

Abstract
The mitochondrial aspartate/glutamate carrier catalyzes an important step in both the urea cycle and the aspartate/malate NADH shuttle. Citrin and aralar1 are homologous proteins belonging to the mitochondrial carrier family with EF-hand Ca2+-binding motifs in their N-terminal domains. Both proteins and their C-terminal domains were overexpressed in Escherichia coli, reconstituted into liposomes and shown to catalyze the electrogenic exchange of aspartate for glutamate and a H+. Overexpression of the carriers in transfected human cells increased the activity of the malate/aspartate NADH shuttle. These results demonstrate that citrin and aralar1 are isoforms of the hitherto unidentified aspartate/glutamate carrier and explain why mutations in citrin cause type II citrullinemia in humans. The activity of citrin and aralar1 as aspartate/glutamate exchangers was stimulated by Ca2+ on the external side of the inner mitochondrial membrane, where the Ca2+-binding domains of these proteins are localized. These results show that the aspartate/glutamate carrier is regulated by Ca2+ through a mechanism independent of Ca2+ entry into mitochondria, and suggest a novel mechanism of Ca2+ regulation of the aspartate/malate shuttle.
Keywords: aralar1/aspartate/glutamate carrier/calcium/citrin/mitochondria
Introduction
Mutations in the human gene SLC25A13 are responsible for adult-onset type II citrullinemia (CTLN2: 603471) (Kobayashi et al., 1999; Yasuda et al., 2000). The gene encodes citrin, a Ca2+-binding mitochondrial solute carrier protein (Kobayashi et al., 1999, 2000). The most characteristic feature of CTLN2 is the liver-specific deficiency of argininosuccinate synthetase (ASS), which catalyzes the formation of argininosuccinate from citrulline and aspartate (Saheki et al., 1987). This deficiency is not accompanied by abnormalities in either the hepatic ASS mRNA or its gene (Kobayashi et al., 1986, 1993), whereas in classical citrullinemia (CTLN1: 215700) the gene for ASS is mutated (Kobayashi et al., 1995), leading to ASS deficiency in all tissues (Saheki et al., 1987).
The overall structure of citrin is very similar to that of aralar1 (del Arco and Satrústegui, 1998), encoded by the gene SLC25A12. Both proteins have four EF-hand Ca2+-binding motifs in their N-terminal domains, the characteristic features of the mitochondrial carrier family in their C-terminal domains, and both proteins localize to mitochondria and bind Ca2+ (del Arco and Satrústegui, 1998; del Arco et al., 2000; Kobayashi et al., 2000; Iijima et al., 2001). Citrin is expressed in many tissues but most abundantly in liver, while aralar1 is found mainly in heart, skeletal muscle and brain (del Arco and Satrústegui, 1998; Kobayashi et al., 1999; del Arco et al., 2000; Iijima et al., 2001). The existence of a subfamily of Ca2+-binding mitochondrial carriers with representatives in different species (del Arco et al., 2000) opens up the possibility that these proteins might be involved in Ca2+-regulated transport and/or signaling to mitochondria.
The functions of nearly 20 members of the mitochondrial carrier family have been identified to date (for references see Dolce et al., 2001). Other transport activities observed in mitochondria have yet to be associated with specific proteins, an example being the aspartate/glutamate carrier (AGC), which catalyzes a 1:1 exchange of aspartate for glutamate and plays an important role in the malate/aspartate shuttle, urea synthesis and gluconeogenesis from lactate (Williamson, 1976; LaNoue and Schoolwerth, 1979). Since aspartate is transported as the anion and glutamate is co-transported with a proton, the aspartate/glutamate antiport is electrogenic (LaNoue et al., 1974) and, therefore, in energized mitochondria, the physiological direction of the exchange (efflux of aspartate and entry of glutamate) is favored. AGC also transports cysteinesulfinate in exchange for either aspartate or glutamate (Palmieri et al., 1979).
Here we have studied the function of citrin and aralar1. These proteins were overexpressed in bacteria, reconstituted into phospholipid vesicles and identified from their transport characteristics as isoforms of the AGC. Citrin and aralar1 were also overexpressed in the human cell line, HEK-293T, where they increased the activity of the malate/aspartate shuttle. In HEK-293T cells, the AGC was stimulated by Ca2+ on the external face of the inner mitochondrial membrane, and this stimulation was increased by overexpression of citrin and aralar1. The function of citrin as the AGC, a key component of the malate/aspartate shuttle, may explain both the inhibition of the ASS reaction by a defective supply of aspartate from mitochondria and many of the characteristic symptoms of CTLN2.
Results
Bacterial expression of citrin and aralar1
Citrin, aralar1 and their C-terminal domains (corresponding to the last 391 and 393 amino acids of citrin and aralar1, respectively, plus a few extra amino acids due to the cloning procedure) were expressed at high levels in Escherichia coli (Figure 1A, lane 2 for citrin). They accumulated as inclusion bodies, and were purified by centrifugation and washing (Figure 1A, lane 3, and C) with a yield of 40–60 mg/l bacterial culture. The proteins were not detected either in bacteria harvested immediately before induction of expression (Figure 1A, lane 1 for citrin) or in control (with only vector) cells harvested after induction (not shown). The identity of the expressed proteins was confirmed by N-terminal sequencing, and by reaction with specific antisera (Figure 1B, D and E).
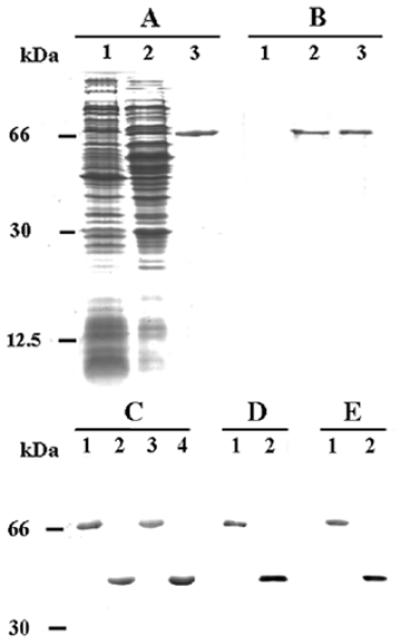
Fig. 1. Bacterial overexpression and purification of citrin, aralar1 and their C-terminal domains. Proteins were separated by SDS–PAGE and either stained with Coomassie Blue dye (A and C) or transferred to nitrocellulose membrane and detected with specific polyclonal antibodies (B, D and E). (A and B) Cells of E.coli CO214(DE3) overexpressing citrin. Samples were taken at induction of expression (lane 1) and 12 h later (lane 2). The same number of bacteria was analyzed in each sample. Lanes 3, 6 µg (A) and 1 µg (B) of purified citrin obtained from bacteria in lane 2 of (A). (C) A 5 µg aliquot of purified citrin (lane 1), its C-terminal domain (lane 2), aralar1 (lane 3) and its C-terminal domain (lane 4). (D) Immunoreaction of 0.2 µg of purified citrin and its C-terminal domain (lanes 1 and 2, respectively) with antiserum to the C-terminal domain. (E) Immunoreaction of 0.2 µg of purified aralar1 and its C-terminal domain (lanes 1 and 2, respectively) with antiserum against the C-terminal domain.
Functional characterization of recombinant citrin and aralar1 in proteoliposomes
Citrin and aralar1 were reconstituted into liposomes, and their transport activities for substrates of known mitochondrial transport systems were tested in homo-exchange (same substrate inside and outside) experiments. Using external and internal substrate concentrations of 1 and 10 mM, respectively, both reconstituted proteins catalyzed an efficient [14C]aspartate/aspartate exchange, and none of the other substrates was transported (phosphate, pyruvate, ADP, ATP, malonate, succinate, malate, oxoglutarate, ketoisocaproate, citrate, carnitine, ornithine, lysine, arginine, histidine, glutathione, choline, spermine, proline and threonine). The [14C]aspartate/aspartate exchange was not observed with recombinant citrin or aralar1 that had been boiled before incorporation into liposomes.
The uptake of 50 µM [14C]aspartate into proteoliposomes reconstituted with either citrin or aralar1 and containing 20 mM aspartate followed first-order kinetics (rate constants 0.12 and 0.04 min–1 and initial rates of 80.3 and 24.7 µmol/min/g of protein, for citrin and aralar1, respectively) (data not shown). Uptake of aspartate required internal substrate, indicating that citrin and aralar1 catalyze only the exchange reaction and not unidirectional aspartate transport. Possible unidirectional transport could be studied more conveniently by efflux measurements of reconstituted proteoliposomes (Palmieri et al., 1995). [14C]aspartate was introduced into the intraliposomal volume. No efflux was observed in the absence of external substrate with both proteins, but an extensive efflux of radioactivity occurred upon addition of external aspartate or glutamate (both 10 mM), and was totally inhibited by pyridoxal 5′-phosphate and bathophenanthroline (data not shown).
The substrate specificity of reconstituted citrin and aralar1 was examined in detail by measuring the uptake of [14C]aspartate and [14C]glutamate into proteoliposomes that had been pre-loaded with various potential substrates (Table I). With both proteins, external l-aspartate and l-glutamate exchanged significantly only in the presence of internal l-aspartate or l-glutamate (but not the d-isomers), or l-cysteinesulfinate, similarly to the natural AGC (LaNoue et al., 1974; Palmieri et al., 1979). Little or no activity was found with any of the other compounds listed in Table I or with taurine, tartrate, cysteine, fumarate, succinate, l-malate, malonate, phosphate, oxoglutarate, citrate, ATP, sulfate, oxaloacetate, pyruv ate, phosphoenolpyruvate, l-carnitine, l-ornithine or l-citrulline. The substrate specificities of the recombinant C-terminal domains of citrin and aralar1 are confined likewise to l-aspartate, l-glutamate and l-cysteine sulfinate (Table I).
Table I. Dependence on internal substrate of the transport properties of proteoliposomes reconstituted with recombinant citrin, aralar1 or their C-terminal domains (CTDs).
| Internal substrate | Substrate transport (µmol/min/g protein) |
|||||
|---|---|---|---|---|---|---|
| Citrin |
Citrin CTD |
Aralar1 |
Aralar1 CTD |
|||
| [14C]Aspartate | [14C]Glutamate | [14C]Aspartate | [14C]Aspartate | [14C]Glutamate | [14C]Aspartate | |
| None (Cl– present) | 1.4 | 1.3 | 1.6 | 0.3 | 0.3 | 0.3 |
| l-Aspartate | 92.8 | 72.5 | 178.4 | 25.2 | 17.1 | 40.5 |
| l-Glutamate | 55.3 | 90.3 | 118.1 | 16.1 | 21.8 | 23.7 |
| l-Cysteinesulfinate | 81.2 | 65.9 | 133.8 | 17.7 | 14.2 | 27.6 |
| l-Aminoadipate | 4.6 | 6.4 | 6.7 | 1.6 | 1.2 | 2.4 |
| d-Aspartate | 8.1 | 8.9 | 13.4 | 1.5 | 1.9 | 3.4 |
| d-Glutamate | 5.9 | 9.7 | 11.3 | 1.6 | 2.3 | 4.1 |
| l-Asparagine | 10.1 | 10.5 | 18.6 | 3.1 | 2.5 | 5.7 |
| l-Glutamine | 1.5 | 4.7 | 2.3 | 0.7 | 1.3 | 1.3 |
| l-Cysteate | 12.9 | 11.2 | 25.5 | 3.1 | 3.2 | 5.5 |
| l-Homocysteinesulfinate | 1.1 | 2.1 | 2.7 | 0.2 | 0.1 | 0.3 |
| l-Homocysteate | 1.8 | 2.0 | 4.5 | 0.1 | 0.3 | 0.9 |
Proteoliposomes were pre-loaded internally with various substrates (concentration, 20 mM). Transport was started by the external addition of 50 µM [14C]aspartate or 200 µM [14C]glutamate and terminated after 30 s (citrin and its CTD) or 1 min (aralar1 and its CTD). Similar results were obtained in at least three independent experiments for each carrier investigated.
The [14C]aspartate/aspartate exchange reaction catalyzed by citrin and aralar1, as well as by their C-terminal domains, was inhibited strongly (94–100% inhibition) by 10 mM pyridoxal 5′-phosphate, 10 mM bathophenanthroline, 10 µM mercurials (HgCl2, mersalyl and p-chloromercuriphenylsulfonate) and 1 mM diethyl pyro carbonate, as is the natural AGC (LaNoue and Schoolwerth, 1979; Bisaccia et al., 1992; Dierks et al., 1992). They were also inhibited (53–62%) by 1 mM N-ethylmaleimide, but not by known inhibitors of mitochondrial carriers [i.e. butylmalonate, phenylsuccinate and 1,2,3-benzenetricarboxylate (2 mM each), 20 µM carboxyatractyloside and 0.1 mM α-cyano-4-hydroxycinnamate].
As the AGC-catalyzed aspartate/glutamate antiport is electrogenic (LaNoue et al., 1974), the influence of membrane potential was investigated on the aspartate/glutamate exchange mediated by the recombinant proteins. A K+ diffusion potential was generated across the proteoliposomal membranes with valinomycin/KCl (calculated value ∼100 mV, positive inside). With citrin, aralar1 and their C-terminal domains, the rates of the [14C]aspartateout/glutamatein and [14C]glutamateout/aspartatein exchanges were stimulated and decreased, respectively (Table II). In the absence of a membrane potential or when homo-exchanges were measured, no effect was observed. Therefore, aralar1 and citrin catalyze an electrogenic exchange of aspartate for glutamate independently of the location of each substrate. The same phenomenon is associated with their C-terminal domains, and therefore these domains are responsible for electrogenic transport.
Table II. Influence of the membrane potential on the activity of reconstituted citrin, aralar1 and their C-terminal domains (CTDs).
| Uptake of | Internal substrate | K+in/K+out (mM/mM) | Exchange activity (µmol/min/g protein) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Citrin |
Citrin CTD |
Aralar1 |
Aralar1 CTD |
|||||||
| –valin | +valin | –valin | +valin | –valin | +valin | –valin | +valin | |||
| [14C]Aspartate | aspartate | 1/1 | 88 | 92 | 205 | 212 | 20 | 22 | 40 | 40 |
| aspartate | 1/50 | 81 | 84 | 208 | 203 | 24 | 21 | 35 | 39 | |
| glutamate | 1/1 | 50 | 52 | 132 | 128 | 16 | 15 | 23 | 24 | |
| glutamate | 1/50 | 56 | 173 | 130 | 386 | 20 | 47 | 22 | 51 | |
| [14C]Glutamate | glutamate | 1/1 | 74 | 70 | 183 | 190 | 26 | 23 | 47 | 43 |
| glutamate | 1/50 | 71 | 68 | 178 | 181 | 25 | 22 | 50 | 42 | |
| aspartate | 1/1 | 55 | 59 | 148 | 144 | 15 | 17 | 36 | 33 | |
| aspartate | 1/50 | 52 | 28 | 140 | 71 | 13 | 8 | 30 | 21 | |
The exchange was started by the addition of 0.05 mM [14C]aspartate or 0.2 mM [14C]glutamate to proteoliposomes which contained 20 mM of the indicated internal substrate. K+in was included as KCl in the reconstitution mixture, whereas K+out was added as KCl together with the labeled substrate. The differences in osmolarity were compensated for by the addition of appropriate concentrations of sucrose in the opposite compartment. Valinomycin (1.5 µg/mg phospholipid) was added in 10 µl ethanol/ml of proteoliposomes (+valin). In the samples without valinomycin (–valin) the solvent alone was added. The exchange reactions were stopped after 30 s for citrin and its CTD and 1 min for aralar1 and its CTD. The data are from representative experiments. Similar results were obtained in at least three independent experiments for each carrier investigated.
Kinetic characteristics
The influence of the proton motive force developed by the [aspartate–]/[glutamate– + H+] exchange on the kinetic parameters of citrin was investigated by analyzing the rate of uptake of [14C]aspartate or [14C]glutamate into proteoliposomes at various external substrate concentrations and constant internal aspartate concentration (20 mM) with and without valinomycin/K+ and nigericin (Figure 2). The combined effect of these two ionophores provides de-energized conditions in the presence of K+ by the collapse of both the ΔΨ and the ΔpH components of the proton motive force generated by aspartate/glutamate exchange. Under both conditions, the half-saturation constants (transport affinities or Km values) of [14C]aspartate and [14C]glutamate for citrin were the same (Figure 2), whereas the maximum transport rates (Vmax) of the [14C]aspartate/aspartate and [14C]glutamate/aspartate exchanges were different in the absence of ionophores (Figure 2A), but identical in their presence (Figure 2B). Without ionophores, the Vmax of the glutamate/aspartate exchange was lower than that of the aspartate/aspartate exchange by 28–40% for four experiments. Thus, from a kinetic point of view, the effect of membrane potential on citrin transport activity depends on an electrogenic modulation of the transport rate constant of the negatively charged carrier–aspartate complex, and is not due to a change in the substrate affinity of the carrier. The kinetic constants of citrin, aralar1 and their C-terminal domains were compared under de-energized conditions for both homo- and hetero-exchange reactions (Table III). For all four recombinant proteins, which were oriented unidirectionally in the liposomal membrane (L.Palmieri and F.M.Lasorsa, unpublished data), the half-saturation constants (Km) for external aspartate and glutamate were ∼50 µM and 0.21 mM, respectively, values that are essentially identical to those determined with natural AGC (Dierks and Krämer, 1988; Dierks et al., 1988). Moreover, the maximal activities (Vmax) of all homo- and hetero-exchanges were the same for each carrier, and so, in the absence of energy, citrin and aralar1 mediate symmetrical exchange of aspartate and glutamate in both directions, as does the natural AGC (Dierks et al., 1988). The standard error of the Vmax values was rather high when different experiments were compared, probably due to variation in the ratio of active and inactive carrier molecules. Nevertheless, in any individual experiment, the Vmax values of the four modes of aspartate/glutamate exchange were very close for each carrier protein under de-energized conditions. It is also notable that the Vmax values of the C-terminal domains of citrin and aralar1 were higher than those of the corresponding intact proteins, whereas the turnover numbers were very similar. Therefore, the C-terminal domains account for the entire transport activity of citrin and aralar1. One distinctive difference between citrin (and its C-terminal domain) and aralar1 (and its C-terminal domain) is that both activity and turnover number are about four times greater for the former.

Fig. 2. Lineweaver–Burk plots of the [14C]aspartate/aspartate and [14C]glutamate/aspartate exchanges in proteoliposomes reconstituted with citrin. Radioactive aspartate (squares) or glutamate (circles) was added at the concentrations indicated to proteoliposomes containing 20 mM aspartate. Valinomycin (1 µg/mg of phospholipids) and nigericin (50 ng/mg of phospholipids) in ethanol (10 µl/ml of liposomes) (B) or ethanol alone (A) were present in the reaction mixture. All data were determined in one experiment with the same preparation of proteoliposomes. Similar results were obtained in three additional independent experiments.
Table III. Kinetic constants of recombinant citrin, aralar1 and their C-terminal domains (CTDs) under de-energized conditions.
| Carrier | External substrate | Internal substrate | Km (µM) | Vmax (µmol/min/g protein) | Turnover number (per min) | Expts (No.) |
|---|---|---|---|---|---|---|
| Citrin | aspartate | aspartate | 56 ± 7 | 194 ± 28 | 14.9 | 6 |
| aspartate | glutamate | 50 ± 7 | 172 ± 31 | 13.2 | 5 | |
| glutamate | glutamate | 228 ± 30 | 169 ± 26 | 13.0 | 3 | |
| glutamate | aspartate | 205 ± 22 | 185 ± 25 | 14.2 | 4 | |
| Citrin CTD | aspartate | aspartate | 51 ± 5 | 350 ± 57 | 15.7 | 3 |
| aspartate | glutamate | 47 | 369 | 16.5 | 2 | |
| glutamate | glutamate | 200 | 344 | 15.4 | 2 | |
| glutamate | aspartate | 182 ± 20 | 328 ± 50 | 14.7 | 3 | |
| Aralar1 | aspartate | aspartate | 51 ± 8 | 43 ± 6 | 3.2 | 5 |
| aspartate | glutamate | 52 ± 9 | 52 ± 8 | 3.9 | 4 | |
| glutamate | glutamate | 220 ± 25 | 56 ± 9 | 4.2 | 4 | |
| glutamate | aspartate | 216 ± 25 | 49 ± 7 | 3.7 | 5 | |
| Aralar1 CTD | aspartate | aspartate | 61 | 79 | 3.4 | 2 |
| aspartate | glutamate | 55 | 96 | 4.1 | 2 | |
| glutamate | glutamate | 220 | 96 | 4.1 | 2 | |
| glutamate | aspartate | 229 | 88 | 3.8 | 2 |
The values were calculated from double reciprocal plots of the rate of [14C]aspartate or [14C]glutamate uptake versus substrate concentrations. The exchange was started by the addition of 20–800 µM [14C]aspartate or [14C]glutamate to proteoliposomes containing 20 mM aspartate or 20 mM glutamate and pre-incubated in the presence of valinomycin (1 µg/mg of phospholipids) and nigericin (50 ng/mg of phospholipids) in ethanol (10 µl/ml of liposomes) for 10 min. Transport was stopped after 30 s (citrin and its C-terminal domain) or 1 min (aralar1 and its CTD). The data represent means ± SE.
Functional characterization of aralar1 and citrin in HEK-293T cells
Aralar1 and citrin were overexpressed individually in the human cell line HEK-293T derived from kidney epithelium. The localization of both proteins in mitochondria was confirmed by the granular cytoplasmic immunostaining and by their co-localization with the mitochondrial-specific fluorescent vital dye MitoTracker Red CMXRos (data not shown). The levels of aralar1 and citrin in mitochondria were estimated from the intensities of the bands on western blots by comparison with known amounts of purified protein, and the values were normalized to the contents of the β-subunit of F1-ATPase. The levels of overexpressed citrin and aralar1 were 2.3 and 0.86 µg of protein per arbitrary unit of the β-subunit, respectively, ∼4-fold higher than in mitochondria from control cells (0.43 and 0.18 µg of protein per arbitrary unit of β-subunit, respectively). The level of citrin was about twice that of aralar1, consistent with the high expression of citrin, but not of aralar1, in kidney (Kobayashi et al., 1999; del Arco et al., 2000; Iijima et al., 2001).
Increased expression of the AGC should stimulate the malate/aspartate shuttle and this should lead to increased intramitochondrial NADH and increased 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by mitochondria (Berridge and Tan, 1993). In order to test this possibility and analyze the behavior of the mitochondrial exchangers in their own cytosolic environment, cells were permeabilized with digitonin (20 µM) and incubated with 1 mM glutamate, 5 mM malate and 10 mM lactate (G + M + L), conditions that boost the aspartate/malate shuttle function by providing the substrates of the shuttle and increasing the cytosolic NADH/NAD ratio. In the absence of added substrates, MTT reduction by mitochondria was low both in control cells and in cells in which citrin or aralar1 were overexpressed (Figure 3A). It was stimulated by G + M + L to a greater extent in cells overexpressing either protein than in control cells (Figure 3A). In the presence of glycerol-3-phosphate, the substrate for the glycerol phosphate NADH shuttle, and lactate (G-P + L), MTT reduction was stimulated in overexpressing and control cells alike (Figure 3A). The normalized MTT reduction (ratio of G + M + L to G-P + L) was ∼1.6- to 2-fold higher with cells overexpressing aralar1 or citrin (Figure 3B). These results confirm that aralar1 and citrin function as AGCs in human cells.

Fig. 3. Reduction of MTT in cells overexpressing aralar1 or citrin. Digitonin (20 µM)-permeabilized HEK-293T cells overexpressing aralar1 (filled bars) or citrin (striped bars) or the empty pIRES vector (open bars) were tested for reduction of MTT in the presence of either 1 mM glutamate, 5 mM malate and 10 mM lactate (G + M + L) or 15 mM glycerol-3-phosphate and 10 mM lactate (G-P + L). Under basal conditions, no substrates were added. (A) Reduction of MTT in the presence of various substrates. (B and C) Normalized MTT reduction. The results (means ± SEM) correspond to a representative experiment performed four times. The differences between controls and cells overexpressing aralar1 or citrin were significant (*P <0.05, ***P <0.001, one-way ANOVA and Bonferroni t-test).
Ca2+ activation of the AGC
In the current model of the topography of mitochondrial transporters, the N- and C-termini are localized on the external surface of the inner membrane (Palmieri, 1994). This means that the long N-terminal domains of citrin and aralar1 may be situated likewise. This arrangement was verified by proteolysis experiments (Figure 4). In rat brain and liver mitoplasts, but not in intact mitochondria, the N-terminal domains of aralar1 and citrin, respectively, were removed by treatment with proteinase K, whereas the β-subunit of F1-ATPase was protected. These results show that the N-terminal domains of citrin and aralar1 containing EF-hand Ca2+-binding motifs are exposed to the intermembrane space and suggest the possibility that the transport activity of these proteins is regulated by Ca2+ ions outside the matrix space.
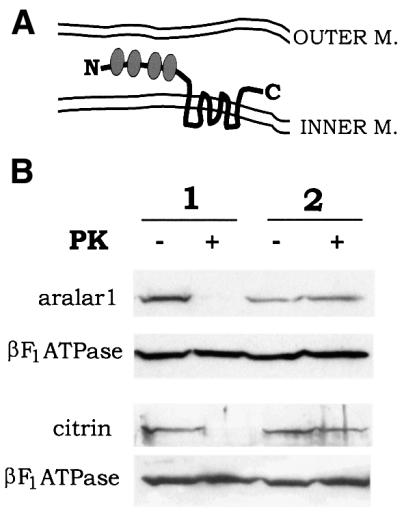
Fig. 4. The N-terminal domains of aralar1 and citrin containing the Ca2+-binding sites are exposed to the cytoplasmic surface of the inner mitochondrial membrane. (A) Schematic representation of the topology of aralar1 or citrin in the inner mitochondrial membrane. In the N-terminal domain, the four EF-hand Ca2+-binding motifs are indicated. (B) Proteolysis of aralar1 and citrin with proteinase K. Mitoplasts (1) and mitochondria (2) isolated from rat brain (aralar1) or liver (citrin) were treated with proteinase K as indicated. Each fraction (20 µg protein/lane) was subjected to reaction with an antibody against the N-terminal domain of aralar1 or citrin. As control, analyses were also performed for the β-subunit of F1-ATPase.
Glutamate is taken up into mitochondria either via the AGC and then transaminated in the matrix by aspartate aminotransferase (mAST), or by the glutamate/OH– carrier (GC) and then deaminated by glutamate dehydrogenase (GDH) (Figure 5A; Schoolwerth et al., 1978; Schoolwerth and LaNoue, 1980). In both instances, the product is α-ketoglutarate (α-KG), which is decarboxylated by α-ketoglutarate dehydrogenase (α-KGDH), an enzyme that is confined to mitochondria. As glutamate decarboxylase activity is very low in kidney (Goodyear et al., 1982), 14CO2 production gives an unequivocal measure of glutamate metabolism via α-KG to succinyl-CoA and carbon dioxide. In intact HEK-293T cells incubated with glutamate alone (G) or glutamate + malate (G + M), 40% of 14CO2 production from [1-14C]glutamate was inhibited by 5 mM aminooxyacetic acid (AOAA), an inhibitor of AST (Palmieri et al., 1979; Schoolwerth and LaNoue, 1980) (Figure 5B), indicating that the AGC/mAST pathway accounts for ∼40% of α-KG decarboxylation with G or G + M as metabolic substrates. When 10 mM lactate was also added to increase the cytosolic NADH/NAD ratio and the aspartate/malate NADH shuttle function, 14CO2 production was almost fully inhibited by AOAA (Figure 5B), indicating that AGC/mAST was now the major pathway for 14CO2 production. Similar effects (not shown) were maintained in permeabilized cells.

Fig. 5. Decarboxylation of l-[1-14C]glutamate and [1-14C]α-ketoglutarate in HEK-293T cells. (A) Pathways for glutamate uptake and decarboxylation in mitochondria. AGC, aspartate/glutamate carrier; OMC, α-ketoglutarate/malate carrier; AOAA, aminooxyacetic acid; GC, glutamate/OH-- carrier; CaU, Ca2+ uniporter; RR, ruthenium red; α-KGDH, α-ketoglutarate dehydrogenase; GDH, glutamate dehydrogenase; LDH, lactate dehydrogenase; mAST, cAST, mMDH and cMDH, mitochondrial and cytosolic aspartate aminotransferases and malate dehydrogenases, respectively. (B) Effects of malate and lactate on the pathway of glutamate decarboxylation. HEK-293T cells (∼10 µg protein/well) were incubated in the presence of 1 mM l-[1-14C]glutamate (circles), and 5 mM malate (squares) or 5 mM malate + 10 mM lactate (triangles) for the times shown. AOAA (5 mM) was added where indicated (filled symbols). The data correspond to a single experiment performed in triplicate. It has been repeated 2–3 times with similar results. (C) Effects of Ca2+ and RR on glutamate decarboxylation in permeabilized cells. HEK-293T cells were incubated with 1 mM l-[1-14C]glutamate, 5 mM malate, 10 mM lactate and 20 µM digitonin for 60 min in the presence (AOAA-resistant CO2 production) or absence (total CO2) of 5 mM AOAA with 20 µM CaCl2 (filled bars) or 20 µM CaCl2 + 1 nmol RR/mg protein (striped bars), or without CaCl2 (open bars). AOAA-sensitive CO2 production was the difference between total and AOAA-resistant CO2 production. Results are means ± SEM of five paired experiments performed in triplicate. The difference between incubations with or without Ca2+ and incubations with Ca2+ with or without RR was significant, where indicated (*P <0.05, Wilcoxon signed rank t-test). (D) Inhibition by RR of Ca2+-stimulated α-ketoglutarate decarboxylation in digitonin-permeabilized cells. HEK-293T cells were incubated with 50 µM [1-14C]α-ketoglutarate, 5 mM malate and 20 µM digitonin for 60 min in the presence of 20 µM CaCl2 alone, or together with 1 or 2 nmol RR/mg protein, or in the absence of Ca2+. Data are means ± SEM of four experiments performed in triplicate. The significance of the difference between incubations with Ca2+ and either no Ca2+ or Ca2+ and RR is indicated (***P <0.001, one-way ANOVA followed by Bonferroni t-test).
In permeabilized cells and in the presence of G + M + L, total 14CO2 production from [1-14C]glutamate was significantly increased by the addition of 20 µM Ca2+ (Figure 5C). This figure also shows that both the AOAA-sensitive and the AOAA-resistant CO2 production (which reflect the respective share of the AGC/mAST/α-KGDH and of the GC/GDH/α-KGDH pathways to the total CO2 production) are clearly activated by Ca2+. Two explanations can be provided for the effect of Ca2+: first, Ca2+ is taken up into mitochondria by the Ca2+ uniporter (CaU) where it activates α-KGDH and hence the (i) GC/ GDH/α-KGDH or (ii) AGC/mAST/α-KGDH pathway of glutamate decarboxylation; or, secondly, Ca2+ activates the AGC via its Ca2+-binding domains. The stimulation by Ca2+ of the residual AOAA-resistant 14CO2 formation (Figure 5C) is consistent with the first explanation (i). The addition of ruthenium red (RR; 1 nmol/mg protein; Figure 5C), at a concentration that fully inhibits the CaU (Reed and Bygrave, 1974; A.Martínez-Serrano and J.Satrústegui, unpublished data) without interfering with Ca2+ binding to EF-hand domains (Sasaki et al., 1992), abolishes the Ca2+ stimulation of the AOAA-resistant glutamate decarboxylation. Moreover, the same RR concentration (1–2 nmol/mg protein) fully inhibited Ca2+ activation of the decarboxylation of [1-14C]α-KG (Figure 5D). In contrast, Ca2+ stimulation of the AOAA-sensitive 14CO2 formation from glutamate was little affected by RR (Figure 5C). This indicates that Ca2+ activation of the AGC/mAST/α-KGDH pathway in HEK-293T cells is not caused by matrix Ca2+ acting on α-KGDH, but is largely (∼90%) caused by its direct action on citrin and aralar1 as the AGC.
Interestingly, overexpression of aralar1 in HEK-293T cells resulted in a substantial increase in Ca2+ stimulation of the total and the AOAA-sensitive (i.e. the AGC/mAST/α-KGDH pathway) CO2 production from glutamate (Figure 6). The percentage stimulation by Ca2+ was almost tripled in cells overexpressing aralar1 as compared with that in control cells, and this difference persisted in the presence of RR. Similar effects, albeit somewhat smaller, were also observed in cells overexpressing citrin (Figure 6). Therefore, overexpression of the AGC isoforms aralar1 and citrin enhances the effects of Ca2+ on the AGC. It should also be noted that [1-14C]α-KG decarboxylation was found to be stimulated by Ca2+ equally in cells expressing aralar1 and in control cells (158 ± 20 and 172 ± 18% stimulation, respectively). This indicates that aralar overexpression does not interfere with Ca2+ entry into mitochondria and with the CaU-dependent Ca2+ activation of α-KGDH.

Fig. 6. Ca2+-induced stimulation of glutamate decarboxylation in HEK-293T cells overexpressing aralar1 or citrin. Cultures of HEK-293T cells expressing aralar1 (filled bars), citrin (striped bars) or empty pIRES vector (open bars) were incubated with 1 mM l-[1-14C]glutamate, 5 mM malate, 10 mM lactate and 20 µM digitonin for 60 min in the presence (AOAA-resistant 14CO2 production) or absence (total 14CO2) of 5 mM AOAA, and either 20 µM CaCl2, 20 µM CaCl2 + 1 nmol RR/mg protein, or in the absence of Ca2+. AOAA-sensitive 14CO2 production was the difference between total and AOAA-resistant 14CO2 production. The results, expressed as percentage of 14CO2 formation in the absence of Ca2+, are means ± SEM of 4–9 experiments performed in triplicate. The significance of the difference between cells expressing aralar1 or citrin and controls is indicated (*P <0.05, **P <0.025, Mann–Whitney t-test, ***P< 0.001, one-way ANOVA followed by Bonferroni t-test).
Discussion
Citrin and aralar1 are two isoforms of the human mitochondrial aspartate/glutamate exchanger
Knowledge about the mitochondrial AGC has existed for a long time, and its main properties have been studied in intact mitochondria (for references see LaNoue and Schoolwerth, 1979) and in reconstituted proteoliposomes (Krämer et al., 1986; Dierks and Krämer, 1988; Dierks et al., 1988; Bisaccia et al., 1992). However, the protein responsible for the exchange had not been identified hitherto. From our present study, it is clear that we have identified citrin and aralar1 as distinct isoforms of AGC because the properties of the reconstituted proteins fully match those of the native AGC regarding substrate specificity, transport affinities, inhibitor sensitivity and voltage dependence. They have molecular masses well above the usual 30 kDa of most mitochondrial carriers, and the partially purified preparation of AGC from bovine heart mitochondria gave a major protein band of 68 kDa on SDS–PAGE (Krämer et al., 1986). A different purification procedure gave rise to a homogeneous smaller fragment of ∼31.5 kDa (Bisaccia et al., 1992). In the light of our findings with the C-terminal domains of citrin and aralar1, both the 68 kDa (the full-length protein) and the 31.5 kDa (the carrier moiety) protein represent the AGC as both are active as aspartate/glutamate exchangers. It is likely that the fragment of 31.5 kDa originated by proteolysis during the solubilization/purification procedure. Consistently, we noted that the recombinant aralar1 is easily cleaved in two halves, one of which (∼32 kDa) is no longer recognized by antibodies against the N-terminal domain of the protein.
Possible roles of the AGC in CTLN2
In liver, the AGC plays an important role in the urea cycle by providing aspartate for incorporation into argininosuccinate (Meijer et al., 1978). In CTLN2, the identified mutations in citrin lead to either truncation of the protein or deletion of a loop between transmembrane spans (Kobayashi et al., 1999; Yasuda et al., 2000). It seems likely that such dramatic changes in the protein would lead to impairment of the function of citrin as an AGC in mitochondria. This impairment would lead to a failure in supply of aspartate from mitochondria for formation of argininosuccinate. As the AGC is an essential component of the malate/aspartate NADH shuttle, they would also prevent its function. Either or both of these effects could lead to the symptoms of CTLN2. Since in CTLN2 hepatic ASS protein is deficient with no alterations to either its gene or mRNA (Kobayashi et al., 1986, 1993), it remains unclear how a defective AGC brings this about. Possible explanations are that critical aspartate levels may be required to make the synthetase active, or that defects in citrin destabilize the enzyme.
The AGC is regulated by Ca2+
The malate/aspartate NADH shuttle function is known to be regulated by Ca2+ and Ca2+-mobilizing hormones in liver (Leverve et al., 1986; Sugano et al., 1988; Sterniczuk et al., 1991) and heart (Scaduto, 1994). The mechanism by which Ca2+ activates this shuttle is, however, still a matter of debate. For example, the stimulation of α-KGDH by matrix Ca2+ (Sterniczuk et al., 1991; Scaduto, 1994) has been shown to decrease α-KG efflux from mitochondria (O’Donnell et al., 1998), an effect that would immediately oppose malate/aspartate shuttle activation. The results reported here indicate a novel mechanism whereby AGC, which catalyzes the irreversible step of the malate/aspartate NADH shuttle (LaNoue and Schoolwerth, 1979), is activated by Ca2+. The N-termini of aralar1 and citrin harbor EF-hand domains shown to bind Ca2+ (del Arco and Satrústegui, 1998; del Arco et al., 2000; Kobayashi et al., 2000), and overexpression of these proteins in human cell lines greatly increases Ca2+ activation of AGC, as reflected in a Ca2+-stimulated increase in AOAA-sensitive glutamate decarboxylation in digitonin-permeabilized cells. This increase is not sensitive to RR concentrations that abolish Ca2+ activation of α-KG decarboxylation, indicating that Ca2+ acts on the external face of the inner mitochondrial membrane to activate AGC. This is consistent with the fact that the N-terminal domains of aralar1 and citrin face the external side of the inner mitochondrial membrane (Figure 4). Taken together, the results clearly demonstrate that the mechanism of Ca2+ regulation of AGC requires interaction of Ca2+ with the N-terminal domain of the AGC molecule at the external side of the inner mitochondrial membrane, strongly suggesting that the N-terminal domain alone or through its interaction with other proteins is responsible for activation by external Ca2+.
The significance of a site for Ca2+ regulation in AGC on the external face of the inner mitochondrial membrane is intriguing. So far, the role of Ca2+ in the regulation of metabolite transport has been related conceptually to the effects of matrix mitochondrial Ca2+ on three dehydrogenases (pyruvate dehydrogenase, NAD-isocitrate dehydrogenase and α-KGDH). Activation by Ca2+ of these enzymes is important to increase NADH levels and regulate Krebs cycle activity (Nichols and Denton, 1995). For mitochondrial metabolite carriers involved in near-equilibrium reactions, increased dehydrogenase flux brought about by matrix Ca2+ results in parallel changes in metabolite uptake/exchange by mitochondria (Bunger and Mallet, 1993; O’Donnell et al., 1998). However, the AGC reaction is far from substrate equilibrium under physiological conditions (LaNoue and Schoolwerth, 1979), so that simple changes in the mass–action ratio may not be sufficient to increase flux through the carrier. This may explain why this carrier is regulated directly by extramitochondrial Ca2+. A member of the second NADH shuttle, glycerol-phosphate dehydrogenase (FAD-GPDH), also has two EF-hand Ca2+-binding domains facing the external side of the inner mitochondrial membrane (Brown et al., 1994). FAD-GPDH is activated by calcium through a decrease in the K0.5 for glycerol-3-phosphate (Rutter et al., 1992). Ca2+ activation of FAD-GPDH leads to increased supply of reducing equivalents to ubiquinone in the respiratory chain, without any net consumption of the metabolites of the glycerol-phosphate shuttle. We suggest that activation of AGC by Ca2+ from the cytosolic side likewise is involved in increased supply of reducing equivalents to mitochondrial NAD without net consumption of the metabolites of the shuttle. In other words, Ca2+ activation ‘pushes’ two reducing equivalents into mitochondria rather than ‘pulling’ glutamate into mitochondria. This entails that under conditions of Ca2+ activation of the shuttle function, the members of the shuttle operate in a coordinated fashion with preferential channeling of substrates along the AGC, mAST and α-KG/malate carrier (OMC) pathway.
The present findings suggest a new mechanism for how Ca2+ regulates the supply of reducing equivalents to the mitochondrial respiratory chain. On the one hand, it activates mitochondrial dehydrogenases after entry into mitochondria and, on the other hand, it increases the activity of the NADH shuttles by acting on the external side of the inner mitochondrial membrane. While some redundancy may be anticipated, these two mechanisms may play separate roles in the control of mitochondrial function by Ca2+. During cell activation, some mitochondria take up Ca2+ from cytosolic domains of high Ca2+ concentration that are generated by the activation of Ca2+ channels, ryanodine receptors or IP3 receptors (Rizzuto et al., 1993; Montero et al., 2000). The response of mitochondria is highly heterogeneous at the cellular level, and optimal activation of mitochondrial Ca2+ uptake is obtained by the proximity and synchronous activation of these channels (Csordás et al., 1999; Montero et al., 2000). Mitochondria located at some distance from these Ca2+ hotspots sense the overall increase in cytosolic Ca2+ and take up much less calcium (Montero et al., 2000; Park et al., 2001). In these mitochondria, Ca2+ activation of AGC and the malate/aspartate NADH shuttle may be involved in increasing mitochondrial NADH without the need for Ca2+ uptake. The relative contribution of these two mechanisms in the regulation of mitochondrial function by Ca2+, in different settings or under different conditions, now needs to be assessed.
Materials and methods
Construction of expression plasmids
The coding sequences for citrin and its C-terminal domain, and for aralar1 and its C-terminal domain were amplified by PCR from human liver or heart cDNAs, respectively. For amplification of citrin and aralar1, forward and reverse oligonucleotide primers corresponded to the extremities of their coding sequences (del Arco and Satrústegui, 1998; Kobayashi et al., 1999). For amplification of C-terminal domains, the primers were nucleotides 989–1010 and 2143–2161 of the citrin cDNA (DDBJ/EMBL/GenBank accession No. AF118838) and nucleotides 879–899 and 2040–2057 of the aralar1 cDNA (DDBJ/EMBL/GenBank accession No. Y14494). The forward and reverse primers had BamHI restriction sites (citrin), NdeI and BamHI (citrin C-terminal domain), and NdeI and HindIII restriction sites (aralar1 and its C-terminal domain) as linkers. For aralar1 and its C-terminal domain, the reverse primers also contained 18 extra bases encoding a His6 tag immediately before the stop codon. The amplified products were cloned into the expression vectors pET-15b (citrin), pET-21b (citrin C-terminal domain) and pMW7 (aralar1 and its C-terminal domain) and the constructs were transformed into E.coli CO214(DE3) cells. Transformants were selected on 2× YT plates containing ampicillin (100 µg/ml) and screened by direct colony PCR and by restriction digestion of purified plasmid DNA. The sequences of inserts were verified.
Bacterial expression of citrin, aralar1 and their C-terminal domains
Proteins were overexpressed at 30°C in E.coli C0214(DE3) (Fiermonte et al., 2001). Inclusion bodies were purified on a sucrose density gradient (Fiermonte et al., 1993), washed at 4°C with TE buffer (10 mM Tris–HCl, 1 mM EDTA pH 6.5), then twice with a buffer containing Triton X-114 (3%, w/v), 1 mM EDTA and 10 mM PIPES–KOH pH 6.5, and once again with TE buffer. Proteins were separated by SDS–PAGE in 17.5% gels and either stained with Coomassie Blue dye or transferred to nitrocellulose membranes for immunodetection with rabbit antibodies raised against the N-terminal domains of citrin (peptide 1–285) (Yasuda et al., 2000) and aralar1 (peptide 12–243) (del Arco and Satrústegui, 1998), peptide 305–319 in the middle part of citrin and peptide 507–520 in the C-terminal domain of aralar1 (A.del Arco and J.Satrústegui, in preparation). The N-termini were sequenced, and the yield of purified proteins was estimated by laser densitometry of stained samples (Fiermonte et al., 1998).
Reconstitution into liposomes and transport measurements
Citrin, aralar1 and their C-terminal domains were solubilized in 1.9% sarkosyl (w/v), and a small residue was removed by centrifugation (258 000 g, 1 h). Solubilized proteins were diluted 9-fold with buffer (10 mM PIPES–KOH pH 6.5, 1 mM EDTA) and then reconstituted into liposomes in the presence of substrates (Palmieri et al., 1995). The amount of protein incorporated into liposomes was measured as described (Dolce et al., 2001). In all cases, it was ∼20% of the protein added to the reconstitution mixture. Transport at 25°C was started by adding l-[14C(U)]aspartate or l-[14C(U)]glutamate (NEN Life Science Products) to the proteoliposomes and terminated by addition of 15 mM pyridoxal 5′-phosphate and 10 mM bathophenanthroline (Palmieri et al., 1995). In controls, inhibitors were added with the labeled substrate. Entrapped radioactivity was counted (Palmieri et al., 1995). The initial transport rate was calculated from the radioactivity taken up by proteoliposomes after 30–60 s (in the initial linear range of substrate uptake). For efflux measurements, proteoliposomes containing 2 mM aspartate were labeled with carrier-free [14C]aspartate by carrier-mediated exchange equilibration (Palmieri et al., 1995). After 40 min, the external radioactivity was removed by passing the proteoliposomes through Sephadex G-75. In some experiments, valinomycin and nigericin were added to the proteoliposomes in order to provide de-energized conditions. K+ diffusion potentials were generated using valinomycin and K+ gradients. In these experiments, substrate and buffer were neutralized with NaOH.
Cell lines and transfections
Citrin was expressed in the human cell line HEK-293T from the vector pIREScitrin (del Arco et al., 2000). The pIRESaralar1 with the coding sequence of aralar1 cloned into the BamHI-digested pIRES1hyg vector and the empty pIRES vector as control were transfected into the same cell line, and stable cells were established. Cloned cell lines were grown and maintained as described (del Arco et al., 2000) in the presence of hygromycin (100 µg/ml). Immunofluorescence and western blotting were carried out with antibodies specific against aralar1 peptide 12–343 or citrin peptide 36–278 (A.del Arco and J.Satrústegui, in preparation). In order to determine the protein levels of aralar1 and citrin, cells were homogenized in 250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol pH 7.4, 1 mM iodoacetate and 1 mM phenylmethylsulfonyl fluoride (PMSF). Mitochondria were isolated by differential centrifugation and analyzed by western blotting with an enhanced chemiluminiscence kit (Amersham). As control, an antibody against the β-subunit of F1-ATPase (generous gift of Professor J.M.Cuezva) was used. The optical densities of bands were measured, and the protein amounts were calculated by using the standard curve obtained from serial dilution of each purified N-terminal domain of aralar1 and citrin.
Reduction of MTT by mitochondria
Cells were seeded at a density of 105 cells/cm2 and, after 24 h, transferred to a glucose-free medium (HCSS: 5.4 mM KCl, 0.12 M NaCl, 0.8 mM MgCl2, 1 mM CaCl2, 25 mM HEPES pH 7.4). About 12 h later, cultures were washed in Ca2+-free HCSS and pre-incubated for 30 min in the same medium containing 20 µM digitonin. This digitonin concentration was sufficient to make Ca2+ available to mitochondria without loss of cytosolic components (Martínez-Serrano and Satrústegui, 1992). Then MTT (1 mg/ml) was added alone or together with either 1 mM glutamate, 5 mM malate and 10 mM lactate, or 15 mM glycerol-3-phosphate and 10 mM lactate, and incubated at 37°C in the dark for 30 min. Cells were lysed in SDS (20%, w/v), in 50% aqueous N,N-dimethyl formamide pH 4.7 and, after incubation for 18 h at 37°C, formazan was estimated from the absorbance at 570 nm, with the lysis solution as blank.
Proteolysis of mitochondria and mitoplasts
Mitoplasts were made from rat brain or liver mitochondria by hypotonic shock in 20 mM Tris–HCl pH 7.4. Mitochondria or mitoplasts (100 µg; protein concentration 200 µg/ml) were kept on ice for 15 min with and without proteinase K (0.1 mg/ml). Following the addition of 2 mM PMSF, mitochondria and mitoplasts were pelleted immediately, and analyzed by western blotting with antibodies against the N-terminal domains of aralar1 or citrin and the β-subunit of F1-ATPase.
Decarboxylation of l-glutamate or α-KG
HEK-293T cells were subcultured in P96 plates (105 cells/cm2). After 24–48 h growth, they were washed with Krebs Ringer phosphate (KRP) pH 7.2, and incubated for 60–120 min at 37°C in KRP (100 µl) containing either 1 mM l-[1-14C]glutamate (1 µCi/ml) or 50 µM [1-14C]α-KG (0.5 µCi/ml) with or without other reagents. Ca2+-induced stimulation of glutamate or α-KG decarboxylation was studied in the cells, permeabilized by 20 µM digitonin, with or without 20 µM CaCl2 and/or 100–200 nM RR (1–2 nmol/mg protein). 14CO2 was trapped in filters (10 mm diameter; Millipore AP 250 1000) soaked in 3.5 M NaOH. At the end of the reaction, 0.4 M citric acid (20 µl) was added and, after 30 min at 37°C, trapped radioactivity was counted. Unless indicated, blank values without cells (∼50 c.p.m.) were not deducted.
Acknowledgments
Acknowledgements
This work was supported by grants from MURST-PRIN, MURST L.488/92 CO3 and CO4, MURST-CNR L.95/95, CEGBA and the CNR target project on Biotechnology (to F.P.), from the Fundacion Ramón Areces to the Centro de Biología Molecular Severo Ochoa, the Spanish Direccion General de Investigación Científica y Técnica, Ministerio de Educación y Cultura, and Química Farmacéutica Bayer, S.A. (to J.S.), from the Ministry of Health and Welfare of Japan (T.S.) and by Grants-in-Aid for Scientific Research from Japan (to K.K. and to M.I.).
References
- Berridge M.V. and Tan,A.S. (1993) Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys., 303, 474–482. [DOI] [PubMed] [Google Scholar]
- Bisaccia F., De Palma,A. and Palmieri,F. (1992) Identification and purification of the aspartate/glutamate carrier from bovine heart mitochondria. Biochim. Biophys. Acta, 1106, 291–296. [DOI] [PubMed] [Google Scholar]
- Brown L.J., MacDonald,M.J., Lehn,D.A. and Moran,S.M. (1994) Sequence of rat mitochondrial glycerol-3-phosphate dehydrogenase cDNA. J. Biol. Chem., 269, 14363–14366. [PubMed] [Google Scholar]
- Bunger R. and Mallet,R.T. (1993) Mitochondrial pyruvate transport in working guinea-pig heart. Work-related vs. carrier-mediated control of pyruvate oxidation. Biochim. Biophys. Acta, 1151, 223–236. [DOI] [PubMed] [Google Scholar]
- Csordás G., Thomas,A.P. and Hajnóczky,G. (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J., 18, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Arco A. and Satrústegui,J. (1998) Molecular cloning of aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J. Biol. Chem., 273, 23327–23334. [DOI] [PubMed] [Google Scholar]
- del Arco A., Agudo,M. and Satrustegui,J. (2000) Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem. J., 345, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks T. and Krämer,R. (1988) Asymmetric orientation of the reconstituted aspartate/glutamate carrier from mitochondria. Biochim. Biophys. Acta, 937, 122–126. [DOI] [PubMed] [Google Scholar]
- Dierks T., Riemer,E. and Krämer,R. (1988) Reaction mechanism of the reconstituted aspartate/glutamate carrier from bovine heart mitochondria. Biochim. Biophys. Acta, 943, 231–244. [DOI] [PubMed] [Google Scholar]
- Dierks T., Stappen,R., Salentin,A. and Krämer,R. (1992) Probing the active site of the reconstituted aspartate/glutamate carrier from bovine heart mitochondria: carbodiimide-catalyzed acylation of a functional lysine residue. Biochim. Biophys. Acta, 1103, 13–24. [DOI] [PubMed] [Google Scholar]
- Dolce V., Fiermonte,G., Runswick,M.J., Palmieri,F. and Walker,J.E. (2001) The human mitochondrial deoxynucleotide carrier and its role in toxicity of nucleoside antivirals. Proc. Natl Acad. Sci. USA, 98, 2284–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiermonte G., Walker,J.E. and Palmieri,F. (1993) Abundant bacterial expression and reconstitution of an intrinsic membrane transport protein from bovine mitochondria. Biochem. J., 294, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiermonte G., Palmieri,L., Dolce,V., Lasorsa,F.M., Palmieri,F., Runswick,M.J. and Walker,J.E. (1998) The sequence, bacterial expression and functional reconstitution of the rat mitochondrial dicarboxylate transporter cloned via distant homologs in yeast and Caenorhabditis elegans. J. Biol. Chem., 273, 24754–24759. [DOI] [PubMed] [Google Scholar]
- Fiermonte G., Dolce,V., Palmieri,L., Ventura,M., Runswick,M.J., Palmieri,F. and Walker,J.E. (2001) Identification of the human mitochondrial oxodicarboxylate carrier: bacterial expression, reconstitution, functional characterization, tissue distribution and chromosomal location. J. Biol. Chem., 276, 8225–8230. [DOI] [PubMed] [Google Scholar]
- Goodyear P.R., Mills,M. and Scriver,C.R. (1982) Properties of γ-aminobutyric acid synthesis by rat renal mitochondria. Biochim. Biophys. Acta, 716, 348–357. [DOI] [PubMed] [Google Scholar]
- Iijima M. et al. (2001) Pathogenesis of adult-onset type II citrullinemia caused by deficiency of citrin, a mitochondrial solute carrier protein: tissue and subcellular localization of citrin. Adv. Enzyme Regul., 41, 325–342. [DOI] [PubMed] [Google Scholar]
- Kobayashi K. et al. (1986) Messenger RNA coding for argininosuccinate synthetase in citrullinemia. Am. J. Hum. Genet., 38, 667–680. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K., Shaheen,N., Kumashiro,R., Tanikawa,K., O’Brien,W.E., Beaudet,A.L. and Saheki,T. (1993) A search for the primary abnormality in adult-onset type II citrullinemia. Am. J. Hum. Genet., 53, 1024–1030. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K., Kakinoki,H., Fukushige,T., Shaheen,N., Terazono,H. and Saheki,T. (1995) Nature and frequency of mutations in the argininosuccinate synthetase gene that cause classical citrullinemia. Hum. Genet., 96, 454–463. [DOI] [PubMed] [Google Scholar]
- Kobayashi K. et al. (1999) The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nature Genet., 22, 159–163. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Iijima,M., Yasuda,T., Sinasac,D.S., Yamaguchi,N., Tsui,L.-C., Scherer,S.W. and Saheki,T. (2000) Type II citrullinemia (citrin deficiency): a mysterious disease caused by a defect of calcium-binding mitochondrial carrier protein. In Pochet,R., Donato,R., Haiech,J., Heizmann,C. and Gerke,V. (eds), Calcium: The Molecular Basis of Calcium Action in Biology and Medicine. Kluwer, Dordrecht, The Netherlands, pp. 557–579.
- Krämer R., Kürzinger,G. and Heberger,C. (1986) Isolation and functional reconstitution of the aspartate/glutamate carrier from mitochondria. Arch. Biochem. Biophys., 251, 166–174. [DOI] [PubMed] [Google Scholar]
- LaNoue K.F. and Schoolwerth,A.C. (1979) Metabolite transport in mitochondria. Annu. Rev. Biochem., 48, 871–922. [DOI] [PubMed] [Google Scholar]
- LaNoue K.F., Meijer,A.J. and Brouwe,A. (1974) Evidence for electrogenic aspartate transport in rat liver mitochondria. Arch. Biochem. Biophys., 161, 544–550. [DOI] [PubMed] [Google Scholar]
- Leverve X.M., Verhoeven,A.J., Groen,A.K., Meijer,A.J. and Tager,J.M. (1986) The malate/aspartate shuttle and pyruvate kinase as targets involved in the stimulation of gluconeogenesis by phenylephrine. Eur. J. Biochem., 155, 551–556. [DOI] [PubMed] [Google Scholar]
- Martínez-Serrano A. and Satrústegui,J. (1992) Regulation of cytosolic free calcium concentration by intrasynaptic mitochondria. Mol. Biol. Cell, 3, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer A.J., Gimpel,J.A., Deleeuw,G., Tischler,M.E., Tager,J.M. and Williamson,J.R. (1978) Interrelationships between gluconeogenesis and ureogenesis in isolated hepatocytes. J. Biol. Chem., 253, 2308–2320. [PubMed] [Google Scholar]
- Montero M., Alonso,M.T., Carnicero,E., Cuchillo-Ibáñez,I., Albillos,A., García,A.G., García-Sancho,J. and Alvarez,J. (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nature Cell Biol., 2, 57–61. [DOI] [PubMed] [Google Scholar]
- Nichols B.J. and Denton,R.M. (1995) Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell. Biochem., 149, 203–212. [DOI] [PubMed] [Google Scholar]
- O’Donnell J.M., Doumen,C., LaNoue,K.F., White,L.T., Yu,X., Alpert,N.M. and Lewandowski,E.D. (1998) Dehydrogenase regulation of metabolite oxidation and efflux from mitochondria in intact hearts. Am. J. Physiol., 274, H467–H476. [DOI] [PubMed] [Google Scholar]
- Palmieri F. (1994) Mitochondrial carrier proteins. FEBS Lett., 346, 48–54. [DOI] [PubMed] [Google Scholar]
- Palmieri F., Stipani,I. and Iacobazzi,V. (1979) The transport of l-cysteinesulfinate in rat liver mitochondria. Biochim. Biophys. Acta, 555, 531–546. [DOI] [PubMed] [Google Scholar]
- Palmieri F., Indiveri,C., Bisaccia,F. and Iacobazzi,V. (1995) Mitochondrial metabolite carrier proteins: purification, reconstitution and transport studies. Methods Enzymol., 260, 349–369. [DOI] [PubMed] [Google Scholar]
- Park M.K., Ashby,M.C., Erdemli,G., Petersen,O.H. and Tepikin,A.V. (2001) Perinuclear, perigranular and subplasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J., 20, 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed K.C. and Bygrave,F.L. (1974) The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem. J., 140, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., Brini,M., Murgia,M. and Pozzan,T. (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science, 262, 744–747. [DOI] [PubMed] [Google Scholar]
- Rutter G.A.A., Pralong,W-F. and Wollheim,C.B. (1992) Regulation of mitochondrial glycerol-phosphate dehydrogenase by Ca2+ within electopermeabilized insulin-secreting cells (INS-1). Biochim. Biophys. Acta, 1175, 107–113. [DOI] [PubMed] [Google Scholar]
- Saheki T., Kobayashi,K. and Inoue,I. (1987) Hereditary disorders of the urea cycle in man: biochemical and molecular approaches. Rev. Physiol. Biochem. Pharmacol., 108, 21–68. [DOI] [PubMed] [Google Scholar]
- Sasaki T., Naka,M., Nakamura,F. and Tanaka,T. (1992) Ruthenium red inhibits the binding of calcium to calmodulin required for enzyme activation. J. Biol. Chem., 267, 21518–21523. [PubMed] [Google Scholar]
- Scaduto R.C. Jr (1994) Calcium and 2-oxoglutarate-mediated control of aspartate formation by rat heart mitochondria. Eur. J. Biochem., 223, 751–758. [DOI] [PubMed] [Google Scholar]
- Schoolwerth A.C. and LaNoue,K.F. (1980) The role of microcompartmentation in the regulation of glutamate metabolism by rat kidney mitochondria. J. Biol. Chem., 255, 3403–3411. [PubMed] [Google Scholar]
- Schoolwerth A.C., Nazar,B.L. and LaNoue,K.F. (1978) Glutamate dehydrogenase activation and ammonia formation by rat kidney mitochondria. J. Biol. Chem., 253, 6177–6183. [PubMed] [Google Scholar]
- Sterniczuk A., Hreniuk,S., Scaduto,R.C.,Jr and LaNoue,K.F. (1991) The mechanism of Ca2+-related control of gluconeogenesis in perfused liver. Eur. J. Biochem., 190, 143–150. [DOI] [PubMed] [Google Scholar]
- Sugano T., Nishimura,K., Sogabe,N., Shiota,M., Oyama,N., Noda,S. and Ohta,M. (1988) Ca2+-dependent activation of the malate–aspartate shuttle by norepinephrine and vasopressin in perfused rat liver. Arch. Biochem. Biophys., 264, 144–154. [DOI] [PubMed] [Google Scholar]
- Williamson J.R. (1976) Role of anion transport in the regulation of metabolism. In Hanson,R.W. and Mehlman,M.A. (eds), Gluconeogenesis: Its Regulation in Mammalian Species. Wiley, New York, NY, pp. 165–238.
- Yasuda T. et al. (2000) Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum. Genet., 107, 537–544. [DOI] [PubMed] [Google Scholar]