2.5-fold increase in the level of gapped minicircles (Figure 6A and B). The gapped species increased even further on day 3, and by day 4 was mostly replaced by a smear species. The latter species is highly sensitive to S1 nuclease (not shown) and probably represents partially degraded minicircles that are heavily gapped. The level of covalently closed minicircles (form I) increased only slightly during the first 3 days, and then dropped sharply at day 4 (Figure 6B). By day 5, all free minicircle species were virtually undetectable, although network fragments were detected in the upper region of the gel and in the slot.
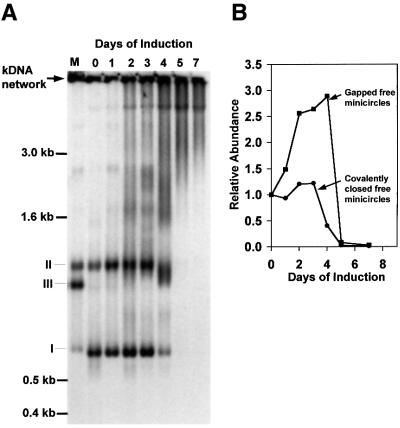
Fig. 6. Effect of topo II RNAi on free minicircle replication intermediates. (A) Total DNA was purified after various times of RNAi and then fractionated (106 cell equivalents/lane) on an agarose gel. After transfer to a membrane, the kDNA was detected by a minicircle probe. Intact kDNA networks (indicated by an arrow) do not enter the gel and are transferred inefficiently to the membrane. Lane M, markers of covalently closed (I), gapped (II) and linearized (III) minicircles (prepared by topo II decatenation of partially degraded kDNA networks). Lanes 0–7, DNA samples from cells at the indicated number of days after induction of RNAi. (B) Levels of covalently closed minicircles (filled circles) and gapped minicircles (filled squares) after induction of RNAi. DNA was estimated by phosphorimaging of the gel shown in (A).
Removal of tetracycline restores topo II mRNA level but not cell growth
To examine whether the topo II mRNA levels could be restored after removal of tetracycline, we induced cells and then removed the drug at day 4. We used a northern blot to examine the levels of topo II mRNA on day 0 and on days 4–9. Figure 7A shows an autoradiogram of the blot and Figure 7B shows a graph of mRNA levels determined by densitometry. Although dsRNA was not detectable 1 day after tetracycline removal (on day 5), only 15% of the initial level of topo II mRNA was present at that time (Figure 7A). By day 6 (2 days after tetracycline removal), ∼50% of the topo II mRNA was present, and it was fully recovered by day 9 (5 days after tetracyline removal).
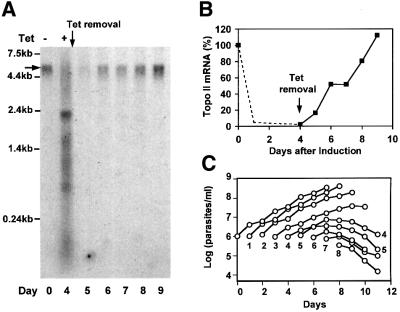
Fig. 7. Effect of tetracycline removal from induced cells. (A) Northern analysis of topo II mRNA (indicated by the horizontal arrow) from uninduced cells (day 0), cells induced for 4 days (day 4) and cells at each subsequent day after removal of tetracycline at day 4. (B) Kinetics of recovery of topo II mRNA, determined by densitometry of the autoradiogram in (A). The dashed line shows typical loss of topo II mRNA from other experiments (e.g. Figure 1B). (C) Growth kinetics after removal of tetracycline. The cells were induced with tetracycline for times ranging from 0 to 8 days (as indicated beneath the growth curve) and were then transferred to tetracycline-free medium. Cell densities were measured daily after tetracycline removal.
We next tested whether recovery of mRNA could restore cell growth (Figure 7C). We induced cultures with tetracycline for times ranging from 0 to 8 days, and then, for each culture, we transferred the cells to a tetracycline-free medium. In each case, we measured cell densities and plotted them as a function of time after initial induction (Figure 7C). We found that when we removed tetracycline 1 or 2 days after induction, the cells recovered normal growth. However, when we removed the drug at 3 days after induction, the cells grew more slowly than normal, and at about day 9 they stopped growth and eventually died. When we removed the drug 4 days after induction, they clearly could not recover. Although <10% of cells were dyskinetoplastic at day 4 when we removed tetracycline (Figure 2B), these cells continued to lose kDNA even in the absence of tetracycline. By day 10 (6 days after tetracycline removal), 80% of these cells had no kinetoplast and 20% of them had abnormally small kinetoplasts, as judged by DAPI staining (not shown). One possible explanation for these experiments is that the RNAi effect persists for several cell divisions after we shut down the synthesis of dsRNA, and the damage to the kDNA network caused by loss of topo II cannot be repaired.
Discussion
The T.brucei topo II studied here seemed likely to be mito chondrial based on its homology to the well characterized C.fasciculata mitochondrial topo II (Melendy et al., 1988; Melendy and Ray, 1989; Pasion et al., 1992; Ray et al., 1992). We have now shown that the T.brucei enzyme does indeed have the same intracellular localization as that of the C.fasciculata enzyme, in the two antipodal sites flanking the kDNA disk (Figure 1E). In fact, this finding is the first report of a protein in the antipodal sites in T.brucei. We also found that RNAi was an easy and effective genetic tool to examine the in vivo function of this topo II. Since the topo II RNAi phenotype developed over several days after induction of dsRNA synthesis, we were able to study the function of this topo II by following the changes in several parameters during the loss of the kDNA network prior to cell death. Similar RNAi methodology should be equally effective in evaluating the function of other essential genes.
We used northern blots to show that the stem–loop RNA was expressed after induction with tetracycline (Figure 1C). We detected a 2.4 kb product that hybridized with probes for the topo II sequence and the stuffer sequence, and it had a size predicted for a full-length stem–loop fragment (whose 3′ end was probably determined by a polyadenylation site downstream of the second topo II sequence). However, most of the stem–loop RNA was fragmented, suggesting that cleavages had occurred near the junctions of the topo II sequences and the stuffer sequences (Figure 1C, lanes 2 and 4). There are also smaller fragments of the topo II sequence (<0.24 kb), and these may include the ∼22 bp mediators of RNAi (Hamilton and Baulcombe, 1999; Hammond et al., 2000; Zamore et al., 2000).
We found that RNAi caused a selective loss of virtually all of the topo II mRNA within 1 day and that this low level was sustained for at least 12 days as long as tetracycline was maintained in the culture. The loss of topo II protein lagged slightly behind that of its mRNA, falling to very low levels within 3 days (Figure 1B and D). Immunofluorescence showed a loss of topo II protein from the antipodal sites (Figure 1E). The loss of topo II protein was accompanied by a dramatic phenotype, dyskinetoplasty, or the loss of the kDNA network (Figure 2). Since the kDNA maxicircles encode proteins required for functioning of the electron transport chain, which is essential for viability of procyclic trypanosomes, dyskinetoplasty results in eventual cell death. The same phenotype, loss of kDNA and inhibition of cell growth, was also observed when we knocked down topo II expression using another RNAi vector, pZJM (Wang et al., 2000). In that case, we did not study these phenomena further. All experiments in this study involved the stem–loop construct.
Upon induction of RNAi, the first indication of kDNA loss appeared at day 4, as visualized by fluorescence microscopy of DAPI-stained cells (Figure 2B). However, automated analysis of DAPI staining intensity (Figure 2C) as well as sedimentation of the kDNA networks in sucrose gradients (Figure 3) suggested that the networks had begun to shrink in size well before this time. The classical method for inducing dyskinetoplasty in bloodstream trypanosomes is to treat with DNA-binding agents such as ethidium bromide, acriflavin and hydroxystilbamidine (reviewed in Simpson, 1972; Hajduk, 1978). Since these agents inhibit trypanosome topoisomerases by promoting cleavable complex formation (Shapiro et al., 1989; Shapiro and Englund, 1990), it is not surprising that RNAi-mediated loss of a topo II could have the same effect. Nevertheless, the question remains as to how loss of this particular topo II contributes to dyskinetoplasty. It is clear a priori that topo II enzymes have multiple roles in kDNA replication (Nenortas et al., 1998). Topo IIs release minicircles from the network for replication, and they reattach the progeny. They may also facilitate the unlinking of parental strands during replication of both minicircles and maxicircles and they segregate the progeny of replication. Finally, they could be involved in unlinking of minicircles during scission of a fully replicated network into two daughter networks. It is possible that there is more than one topo II in the mitochondrion (e.g. see Shlomai, 1994) and that they have distinct intramitochondrial locations to carry out these different functions.
All our data suggest that a major consequence of RNAi knockdown of this mitochondrial topo II is that progeny minicircles cannot be reattached to the network. The fact that the network shrinks gradually in size after expression of topo II dsRNA is consistent with this possibility (Figures 2C and 3), and so is the gradual decline in kDNA level as measured by dot-blotting with minicircle and maxicircle probes (Figure 5D). The most compelling evidence favoring this hypothesis is the rise in level of gapped free minicircles between days 1 and 4 (Figure 6B). The gapped minicircles are the progeny of replication, and they are the species that are reattached to the network by a topo II. The absence of this topo II thus explains their rise in concentration. In contrast, there is only a relatively small change in the level of covalently closed free minicircles between day 0 and 3, consistent with the possibility that there is not much change in the rate of minicircle release from the network or in their replication to form gapped progeny. By day 5, there are few if any detectable free minicircle species, indicating that replication has stopped completely, possibly because the remnant kDNA network is too small to interact properly with the replication machinery. Nearly half the kDNA still remains at day 5, detectable either by sucrose gradients (Figure 3) or by minicircle hybridization (Figure 5D). These small network fragments may be lost by dilution when the cells divide (although there are only about two more doublings after day 5; Figure 1B), or they may be degraded.
Although our data provide strong support for the role of topo II in attaching minicircles to the network, this enzyme may have other functions as well. For example, in Figure 6A, there is an increase in intensity of several bands between the 1.6 and 3 kb markers during days 1–3. Like the gapped circles, these components form a smear at day 4, suggesting that they have undergone some degradation. Based on their electrophoretic mobility, these species could be catenated dimers (with different numbers of interlocks) that are formed in the final stages of replication of free minicircles (Ryan and Englund, 1989b; Shapiro, 1994). If this interpretation is correct, it would imply that this topo II is also involved in segregating minicircle dimers formed by replication. This topo II may also have some functions associated with maxicircles. Ray et al. (1992) found that the homologous C.fasciculata topo II cross-links with maxicircles as well as minicircles when the cells are treated with the topo inhibitor VP16. After RNAi of T.brucei topo II, we found a loss of maxicircles parallel with that of minicircles (Figure 5D). The maxicircle loss could be due to a requirement for topo II in their replication. Although maxicircles are not released from the network during replication (Hajduk et al., 1984; Carpenter and Englund, 1995), the topo II could have a role in removing supercoils generated by replication or in segregating the progeny. The loss of maxicircles during topo II RNAi (Figure 5B) could also be indirect, due to decatenation from the shrinking network and eventual loss by dilution during cell division or degradation.
Although it took less than 1 day for topo II mRNA degradation induced by RNAi (Figure 1B), the restoration of the topo II mRNA level took 5 days after removal of tetracycline from the culture (Figure 7B). This partial inheritance of the RNAi effect is also found in other organisms such as C.elegans (Fire et al., 1998; Grishok et al., 2000), presumably because the ∼22 bp dsRNA is very stable or because there is some RNA-dependent RNA polymerase activity that propagates its effect (Cogoni and Macino, 1999). The persistence of the RNAi effect for several days after tetracycline removal also explains the fact that although cells initially grew normally, when we removed tetracycline at days 3–5 (Figure 7C), they eventually became dyskinetoplastic and died.
In addition to providing insight into the mechanism of kDNA replication, RNAi of this topo II may also be useful in addressing other interesting questions. For example, it could be used to determine the minimal size of the kDNA network that is compatible with the viability of T.brucei. Knockdown of topo II expression by RNAi, as described herein, may provide an excellent way to shrink the size of the network in a relatively controlled manner. This in turn could lead to explorations of the regulation of the size and structure of the kDNA network as well as its minicircle composition.
Materials and methods
Trypanosome growth
Procyclic T.brucei strain 29-13 (a gift from Elizabeth Wirtz and George Cross, Rockefeller University), which harbors integrated genes for T7 RNA polymerase and tetracycline repressor (Wirtz et al., 1999), were grown in SDM-79 supplemented with 15% fetal bovine serum (Wang et al., 2000). Cell densities were determined by a hemocytometer and the cultures were diluted 1:10 when the cell density reached 1 × 107 cells/ml. Growth curves were plotted as the product of the cell density and the dilution factor. The doubling time of both 29-13 cells and uninduced cells transfected with the stem–loop construct was 16 h.
Plasmid constructs, transfections and induction of RNAi
The stem–loop construct (Figure 1A) contained two opposing copies of a 584 bp segment of the topo II-coding sequence (starting at 5′ TAGGCTTTCA…) and 550 bp of an unrelated mouse gene (the stuffer) (Wang et al., 2000) in the pLew100 vector (Wirtz et al., 1999). The trypanosomes transfected with the stem–loop construct were selected with 2.5 µg/ml phleomycin (Wang et al., 2000) and cloned by limiting dilution. Cells were grown for at least 2–3 weeks in the presence of phleomycin prior to induction of RNAi with tetracycline (1 µg/ml).
RNA purification and northern analysis
Cells were lysed with Trizol reagent (Life Technologies) and then extracted with phenol/chloroform (Ngo et al., 1998). Total RNA was then treated with DNase I and fractionated on a 1.5% agarose–7% formaldehyde gel. To determine if there was equal loading of each lane, the rRNA level was estimated by ethidium bromide staining (not shown). The topo II mRNA was detected with a 32P-labeled probe made by random priming of the same 584 bp fragment used in the stem–loop construct.
Western blotting and immunofluorescence
Total trypanosome protein (1 × 106 cell equivalents/lane) was fractionated by SDS–PAGE (8% gel), transferred onto Immobilon-P and probed with a 1:2000 dilution of a mouse anti-topo II polyclonal antibody prepared against recombinant T.brucei topo II (a generous gift from Tom Kulikowicz and Terry Shapiro, Johns Hopkins University). Anti-topo II antibodies were detected with goat anti-mouse IgG labeled with horseradish peroxidase and visualized using ECL western blotting detection reagents (Amersham Pharmacia Biotech). Polyclonal antibodies against C.fasciculata topo II (a gift of Dan Ray, UCLA, diluted 1:2000) were detected by the alkaline phosphatase system (Roche Diagnostics GmbH). The recombinant topo II with a His tag at its C-terminus (20 residues longer than the primary translation product), also provided by Tom Kulikowicz and Terry Shapiro, was fractionated in the same gel as a positive control. For immunofluorescence, we used antibody against C.fasciculata topo II (T.brucei topo II antibody did not work for immunofluorescence). The cells were harvested by centrifugation, washed once and resuspended at 108 cells/ml in phosphate-buffered saline (PBS; 138 mM NaCl, 3 mM KCl, 2 mM KH2PO4, 10 mM Na2HPO4 pH 7.2) prior to spotting on polylysine-coated slides (Guilbride and Englund, 1998). After having adhered to the slide for 10 min, the cells were fixed in 4% paraformaldehyde in PBS for 5 min, washed twice in PBS containing 0.1 M glycine for 5 min each, washed once in PBS for 5 min and then permeabilized by methanol for 1 h at –20°C. The slides were rehydrated with three 10 min washes in PBS, and then incubated for 60 min with antibody against the C.fasciculata topo II [1:50 diluted in PBS with 1% bovine serum albumin (BSA)]. The slides were washed three times (10 min each) in PBS with 0.1% Tween-20. Texas Red-conjugated anti-rabbit secondary antibody (Sigma Co., 1:400 diluted in PBS with 1% BSA) was then applied for 30 min and washed three times in PBS with 0.1% Tween-20 (10 min each). The slides were stained with DAPI (0.2 µg/ml for 1 min), washed twice with PBS (5 min each) and mounted with Vectashield (Vector Lab. Inc.). All operations were performed at room temperature unless otherwise noted. The fluorescence microscopy and digital image acquisition were carried out as described (Drew and Englund, 2001) with a Zeiss Axioskop microscope (Carl Zeiss, Inc.).
DNA purification and analysis
Mid-log phase cells (∼5 × 106 cells/ml) were harvested and washed once with 10 mM Tris–HCl pH 8.0, containing 100 mM NaCl and 100 mM EDTA (NET-100) and then resuspended in NET-100 at a density of 2 × 108 cells/ml. To determine kDNA size and content (Figures 3 and 5), cells were lysed in 0.5% SDS containing 0.2 mg/ml proteinase K (56°C, 4 h), and then treated with 0.1 mg/ml RNase A (37°C, 15 min). For sucrose gradient centrifugation (Figure 3), the cell lysate was passed once through a 16 gauge needle with maximal finger pressure to shear nuclear DNA mildly. Cell lysate (0.2 ml, 4 × 107 cell equivalents) was then layered on the top of 5 ml sucrose gradients (Englund, 1979). Centrifugation was at 7000 r.p.m. for 100 min at 20°C with a Beckman SW55Ti rotor. After collecting fractions, they were analyzed by dot-blotting with a probe made from a cloned full-length T.equiperdum minicircle sequence (Ntambi and Englund, 1985). The ‘conserved region’ of the T.equiperdum minicircle is virtually identical to that of T.brucei. For measurements of the minicircle and maxicircle content (Figure 5), the cell lysate was extracted with phenol/chloroform. The total DNA was then digested with XbaI–HindIII, and fractionated on a 1% agarose gel (11 × 14 × 0.5 cm, 25 V for 16 h) with 1 µg/ml ethidium bromide in both the gel and the 1× TBE buffer.
To detect free minicircles (Figure 6A), cells were lysed in 0.5% SDS containing 0.2 mg/ml proteinase K (56°C, 30 min), treated with 0.1 mg/ml RNase A (37°C, 15 min) and then extracted with phenol/chloroform. Total DNA was then fractionated on a 1.5% agarose gel (20 × 25 × 0.5 cm, 70 V, 18 h) with 1 µg/ml ethidium bromide in both the gel and the 1× TBE buffer (Ryan et al., 1988). All hybridizations (Figures 3, 5 and 6) were conducted as described (Wang et al., 2000). kDNA network purification and EM (Figure 4) were conducted as described (Pérez-Morga and Englund, 1993a).
Automated measurement of the DAPI-staining intensity of kDNA networks in situ
Cells were treated with tetracycline to induce RNAi (for 1, 2 and 3 days), and the three cultures were timed so they finished the experiment simultaneously. After washing in PBS, these cells and uninduced cells (all at 108/ml) were spotted on the same polylysine-coated slide. The cells were fixed in 4% paraformaldehyde (5 min) and washed twice in PBS. After staining with DAPI (0.2 µg/ml, 1 min), they were washed in PBS and then mounted in Vectashield. Fluorescent images were captured under identical conditions by a CCD camera operated with IPLab Spectrum software. A computer-based assay, developed by M.E.Drew, was applied to evaluate each kDNA network for DAPI staining intensity and for size (area in µm2) using scripts written with IPLab software (Drew and Englund, 2001). Briefly, the kDNA networks were identified automatically from all images by limiting the range of pixel intensity and the size. These limits exclude nuclei because their area is too large. Thus each kDNA network is defined as a segment comprised of pixels. Each segment was measured for its total intensity, size (area) and standard deviation of intensity. The out-of-focus networks were eliminated automatically because they have a much smaller standard deviation value (<100 intensity units) compared with an in-focus network segment (∼200–800 intensity units). The DAPI intensity and the size of each segment were exported to spreadsheet software for further analysis.
Acknowledgments
Acknowledgements
We thank Mark Drew, James Morris and Andy Fire for thoughtful discussion, and Viiu Klein for outstanding technical support. We thank Elisabetta Ullu and Chris Tschudi for introducing Zefeng Wang to the study of T.brucei RNAi at the Biology of Parasitism course at the Marine Biological Laboratory, Woods Hole. We appreciate comments on the manuscript by Dr Terry Shapiro, Dr Barbara Sollner-Webb and members of our lab. This work was supported by grant GM 27608 from the NIH.
References
- Abu-Elneel K., Robinson,D.R., Drew,M.E., Englund,P.T. and Shlomai,J. (2001) Intramitochondrial localization of universal minicircle sequence-binding protein, a trypanosomatid protein that binds kinetoplast minicircle replication origins. J. Cell Biol., 153, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin P., Ellis,K., Kohl,L. and Gull,K. (2000) Flagellum ontogeny in trypanosomes studied via an inherited and regulated RNA interference system. J. Cell Sci., 113, 3321–3328. [DOI] [PubMed] [Google Scholar]
- Birkenmeyer L. and Ray,D.S. (1986) Replication of kinetoplast DNA in isolated kinetoplasts from Crithidia fasciculata. Identification of minicircle DNA replication intermediates. J. Biol. Chem., 261, 2362–2368. [PubMed] [Google Scholar]
- Birkenmeyer L., Sugisaki,H. and Ray,D.S. (1985) The majority of minicircle DNA in Crithidia fasciculata strain CF-C1 is of a single class with nearly homogeneous DNA sequence. Nucleic Acids Res., 13, 7107–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeyer L., Sugisaki,H. and Ray,D.S. (1987) Structural characterization of site-specific discontinuities associated with replication origins of minicircle DNA from Crithidia fasciculata. J. Biol. Chem., 262, 2384–2392. [PubMed] [Google Scholar]
- Bringaud F. and Baltz,T. (1993) Differential regulation of two distinct families of glucose transporter genes in Trypanosoma brucei. Mol. Cell. Biol., 13, 1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringaud F., Robinson,D.R., Barradeau,S., Biteau,N., Baltz,D. and Baltz,T. (2000) Characterization and disruption of a new Trypanosoma brucei repetitive flagellum protein, using double-stranded RNA inhibition. Mol. Biochem. Parasitol., 111, 283–297. [DOI] [PubMed] [Google Scholar]
- Carpenter L.R. and Englund,P.T. (1995) Kinetoplast maxicircle DNA replication in Crithidia fasciculata and Trypanosoma brucei. Mol. Cell. Biol., 15, 6794–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoni C. and Macino,G. (1999) Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature, 399, 166–169. [DOI] [PubMed] [Google Scholar]
- Drew M.E. and Englund,P.T. (2001) Intramitochondrial location and dynamics of Crithidia fasciculata kinetoplast minicircle replication intermediates. J. Cell Biol., 153, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed N.M., Hegde,P., Quackenbush,J., Melville,S.E. and Donelson,J.E. (2000) The African trypanosome genome. Int. J. Parasitol., 30, 329–345. [DOI] [PubMed] [Google Scholar]
- Engel M.L. and Ray,D.S. (1999) The kinetoplast structure-specific endonuclease I is related to the 5′ exo/endonuclease domain of bacterial DNA polymerase I and colocalizes with the kinetoplast topoisomerase II and DNA polymerase β during replication. Proc. Natl Acad. Sci. USA, 96, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund P.T. (1979) Free minicircles of kinetoplast DNA in Crithidia fasciculata. J. Biol. Chem., 254, 4895–4900. [PubMed] [Google Scholar]
- Estevez A.M. and Simpson,L. (1999) Uridine insertion/deletion RNA editing in trypanosome mitochondria—a review. Gene, 240, 247–260. [DOI] [PubMed] [Google Scholar]
- Ferguson M., Torri,A.F., Ward,D.C. and Englund,P.T. (1992) In situ hybridization to the Crithidia fasciculata kinetoplast reveals two antipodal sites involved in kinetoplast DNA replication. Cell, 70, 621–629. [DOI] [PubMed] [Google Scholar]
- Ferguson M.F., Torri,A.F., Pérez-Morga,D., Ward,D.C. and Englund,P.T. (1994) Kinetoplast DNA replication: mechanistic differences between Trypanosoma brucei and Crithidia fasciculata. J. Cell Biol., 126, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Grishok A., Tabara,H. and Mello,C.C. (2000) Genetic requirements for inheritance of RNAi in C.elegans. Science, 287, 2494–2497. [DOI] [PubMed] [Google Scholar]
- Guilbride D.L. and Englund,P.T. (1998) The replication mechanism of kinetoplast DNA networks in several trypanosomatid species. J. Cell Sci., 111, 675–679. [DOI] [PubMed] [Google Scholar]
- Hajduk S.L. (1978) Influence of DNA complexing compounds on the kinetoplast of trypanosomatids. Prog. Mol. Subcell. Biol., 6, 158–200. [Google Scholar]
- Hajduk S.L., Klein,V.A. and Englund,P.T. (1984) Replication of kinetoplast DNA maxicircles. Cell, 36, 483–492. [DOI] [PubMed] [Google Scholar]
- Hamilton A.J. and Baulcombe,D.C. (1999) A species of small antisense RNA in posttranscriptional gene silencing in plants. Science, 286, 950–952. [DOI] [PubMed] [Google Scholar]
- Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers J.H.J. and Weijers,P.J. (1980) The segregation of kinetoplast DNA networks in Trypanosoma brucei. Plasmid, 4, 97–116. [DOI] [PubMed] [Google Scholar]
- Kennerdell J.R. and Carthew,R.W. (1998) Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell, 95, 1017–1026. [DOI] [PubMed] [Google Scholar]
- Kitchin P.A., Klein,V.A., Fein,B.I. and Englund,P.T. (1984) Gapped minicircles. A novel replication intermediate of kinetoplast DNA. J. Biol. Chem., 259, 15532–15539. [PubMed] [Google Scholar]
- Kitchin P.A., Klein,V.A. and Englund,P.T. (1985) Intermediates in the replication of kinetoplast DNA minicircles. J. Biol. Chem., 260, 3844–3851. [PubMed] [Google Scholar]
- LaCount D.J., Bruse,S., Hill,K.L. and Donelson,J.E. (2000) Double-stranded RNA interference in Trypanosoma brucei using head-to-head promoters. Mol. Biochem. Parasitol., 111, 67–76. [DOI] [PubMed] [Google Scholar]
- Melendy T. and Ray,D.S. (1989) Novobiocin affinity purification of a mitochondrial type II topoisomerase from the trypanosomatid Crithidia fasciculata. J. Biol. Chem., 264, 1870–1876. [PubMed] [Google Scholar]
- Melendy T., Sheline,C. and Ray,D.S. (1988) Localization of a type II DNA topoisomerase to two sites at the periphery of the kinetoplast DNA of Crithidia fasciculata. Cell, 55, 1083–1088. [DOI] [PubMed] [Google Scholar]
- Morris J.C., Drew,M.E., Klingbeil,M.M., Motyka,S.A., Saxowsky,T.T., Wang,Z. and Englund,P.T. (2001) Replication of kinetoplast DNA: an update for the new millennium. Int. J. Parasitol., 31, 453–458. [DOI] [PubMed] [Google Scholar]
- Nenortas E.C., Bodley,A.L. and Shapiro,T.A. (1998) DNA topo isomerases: a new twist for antiparasitic chemotherapy? Biochim. Biophys. Acta, 1400, 349–354. [DOI] [PubMed] [Google Scholar]
- Ngo H., Tschudi,C., Gull,K. and Ullu,E. (1998) Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl Acad. Sci. USA, 95, 14687–14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntambi J.M. and Englund,P.T. (1985) A gap at a unique location in newly replicated kinetoplast DNA minicircles from Trypanosoma equiperdum. J. Biol. Chem., 260, 5574–5579. [PubMed] [Google Scholar]
- Ntambi J.M., Shapiro,T.A., Ryan,K.A. and Englund,P.T. (1986) Ribonucleotides associated with a gap in newly replicated kinetoplast DNA minicircles from Trypanosoma equiperdum. J. Biol. Chem., 261, 11890–11895. [PubMed] [Google Scholar]
- Pasion S.G., Hines,J.C., Aebersold,R. and Ray,D.S. (1992) Molecular cloning and expression of the gene encoding the kinetoplast-associated type II DNA topoisomerase of Crithidia fasciculata. Mol. Biochem. Parasitol., 50, 57–68. [DOI] [PubMed] [Google Scholar]
- Pérez-Morga D. and Englund,P.T. (1993a) The attachment of minicircles to kinetoplast DNA networks during replication. Cell, 74, 703–711. [DOI] [PubMed] [Google Scholar]
- Pérez-Morga D. and Englund,P.T. (1993b) The structure of replicating kinetoplast DNA networks. J. Cell Biol., 123, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D.S., Hines,J.C. and Anderson,M. (1992) Kinetoplast-associated DNA topoisomerase in Crithidia fasciculata: crosslinking of mitochondrial topoisomerase II to both minicircles and maxicircles in cells treated with the topoisomerase inhibitor VP16. Nucleic Acids Res., 20, 3353–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.A. and Englund,P.T. (1989a) Replication of kinetoplast DNA in Trypanosoma equiperdum. Minicircle H strand fragments which map at specific locations. J. Biol. Chem., 264, 823–830. [PubMed] [Google Scholar]
- Ryan K.A. and Englund,P.T. (1989b) Synthesis and processing of kinetoplast DNA minicircles in Trypanosoma equiperdum. Mol. Cell. Biol., 9, 3212–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.A., Shapiro,T.A., Rauch,C.A., Griffith,J.D. and Englund,P.T. (1988) A knotted free minicircle in kinetoplast DNA. Proc. Natl Acad. Sci. USA, 85, 5844–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro T.A. (1994) Mitochondrial topoisomerase II activity is essential for kinetoplast DNA minicircle segregation. Mol. Cell. Biol., 14, 3660–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro T.A. and Englund,P.T. (1990) Selective cleavage of kinetoplast DNA minicircles promoted by antitrypanosomal drugs. Proc. Natl Acad. Sci. USA, 87, 950–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro T.A. and Englund,P.T. (1995) The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol., 49, 117–143. [DOI] [PubMed] [Google Scholar]
- Shapiro T.A., Klein,V.A. and Englund,P.T. (1989) Drug promoted cleavage of kinetoplast DNA minicircles: evidence for type II topoisomerase activity in trypanosome mitochondria. J. Biol. Chem., 264, 4173–4178. [PubMed] [Google Scholar]
- Shi H., Djikeng,A., Mark,T., Wirtz,E., Tschudi,C. and Ullu,E. (2000) Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA, 6, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomai J. (1994) The assembly of kinetoplast DNA. Parasitol. Today, 10, 341–346. [DOI] [PubMed] [Google Scholar]
- Simpson A.M. and Simpson,L. (1976) Pulse-labeling of kinetoplast DNA: localization of 2 sites of synthesis within the networks and kinetics of labeling of closed minicircles. J. Protozool., 23, 583–587. [DOI] [PubMed] [Google Scholar]
- Simpson L. (1972) The kinetoplast of the hemoflagellates. Int. Rev. Cytol., 32, 139–207. [Google Scholar]
- Simpson L. (1987) The mitochondrial genome of kinetoplastid protozoa: genomic organization, transcription, replication and evolution. Annu. Rev. Microbiol., 41, 363–382. [DOI] [PubMed] [Google Scholar]
- Steinert M. and Van Assel,S. (1980) Sequence heterogeneity in kinetoplast DNA: reassociation kinetics. Plasmid, 3, 7–17. [DOI] [PubMed] [Google Scholar]
- Strauss P.R. and Wang,J.C. (1990) The TOP2 gene of Trypanosoma brucei: a single-copy gene that shares extensive homology with other TOP2 genes encoding eukaryotic DNA topoisomerase II. Mol. Biochem. Parasitol., 38, 141–150. [DOI] [PubMed] [Google Scholar]
- Wang Z., Morris,J.C., Drew,M.E. and Englund,P.T. (2000) Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem., 275, 40174–40179. [DOI] [PubMed] [Google Scholar]
- Wirtz E. and Clayton,C. (1995) Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science, 268, 1179–1183. [DOI] [PubMed] [Google Scholar]
- Wirtz E., Leal,S., Ochatt,C. and Cross,G.A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol., 99, 89–101. [DOI] [PubMed] [Google Scholar]
- Zamore P.D., Tuschl,T., Sharp,P.A. and Bartel,D.P. (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 101, 25–33. [DOI] [PubMed] [Google Scholar]