

Abstract
OmpT from Escherichia coli belongs to a family of highly homologous outer membrane proteases, known as omptins, which are implicated in the virulence of several pathogenic Gram-negative bacteria. Here we present the crystal structure of OmpT, which shows a 10-stranded antiparallel β-barrel that protrudes far from the lipid bilayer into the extracellular space. We identified a putative binding site for lipopolysaccharide, a molecule that is essential for OmpT activity. The proteolytic site is located in a groove at the extracellular top of the vase-shaped β-barrel. Based on the constellation of active site residues, we propose a novel proteolytic mechanism, involving a His–Asp dyad and an Asp–Asp couple that activate a putative nucleophilic water molecule. The active site is fully conserved within the omptin family. Therefore, the structure described here provides a sound basis for the design of drugs against omptin-mediated bacterial pathogenesis. Coordinates are in the Protein Data Bank (accession No. 1I78)
Keywords: His–Asp dyad/lipopolysaccharide/OmpT/omptin/protease
Introduction
Omptins are outer membrane proteases found in several Gram-negative bacteria and include OmpT of Escherichia coli (Sugimura and Nishihara, 1988), PgtE of Salmonella typhimurium (mature part 49% identical to OmpT) (Yu and Hong, 1986), Pla of Yersinia pestis (50% identical) (Sodeinde and Goguen, 1989), SopA of Shigella flexneri (60% identical) (Egile et al., 1997) and OmpP of E.coli (72% identical) (Kaufmann et al., 1994). Several studies have implicated the omptin family in the pathogenicity of these bacteria (Stathopoulos, 1998). The presence of the ompT gene in clinical isolates of E.coli has been associated with complicated urinary tract disease (Webb and Lundigran, 1996), a notion supported by the observation that OmpT cleaves protamine, a highly basic antimicrobial peptide that is excreted by epithelial cells of the urinary tract (Stumpe et al., 1998). Inactivation of the gene encoding Pla in Y.pestis, the causative agent of plague, increased the median lethal dose of the bacterium for mice by 106-fold (Sodeinde et al., 1992). The role of Pla in pathogenicity might be related to its ability to activate plasminogen, a feature shared with OmpT (Lundrigan and Webb, 1992). SopA from S.flexneri, the causative agent of bacillary dysentery, cleaves the endogenous autotransporter IcsA that has an essential role in the formation of actin tails in host cells, and therefore SopA might be involved in actin-based motility inside infected cells (Egile et al., 1997; Shere et al., 1997). Thus, the proteolytic activity of the omptins is probably involved in a variety of ways in the pathogenicity of these bacteria, ranging from bacterial defence and plasmin-mediated tissue infiltration to motility inside infected cells.
OmpT is biochemically the best characterized member of the omptins. It preferentially cleaves substrates between two consecutive basic amino acids (Dekker et al., 2001). The protease displays optimal activity at alkaline pH and it is dependent on lipopolysaccharide (LPS), showing no detectable enzymatic activity towards peptide substrates in the absence of LPS (Kramer et al., 2000b). Hydrolysis of a fluorogenic peptide substrate by OmpT was characterized by a specificity constant kcat/Km of 108 s–1 M–1, indicating a cleavage efficiency comparable with that of water-soluble proteases such as chymotrypsin (Kramer et al., 2000b). The enzyme does not contain any conserved active site sequence found in other known protease families. In addition, commonly used serine protease inhibitors have little or no effect on the activity of OmpT (Sugimura and Nishihara, 1988). However, because some serine protease inhibitors weakly affect OmpT activity, the omptins have been classified as novel serine proteases (family S18) (Rawlings and Barrett, 1994). Site-directed mutagenesis studies appeared to support this classification, since Ser99 and His212 have been found to be important for the activity of OmpT (Kramer et al., 2000a).
Here we describe the first structure of an integral outer membrane protease, OmpT from E.coli, at 2.6 Å resolution. The crystallized protein contained three mutations to prevent autoproteolytic degradation. Based on the structure, we identified a putative LPS-binding site and provide support for the omptins constituting a novel class of proteases.
Results and discussion
Structure determination
The OmpT structure was determined with the single-wavelength anomalous diffraction (SAD) phasing method using a seleno-methionine (Se-Met)-containing OmpT crystal, which diffracted to 3.1 Å resolution. The final structure was refined against data to 2.6 Å resolution using a crystal of OmpT containing natural methionines. The enzyme contained the mutations S99A, G216K and K217G, to abolish autoproteolytic activity. Additionally, crystals of a S99A mutant were obtained that diffracted to a lower resolution of 3.2 Å, yielding a map indicating an identical structure (unpublished results). OmpT was crystallized using 1% (w/v) octyl-β-d-glucopyranoside (OG), 30% (v/v) 2-methyl-2,4-pentanediol (MPD) and 0.3 M sodium citrate pH 5.5. It crystallized in the space group P3221 with unit cell dimensions a = b = 97.8 Å and c = 165.3 Å. Two OmpT molecules are present in the asymmetric unit that show a high degree of structural similarity, with a root mean square deviation (r.m.s.d.) of 0.50 Å for main-chain atoms. The refined model consists of 584 amino acid residues, four OG and 29 water molecules, and has a crystallographic R-factor of 23.7% and an Rfree of 28.0% for data in the 20–2.6 Å resolution range. Table I summarizes the statistics of the crystallographic data and the refinement. Coordinates and structure factors have been deposited in the Protein Data Bank with accession No. 1I78.
Table I. Summary of data and refinement statistics.
| Native | Se-Met | |
|---|---|---|
| Data set statistics |
|
|
| resolution limits (outer shell) (Å) | 20–2.6 (2.69–2.6) | 40–3.1 (3.21–3.1) |
| space group | P3221 | P3221 |
| unit cell parameters (Å, °) | a = 98.39 | a = 97.79 |
| b = 98.39 | b = 97.79 | |
| c = 165.70 | c = 165.86 | |
| α = 90, β = 90, γ = 120 | α = 90, β = 90, γ = 120 | |
| mosaicity (°) | 0.1 | 0.8 |
| oscillation range (°) | 0.2 | 0.5 |
| total oscillation for data set (°) | 25 | 100 |
| total no. of reflections (outer shell) | 43 471 (4352) | 107 336 (10 902) |
| no. of unique reflections (outer shell)a | 27 400 (2766) | 17 252 (1705) |
| redundancy | 1.59 (1.57) | 6.22 (6.39) |
| Rmerge (%) (outer shell)b | 7.4 (26.3) | 9.7 (39.5) |
| completeness (%) (outer shell) | 94.3 (97.7) | 99.8 (100) |
| I/σ (I) (outer shell) |
8.82 (2.54) |
15.48 (4.56) |
| |
||
| Refinement statistics | ||
| resolution range (Å) | 20–2.6 | |
| total no. of non-hydrogen atoms | 4734 | |
| no. of water molecules | 29 | |
| no. of β-OG molecules | 4 | |
| Rwork (%) | 23.7 | |
| Rfree (%) | 28.0 | |
| r.m.s.d. bond lengths (Å) | 0.007 | |
| r.m.s.d. bond angles (°) | 1.35 | |
| average B-factor (all protein atoms) (Å2) | 52.6 |
aFor the Se-Met data set, the Friedel mates have been counted separately.
bRmerge = Σ(|I – <I>|)/Σ(I).
Overall structure
The overall structure of OmpT consists of an exceptionally long 10-stranded vase-shaped antiparallel β-barrel of ∼70 Å in its longest dimension (Figure 1). The number, as well as the approximate position of the 10 β-strands, was predicted correctly by Kramer et al. (2000b). The β-strands have an average length of 23 residues, have a tilt angle of 37–45° with respect to the barrel axis and have a shear number of 12 (Figure 2). Like other outer membrane proteins (Weiss et al., 1990; Cowan et al., 1992; Kreusch et al., 1994; Schirmer et al., 1995; Ferguson et al., 1998; Locher et al., 1998; Pautsch and Schulz, 1998; Buchanan et al., 1999; Snijder et al., 1999; Vogt and Schulz, 1999; Koronakis et al., 2000), OmpT contains short turns at the periplasmic side of the barrel, long, more mobile loops (Figure 3) at the extracellular part and a hydrophobic band of 25 Å in height flanked by aromatic residues that determine the position of the molecule in the membrane. On the extracellular side, OmpT protrudes ∼40 Å from the lipid bilayer with the outermost loops located just above the rim of the LPS core region. Near the top of OmpT, the β-barrel has a circular diameter of ∼32 Å, whereas in the central part the molecular cross-section is elliptical with dimensions of ∼13 × ∼26 Å, determined using the Cα positions.

Fig. 1. Overall structure of OmpT. (A) Ribbon representation of OmpT. The extracellular space is located at the top of the figure, and the periplasmic space is at the bottom. Extracellular loops are labelled L1–L5. The position of the membrane bilayer is delineated by horizontal lines. Aromatic residues that are located at the boundary of the hydrophobic and hydrophilic area on the molecular surface are coloured yellow. The proposed catalytic residues are depicted in red, and the purple-coloured residues show the putative LPS-binding site. (B) Stereo representation of a modelled LPS molecule at the putative binding site. The orientation of the OmpT molecule is rotated 90° along the barrel axis, with respect to (A). The OmpT–LPS model was obtained by superimposing the putative LPS-binding site of OmpT onto the LPS-binding site of FhuA (Ferguson et al., 2000). LPS (from the FhuA structure) is shown by thin grey lines. The putative LPS-binding residues in purple are labelled. This figure, and Figures 5 and 7 were prepared using Bobscript (Esnouf, 1997) and Raster3D (Merritt and Bacon, 1997).

Fig. 2. Topology model of the OmpT β-barrel. Amino acid residues are given in one-letter code. Squares represent residues that are present in the β-strands. Side chains of amino acids that are shaded grey point to the outside of the barrel. Extracellular loops are labelled L1–L5 and periplasmic turns are labelled T1–T4. Every 25th residue is marked with the corresponding residue number. The grey area indicates the approximate position of the outer membrane.

Fig. 3. B-factors of the Cα atoms of the two OmpT molecules in the asymmetric unit, i.e. molecule A (solid line) and B (dashed line). β-strands are shown by bars that are black within the membrane-spanning region and grey outside this region. The positions of loops and turns are marked with L1–L5 and T1–T5, respectively.
Within the membrane region, the barrel is highly regular, hollow and positively charged on the inside (Figure 4). Above the membrane, the barrel is constricted and distorted in its hydrogen bonding pattern; seven side chains, i.e. Ser22, Thr73, Arg77, Gln228, Asn258, Lys259 and Lys260, form hydrogen bonds to main chain atoms in the β-barrel. This constricted area, consisting of a few layers of predominantly conserved aromatic residues, forms the floor of a negatively charged groove that harbours the active site (discussed below).
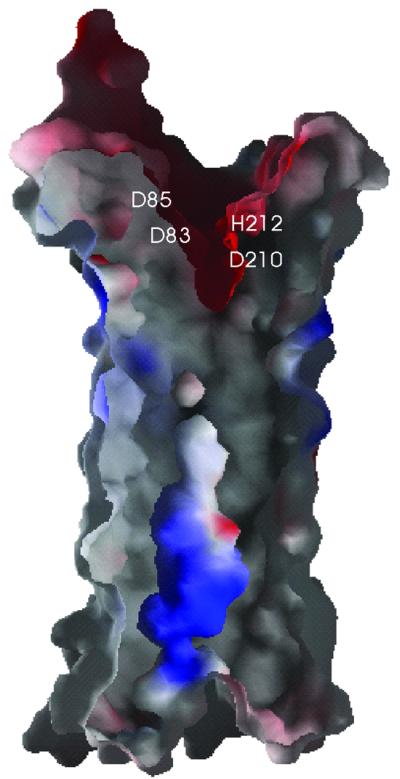
Fig. 4. Electrostatic surface potential of a cross-section of the β-barrel (orientation identical to Figure 1B). Negatively charged areas are shown in red, positively charged areas in blue. The positions of the proposed catalytic residues are labelled. This figure was produced by the program GRASP (Nicholls et al., 1991).
Putative LPS-binding site
In vitro, OmpT displays enzymatic activity only in the presence of LPS (Kramer et al., 2000b). It is as yet unclear how LPS contributes to OmpT activity, but preliminary experiments suggest that one LPS molecule per OmpT is sufficient for activity (unpublished results). Based on the structure of outer membrane protein FhuA from E.coli in complex with LPS, Ferguson et al. (1998, 2000) identified a structural motif for LPS binding, consisting of four basic amino acids, which is conserved among pro- and eukaryotic LPS-binding proteins. Three of the four basic residues are found in a similar constellation in OmpT (Arg138, Arg175 and Lys226) (Figure 1B). The fourth conserved amino acid (Arg382 in FhuA) is lacking, possibly due to strong bending of OmpT at this position. Two additional amino acids that bind LPS in FhuA (Glu304 and Phe302) also have counterparts in OmpT (Glu136 and Tyr134). The five similar residues in FhuA and OmpT are at the same height in the barrel and have an r.m.s.d. of 1.1 Å for all atoms. Based on these similarities, we propose that the above-mentioned residues constitute an LPS-binding site in OmpT. A model of OmpT with an LPS molecule at the proposed binding site is shown in Figure 1B. The structure of OmpT enables us to test, using mutagenesis studies, whether these residues are indeed responsible for LPS binding and if they contribute to OmpT activity.
Active site
OmpT exerts its proteolytic activity in the extracellular medium (Kramer et al., 2000b). The extracellular part of the molecule contains a large negatively charged groove, which is consistent with the substrate specificity of OmpT for two consecutive basic residues (Figure 4). All 18 residues in this proposed active site groove are fully conserved within the omptin family (Figures 5 and 6). Substitution of serines, histidines and acidic residues by alanines have resulted in ∼10-fold reduced activity for Glu27, Asp97, Asp208 or His101, ∼500-fold reduced activity for Ser99, and ∼10 000-fold reduced activity for Asp83, Asp85, Asp210 or His212 (Kramer et al., 2000a; R.A.Kramer, L.Vandeputte-Rutten, G.J.De Roon, P.Gros, N.Dekker and M.R.Egmond, submitted).

Fig. 5. Stereo view of a space-filling representation of the active site groove of OmpT, viewed down the β-barrel axis. All conserved residues in the active site are distinctly coloured and labelled. Conserved residues outside the active site are depicted in dark grey and non-conserved residues in light grey. Conserved serines and threonines in the active site are coloured purple, acidic residues red, histidines blue, a tyrosine brown and all hydrophobic residues green. Residues S99, H101 and D210 are labelled with an asterisk, since they are hidden behind other residues. The three mutated residues S99A, G216K and K217G are labelled in black.

Fig. 6. Sequence alignment of OmpT with other members of the omptin family, i.e. OmpP of E.coli, SopA of S.flexneri, PgtE of S.typhimurium and Pla of Y.pestis. All fully conserved residues are shaded grey. The four proposed catalytic residues are depicted with a black background. The approximate position of the 10 β-strands are indicated by arrows and the extracellular loops are labelled L1–L5.
The structure contradicts the classification of OmpT as a serine protease, since the previously proposed catalytic residues Ser99 (alanine in the crystal structure) and His212 are too far apart, with a distance of 9 Å between Cβ of Ser99 and Nε2 of His212. Furthermore, the single histidine located close to Ser99, His101, is only moderately important for catalysis. The four residues most important for activity form pairs, Asp83–Asp85 and His212–Asp210, which are located on opposite sides of the active site groove (Figure 7). The distance between the two couples is ∼5 Å, determined by the closest distance between Asp83 and His212. The Asp83–Asp85 couple resembles the catalytic site of aspartic proteases. On the other hand, the His212–Asp210 couple resembles the His–Asp pair found in Ser–His–Asp triads of serine proteases. The observation that OmpT is active at high pH with a pKa of 6.2 (Kramer et al., 2000b) suggests the involvement of an active site histidine. However, a putative nucleophilic residue is not present near the His212–Asp210 couple. We propose a mechanism in which a water molecule (invisible in the electron density map at the current resolution), positioned between Asp83 and His212, is activated by the His212–Asp210 dyad and subsequently performs the nucleophilic attack on the scissile peptide bond. A schematic model of the peptide substrate in the active site is given in Figure 7B.

Fig. 7. Stereo view of the proposed catalytic site. (A) The final 2Fo – Fc electron density map, at 2.6 Å resolution and contoured at 1σ (orientation identical to Figure 1B). Residues are shown as sticks. (B) The catalytic site viewed from the top down the β-barrel axis. Active site residues are depicted by ball-and-sticks. Proposed catalytic residues are coloured dark grey and the other residues that are probably involved in substrate binding are coloured light grey. The proposed proteolytic mechanism, in which the His212–Asp210 couple abstract a proton from a water molecule which then attacks the main chain carbon, is represented schematically.
OmpT is highly specific for lysine and arginine at position P1 (nomenclature according to Schechter and Berger, 1967) and is less specific for residues at P1′, with a decreasing preference for lysine, isoleucine, histidine and arginine (Dekker et al., 2001). Furthermore, OmpT shows a high specificity for small hydrophobic residues (isoleucine, valine and alanine) at P2′, and has a broad tolerance for residues at the P2 position (Dekker et al., 2001). Given the high specificities for P1 and P2′, we expect well-defined S1 and S2′ subsites. A deep negative pocket containing Glu27 and Asp208 probably forms the S1 subsite, which determines the specificity for lysine and argine at P1 (Figure 5). Approximately 7 Å away from this pocket, the groove is shallow and hydrophobic, with Met81 and Ile170 located at the bottom of the groove. This region may well form the S2′ subsite, explaining the high specificity for small hydrophobic residues at P2′. Interactions between the substrate and the putative S1 and S2′ subsites would position the scissile peptide bond between the Asp83–Asp85 and His212–Asp210 couples, in agreement with our proposed catalytic mechanism.
The current model implies a novel proteolytic mechanism, consistent with the observation that commonly used protease inhibitors do not or weakly affect the activity of OmpT. Solving the X-ray structure of OmpT in complex with a substrate analogue is needed to confirm our active site model and to provide more details on the catalytic mechanism. Since the active site is fully conserved among the omptins, the structure of OmpT may serve as a template to develop new types of antimicrobial compounds against omptin-mediated pathogenicity of these Gram-negative bacteria.
Materials and methods
Protein expression and purification
OmpT, containing the mutations S99A, G216K and K217G in order to abolish autoproteolysis (Kramer et al., 2000b), was overexpressed without its signal sequence (of 20 residues) in E.coli and refolded as described previously (Kramer et al., 2000b). For expression of Se-Met OmpT, E.coli strain B834(DE3) (Met-auxotroph, Novagen) containing the plasmid that encodes OmpT (S99A, G216K, K217G) was grown in new minimal medium (Budisa et al., 1995) supplemented with 0.3 mM l-Se-Met (Acros). Refolded native OmpT and Se-Met OmpT (∼200 mg) were loaded onto a 100 ml Fast Flow S-Sepharose column (Amersham Pharmacia) equilibrated with buffer A [10 mM Zwittergent 3–12 (Fluka, Switzerland), 20 mM sodium acetate pH 4.0]. The column was washed with buffer A and proteins were eluted with a linear gradient of NaCl to 1 M in buffer A. The native protein and the Se-Met derivative were concentrated to 25 mg/ml in buffer B (1% OG, 5 mM sodium acetate pH 4.0) using a second S-Sepharose column and centricon concentrators (Amicon). After dialysis, samples used for crystallization consisted of 20 mg/ml OmpT in buffer B containing 2.5 mM potassium chloride.
Crystallization
Crystallization conditions for native OmpT were screened using the hanging drop vapour diffusion method at 4 and 20°C. Droplets containing equal amounts (1 µl) of protein and mother liquor were equilibrated against 0.5 ml of reservoir solution. Sparse-matrix screens for initial trials were performed using Hampton research screen kits (Hampton Research, CA). After 6 months, one small crystal with dimensions of 0.08 × 0.05 × 0.05 mm appeared at 4°C in 30% (v/v) MPD (Fluka), 0.2 M ammonium acetate, 0.1 M sodium citrate pH 5.6. A narrower screening around this crystallization condition was performed: 28% (v/v) MPD, 0.5 M NaCl and 0.1 M sodium citrate pH 5.5 yielded one small crystal per drop within a month. Se-Met OmpT was produced in order to obtain phases for structure determination. Se-Met OmpT appeared to crystallize faster and yielded larger crystals than the native protein. After 2 weeks, crystals occurred in 1% OG, 30% (v/v) MPD and 0.3 M sodium citrate pH 5.5. These crystals grew to maximum crystal dimensions of ∼0.3 × 0.2 × 0.2 mm in 3 months.
X-ray diffraction analysis
Crystals were harvested from the droplets with cryo-loops and directly frozen into liquid nitrogen. X-ray data were collected at 100 K on a CCD detector at the ID-14 EH4 beamline at the European Synchrotron Radiation Facility (ESRF) in Grenoble. Native data were collected to 2.6 Å, using an oscillation range of 0.2°. The crystal belongs to the trigonal space group P3221, with unit cell parameters a = b = 98.4 Å, c = 165.7 Å, α = β = 90° and γ = 120°. Crystals have a solvent plus detergent content of 63% (v/v) and two OmpT molecules in the asymmetric unit. A data set of a Se-Met OmpT crystal was collected at λpeak = 0.9790 Å to a resolution of 3.1 Å. Data were indexed using DENZO merged with SCALEPACK (Otwinowski and Minor, 1997) and processed further using truncate from the CCP4 suite (CCP4, 1994). A summary of the data collection and final processing statistics is provided in Table I.
Structure determination and refinement
Using the peak data set of Se-Met OmpT, all 10 selenium sites were found by direct methods with the DREAR/SnB package (Weeks and Miller, 1999). Phases subsequently were calculated using MLPHARE (Otwinowski, 1991) to 3.7 Å resolution. This yielded an Rcullis (ano) of 0.78 [Rcullis (ano) = Σ(|ΔPHobs – ΔPHcalc|/Σ|ΔPHobs| with ΔPH = FPH(+) – FPH(–)]. With density modification in CNS (Brünger et al., 1998), phases were extended to 3.1 Å resolution. The resulting electron density map allowed the placement of 80% of the residues using the program O (Jones et al., 1991). The native data set was used for refinement from 20 to 2.6 Å resolution. After a rigid body refinement, iterative model refinement was performed by model building in O, followed by simulated annealing and restrained individual B-factor refinement in CNS. The final model consists of all residues of OmpT in molecule A, residues 11–297 in molecule B, four OG molecules and 29 water molecules. For the side chains of residues Glu3, Thr4, Glu154, Glu167, Arg168, Lys216 and Tyr266 of molecule A and Glu33, Glu154, Lys216 and Tyr266 of molecule B, no electron density was observed. Therefore, these side chains were omitted from the model. The two monomers in the asymmetric unit pack in a perpendicular orientation to each other and interact through their hydrophobic transmembrane regions. Intermolecular protein–protein interactions in the crystal are predominantly of a hydrophobic nature, with only one unique hydrogen bond (involving Arg100 in molecule A and the main chain oxygen of Ile160 in molecule B) and a salt bridge (involving Asp10 in molecule A and Lys277 in molecule B). In the Ramachandran plot, 85.4% of the residues are in most favoured regions, with 13.2% in additionally allowed regions, 1% in the generously allowed regions and one residue (Glu190) in a disallowed region. Nε of Arg188 is hydrogen bonded to the main chain oxygen of Glu190, generating a type II′ β-turn, explaining the perturbed ϕ,φ-combination of Glu190. The overall temperature factor of the total structure is 53.2 Å2, with the highest B-factors in the extracellular loops (Figure 3).
Acknowledgments
Acknowledgements
We wish to thank Drs R.B.G.Ravelli, S.McSweeney and M.J.van Raaij for help with data collection at beamline ID14-EH4 at the ESRF in Grenoble. We also thank Dr D.A.A.Vandeputte for carefully reading the manuscript. This research has been supported financially by the council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW).
References
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Buchanan S.K., Smith,B.S., Venkatramani,L., Xia,D., Esser,L., Palnitkar,M., Chakraborty,R., van der Helm,D. and Deisenhofer,J. (1999) Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nature Struct. Biol., 6, 56–63. [DOI] [PubMed] [Google Scholar]
- Budisa N., Steipe,B., Demange,P., Eckerskorn,C., Kellermann,J. and Huber,R. (1995) High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur. J. Biochem., 230, 788–796. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cowan S.W., Schirmer,T., Rummel,G., Steiert,M., Ghosh,R., Pauptit, R.A., Jansonius,J.N. and Rosenbusch,J.P. (1992) Crystal structures explain functional properties of two E.coli porins. Nature, 358, 727–733. [DOI] [PubMed] [Google Scholar]
- Dekker N., Cox,R.C., Kramer,R.A. and Egmond,M.R. (2001) Substrate specificity of the integral membrane protease OmpT determined by spatially addressed peptide libraries. Biochemistry, 40, 1694–1701. [DOI] [PubMed] [Google Scholar]
- Egile C., d’Hauteville,H., Parsot,C. and Sansonetti,P.J. (1997) SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol. Microbiol., 23, 1063–1073. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 132–134, 112–113. [DOI] [PubMed] [Google Scholar]
- Ferguson A.D., Hofmann,E., Coulton,J.W., Diederichs,K. and Welte,W. (1998) Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science, 282, 2215–2220. [DOI] [PubMed] [Google Scholar]
- Ferguson A.D., Welte,W., Hofmann,E., Lindner,B., Holst,O., Coulton,J.W. and Diederichs,K. (2000) A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure Fold. Des., 8, 585–592. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kaufmann A., Stierhof,Y.D. and Henning,U. (1994) New outer membrane-associated protease of Escherichia coli K-12. J. Bacteriol., 176, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V., Sharff,A., Koronakis,E., Luisi,B. and Hughes,C. (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature, 405, 914–919. [DOI] [PubMed] [Google Scholar]
- Kramer R.A., Dekker,N. and Egmond,M.R. (2000a) Identification of active site serine and histidine residues in Escherichia coli outer membrane protease OmpT. FEBS Lett., 468, 220–224. [DOI] [PubMed] [Google Scholar]
- Kramer R.A., Zandwijken,D., Egmond,M.R. and Dekker,N. (2000b) In vitro folding, purification and characterization of Escherichia coli outer membrane protease ompT. Eur. J. Biochem., 267, 885–893. [DOI] [PubMed] [Google Scholar]
- Kreusch A., Neubuser,A., Schiltz,E., Weckesser,J. and Schulz,G.E. (1994) Structure of the membrane channel porin from Rhodopseudomonas blastica at 2.0 Å resolution. Protein Sci., 3, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K.P., Rees,B., Koebnik,R., Mitschler,A., Moulinier,L., Rosenbusch,J.P. and Moras,D. (1998) Transmembrane signaling across the ligand-gated FhuA receptor: crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell, 95, 771–778. [DOI] [PubMed] [Google Scholar]
- Lundrigan M.D. and Webb,R.M. (1992) Prevalence of ompT among Escherichia coli isolates of human origin. FEMS Microbiol. Lett., 76, 51–56. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. (1991) Maximum likelihood refinement of heavy atom parameters in isomorphous replacement and anomalous scattering. Proceedings of the CCP4 Study Weekend, 25–26 January, 1991.
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pautsch A. and Schulz,G.E. (1998) Structure of the outer membrane protein A transmembrane domain. Nature Struct. Biol., 5, 1013–1017. [DOI] [PubMed] [Google Scholar]
- Rawlings N.D. and Barrett,A.J. (1994) Families of serine peptidases. Methods Enzymol., 244, 19–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I. and Berger,A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun., 27, 157–162. [DOI] [PubMed] [Google Scholar]
- Schirmer T., Keller,T.A., Wang,Y.F. and Rosenbusch,J.P. (1995) Structural basis for sugar translocation through maltoporin channels at 3.1 Å resolution. Science, 267, 512–514. [DOI] [PubMed] [Google Scholar]
- Shere K.D., Sallustio,S., Manessis,A., D’Aversa,T.G. and Goldberg,M.B. (1997) Disruption of IcsP, the major Shigella protease that cleaves IcsA, accelerates actin-based motility. Mol. Microbiol., 25, 451–462. [DOI] [PubMed] [Google Scholar]
- Snijder H.J., Ubarretxena-Belandia,I., Blaauw,M., Kalk,K.H., Verheij,H.M., Egmond,M.R., Dekker,N. and Dijkstra,B.W. (1999) Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature, 401, 717–721. [DOI] [PubMed] [Google Scholar]
- Sodeinde O.A. and Goguen,J.D. (1989) Nucleotide sequence of the plasminogen activator gene of Yersinia pestis: relationship to ompT of Escherichia coli and gene E of Salmonella typhimurium. Infect. Immun., 57, 1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde O.A., Subrahmanyam,Y.V., Stark,K., Quan,T., Bao,Y. and Goguen,J.D. (1992) A surface protease and the invasive character of plague. Science, 258, 1004–1007. [DOI] [PubMed] [Google Scholar]
- Stathopoulos C. (1998) Structural features, physiological roles and biotechnological applications of the membrane proteases of the OmpT bacterial endopeptidase family: a micro-review. Membr. Cell Biol., 12, 1–8. [PubMed] [Google Scholar]
- Stumpe S., Schmid,R., Stephens,D.L., Georgiou,G. and Bakker,E.P. (1998) Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J. Bacteriol., 180, 4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura K. and Nishihara,T. (1988) Purification, characterization and primary structure of Escherichia coli protease VII with specificity for paired basic residues: identity of protease VII and OmpT. J. Bacteriol., 170, 5625–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J. and Schulz,G.E. (1999) The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure Fold. Des., 7, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Webb R.M. and Lundigran,M.D. (1996) OmpT in Escherichia coli correlates with severity of disease in urinary tract infections. Med. Microbiol. Lett., 5, 8–14. [Google Scholar]
- Weeks C.M. and Miller,R. (1999) Optimizing shake-and-bake for proteins. Acta Crystallogr. D, 55, 492–500. [DOI] [PubMed] [Google Scholar]
- Weiss M.S., Wacker,T., Weckesser,J., Welte,W. and Schulz,G.E. (1990) The three-dimensional structure of porin from Rhodobacter capsulatus at 3 Å resolution. FEBS Lett., 267, 268–272. [DOI] [PubMed] [Google Scholar]
- Yu G.Q. and Hong,J.S. (1986) Identification and nucleotide sequence of the activator gene of the externally induced phosphoglycerate transport system of Salmonella typhimurium. Gene, 45, 51–57. [DOI] [PubMed] [Google Scholar]