

Abstract
Although in vitro evidence suggests two c-Jun N-terminal kinase (JNK) kinases, MKK4 and MKK7, transactivate JNK, in vivo confirmation is incomplete. In fact, JNK deficiency may differ from the composite deficiency of MKK4 and MKK7 in Drosophila and mice. Recently, the Caenorhabditis elegans homolog of human JNK, jnk-1, and two MKK-7s, mek-1 and jkk-1, were cloned. Here we characterize jnk-1, which encodes two isoforms JNK-1α and JNK-1β. A null allele, jnk-1(gk7), yielded worms with defective body movement coordination and modest mechanosensory deficits. Similarly to jkk-1 mutants, elimination of GABAergic signals suppressed the jnk-1(gk7) locomotion defect. Like mek-1 nulls, jnk-1(gk7) showed copper and cadmium hypersensitivity. Conditional expression of JNK-1 isoforms rescued these defects, suggesting that they are not due to developmental errors. While jkk-1 or mek-1 inactivation mimicked jnk-1(gk7) locomotion and heavy metal stress defects, respectively, mkk-4 inactivation did not, but rather yielded defective egg laying. Our results delineate at least two different JNK pathways through jkk-1 and mek-1 in C.elegans, and define interaction between MKK7, but not MKK4, and JNK.
Keywords: body movement/Caenorhabditis elegans/heavy metals/jkk-1/mek-1
Introduction
Mitogen-activated protein kinases (MAPKs) are components of signaling cascades that regulate normal development, mitogenesis and stress responses (Robinson and Cobb, 1997). Further, these pathways are co-opted during oncogenic transformation and tumor progression (Kyriakis and Avruch, 1996). Three subgroups of MAPKs have been identified: the extracellular signal-regulated kinase (ERK) (Davis, 1993), c-Jun N-terminal (JNK also referred to as stress-activated protein kinase) (Derijard et al., 1994) and p38 kinases (also called MpK2) (Han et al., 1994). MAPKs are activated via dual phosphorylation of threonine and tyrosine residues in a Thr–X–Tyr motif present within their activation loops (kinase subdomain VIII). X corresponds to glutamic acid in ERK, proline in JNK and glycine in p38 (Derijard et al., 1994; Kyriakis et al., 1994). The specific MAPK kinases (MAPKKs or MEKs) that carry out this reaction are themselves phosphorylated and activated by specific MAPKK kinases (MAPKKKs). In mammalian cells, evidence suggests that two MAPKKs, MKK4 and MKK7, activate JNK. While MKK4 also functions as an activator of p38, MKK7 probably functions as a specific activator of JNK (Cavigelli et al., 1995; Derijard et al., 1995; Tournier et al., 1997).
The ERK cascade is highly responsive to signals initiated by the binding of growth factors, such as epidermal growth factor or platelet-derived growth factor, to their tyrosine kinase receptors (Marshall, 1995; Whitmarsh et al., 1995). JNK activity is strongly stimulated in vertebrate cell culture by inhibitors of protein biosynthesis, such as cycloheximide and anisomycin, by inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and by heat, osmotic shock, UV light or other DNA-damaging agents (Derijard et al., 1994; Cavigelli et al., 1995; Coso et al., 1995). When JNK is activated, it migrates from the cytoplasmic compartment into the nucleus and stimulates the activity of several transcription factors including c-Jun, ATF-2, ELK-1 and p53 (Hibi et al., 1993; Derijard et al., 1994; Cavigelli et al., 1995; Gupta et al., 1995; Whitmarsh et al., 1995; Fuchs et al., 1998).
Although a substantial body of biochemical and cell biological evidence supports this model, genetic proof remains incomplete. Based on the current hypothesis, it is anticipated that JNK knockouts in developmental models would represent a composite of the cell type-specific expression of MKK4 and MKK7. The best defined system is that of Drosophila. Conserved elements of the Drosophila JNK pathway include D-jun, basket (bsk) and hemipterous (hep), which encode homologs of c-Jun, JNK and MKK7, respectively (Glise et al., 1995; Riesgo-Escovar et al., 1996; Sluss et al., 1996; Noselli, 1998). These gene products are all required for the normal morphogenetic process of dorsal closure beginning 12 h after fertilization. Biochemical evidence shows that HEP phosphorylates D-JNK, which in turn phosphorylates D-JUN (Noselli, 1998). Further support for these genes acting in a linear pathway is derived from epistasis studies which show that activated D-Jun can rescue the dorsal closure defect caused by hep and bsk. In contrast, the Drosophila p38 MAPKs (D-p38a and -b) may be involved in insect immunity, as well as in the response to environmental stresses (Han et al., 1998). Drosophila also contains a homolog of MKK4, D-mkk4, for which mutations have not yet been isolated. Whatever its role in development, it is clear that Drosophila MKK4 is not sufficient to substitute for MKK7 and prevent the lethal dorsal closure phenotype (Noselli, 1998).
Recent investigations in the mouse have extended our understanding of the role of the JNK pathway in development. The mammalian JNK protein kinases are encoded by three genes, which are alternatively spliced to create 10 JNK isoforms. While jnk1 and jnk2 are ubiquitously expressed, jnk3 has a limited pattern of expression and is largely restricted to brain, heart and testis (Davis, 2000). Disruption of the jnk1 (Dong et al., 1998), jnk2 or jnk3 (Yang et al., 1998) genes in mice causes no obvious phenotypic abnormalities. However, jnk1–/– or jnk2–/– mice are immunodeficient due to defects in T-cell function (Constant et al., 2000), and jnk3–/– animals are defective in stress-induced AP-1 transcriptional activity in the hippocampus (Yang et al., 1997b). Mice with compound deletion in jnk1 plus jnk3 or jnk2 plus jnk3 alleles are also viable (Kuan et al., 1999). In contrast, jnk1–/–/jnk2–/– double mutant animals display dysregulated developmental apoptosis in the brain and die by day E11.5 (Kuan et al., 1999).
In mice, both mkk4/sek1 (Yang et al., 1997a) and mkk7 (Dong et al., 2000) are required for embryonic development. While a mkk4/sek1 deficiency in mice results in early embryonic lethality due to defective hepatogenesis (Ganiatsas et al., 1998), the cause of embryonic death in mkk7–/– mice is unclear (Dong et al., 2000). As mkk4/sek1–/– mice do not manifest the central nervous system (CNS) abnormalities observed in jnk1–/–/jnk2–/– mice (Lenord Zon, personal communication), normal CNS development probably requires MKK7. Consistent with the observation that deficiency of MKK7 or JNK delivers similar phenotypes, Flavell and co-workers reported that T cells from both mkk7–/– and jnk1–/–/jnk2–/– mutant mice display increased IL-2 production and proliferation after anti-CD3/CD28, and stimulated mkk7–/– T cells display low JNK activity (Dong et al., 2000). Further, mkk7–/– mice did not manifest defective hepatogenesis, and mkk4–/– T cells displayed normal JNK activation and defective IL-2 production after anti-CD3/CD28 (Dong et al., 2000). These developmental models suggest association of JNK with MKK7 signaling but not with MKK4.
In Caenorhabditis elegans, jnk-1, a jnk homolog, mek-1, which is highly homologous to mammalian mkk7, and jkk-1, a new member of the MAPKK superfamily which has 41.6% amino acid identity with MKK7 in the kinase domain, have recently been identified (Kawasaki et al., 1999; Koga et al., 2000). While jnk-1 and jkk-1 are co-expressed in the cell bodies and the axons of most neurons (Kawasaki et al., 1999), mek-1 is expressed in pharyngeal muscles, uterus, a portion of intestine and neurons in the ring, ventral and anal ganglia (Koga et al., 2000). Although jkk-1 or mek-1 mutants did not show developmental defects, jkk-1 disruption altered coordination of body movement via type-D GABAergic motor neurons (Kawasaki et al., 1999), while mek-1 disruption resulted in hypersensitivity to heavy metals and starvation (Koga et al., 2000). The C.elegans genome also contains mkk-4 (corresponding to the cosmid F42G10.2), which is highly homologous to mammalian mkk4/sek1, about which nothing presently is known.
To determine whether JNK-1 acted genetically distal to these gene products, we isolated a jnk-1 null allele, jnk-1(gk7). Worms homozygous for this mutant allele did not show obvious development defects. However, jnk-1(gk7), showed defects in body movement coordination similar to jkk-1(km2) and, like mek-1(ks54), were hypersensitive to copper and cadmium ions. In contrast, inactivation of mkk-4 in N2 or jnk-1(gk7) worms resulted in an egg-laying defect in adult hermaphrodites. Our results delineate at least two different JNK signaling pathways through jkk-1 and mek-1 in C.elegans. Further, three distinct genetic models, C.elegans, Drosophila and mice, now define interaction between MKK7, but not MKK4, and JNK.
Results
Caenorhabditis elegans jnk-1 encodes two different transcripts generated by SL1 trans-splicing
The mammalian JNK genes, jnk1, jnk2 and jnk3 encode at least 10 different JNK isoforms (four JNK1, four JNK2 and two JNK3) generated by two different mechanisms: alternative cis-splicing that yields different C-termini or the utilization of alternate sequences within protein kinase subdomains IX and X (Gupta et al., 1996). Recently, C.elegans jnk-1 has been cloned (Kawasaki et al., 1999). It is localized to the left arm of chromosome IV. To determine whether jnk-1 encodes different isoforms or whether C.elegans contains related genes, we reverse transcribed total RNA from mixed stages N2 animals to cDNA, applied the rapid amplification of cDNA ends (RACE) technique, and detected and sequenced two transcripts (Figure 1A and B). Both transcripts contained 5′ SL1 trans-splicing leader sequences (Bektesh et al., 1988), defining the limits of their 5′ ends. In jnk-1α, the SL1 leader sequence was trans-spliced to exon 1, whereas in jnk-1β, trans-splicing occurred at the beginning of exon 2. The larger transcript jnk-1α is identical to the transcript of jnk-1 reported by Kawasaki and co-workers (Kawasaki et al., 1999), and contains an open reading frame coding for a 463 amino acid protein (Figure 2). The smaller transcript jnk-1β is predicted to use an in-frame ATG in exon 3 (corresponding to amino acid 92 of JNK-1α) as the translation start site, coding for a protein of 372 amino acids (Figures 1A and B, and 2). The 91 amino acid N-terminal sequence that distinguishes JNK-1α from JNK-1β is unique, while the amino acid sequence comprising JNK-1β is conserved almost entirely in human JNK1 (70% identity, 83% homology; Figure 2). Both JNK-1 isoforms contain the conserved protein kinase subdomains I–XI found in all kinases, as well as the TPY motif located in subdomain VIII of all JNKs (Galcheva-Gargova et al., 1994; Kyriakis et al., 1994; Gupta et al., 1996). Cis-spliced isoforms that might yield different C-termini in the jnk-1 gene were not detected by RACE, nor were alternative sequences in kinase subdomains IX and X detected by RT–PCR/single strand conformation polymorphism (SSCP). In both jnk-1 transcripts, the polyadenylation consensus site (AATAAA) was detected, indicating that we cloned complete cDNAs.

Fig. 1. jnk-1 encodes two different transcripts. (A) Structure of C.elegans jnk-1. Exons are indicated by boxes, while introns are represented as lines. Numbers refer to amino acids. RACE was performed using two different sets of primers as described in Materials and methods. In 11/15 clones (using primers based on sequences from exons 7 and 8) and in 6/10 clones (using primers from exon 12), we detected two transcripts, which we termed jnk-1α and jnk-1β containing 5′ SL1 trans-splicing leader sequences (Bektesh et al., 1988). For jnk-1α, the SL1 trans-spliced sequence was followed by exon 1, whereas for jnk-1β the SL1 trans-spliced sequence was followed by exon 2. The translation initiation site, ATG (in bold and italicized) of jnk-1α is located in exon 1 while the translation initiation site of jnk-1β is located in exon 3 (in bold and marked by a box). JNK-1α has a 91 amino acid extended N-terminal region (in gray), fused in-frame with 372 amino acid residues (in black) that are identical in JNK-1α and JNK-1β. Poly(A), polyadenylation site; SL1, trans-splicing leader sequence; E, EcoRI and H, HindIII restriction sites; NH2, N-terminus; COOH, C-terminus. (B) Nucleotide sequence of the 5′ regions of jnk-1α and jnk-1β transcripts. SL1 trans-splicing leader sequences (underlined and italicized) are located at the beginning of both jnk-1 transcripts. Non-translated sequences are in lower case and translated sequences are in upper case. (C) Southern blot analysis using full-length [α32-P]dCTP-labeled jnk-1α cDNA as a probe shows a pattern of DNA restriction compatible with that predicted from cosmid B0478.

Fig. 2. Primary structure of the C.elegans JNK-1α and JNK-1β proteins. Alignment of amino acid sequences of human JNK1 and JNK3 (DDBJ/EMBL/GenBank accession Nos L26318 and U34820, respectively), Drosophila D-JNK (DDBJ/EMBL/GenBank accession No. U49180), and JNK-1α (DDBJ/EMBL/GenBank accession No. AB024085) and JNK-1β protein kinases using the PILE-UP program. Identical amino acid residues are highlighted and similar residues are shaded. A consensus sequence is presented. The conserved TPY motif is marked with asterisks and the kinase subdomains are marked with Roman numerals. Gaps (–) were introduced into the sequences to optimize alignment. Note that not all groups have reported the long N-terminus of JNK3 (Kyriakis et al., 1994).
Screening the C.elegans database using the BLAST program did not identify any other potential jnk genes. Furthermore, Southern blot analysis of C.elegans genomic DNA hybridized with full-length jnk-1α cDNA at high and low stringency yielded a pattern of digestion identical to that predicted from the B0478 cosmid, which contains the jnk-1 gene (Figure 1C). Together, these results suggest that C.elegans generates two JNK-1 isoforms by alternative transcription or translation of a single jnk gene.
jnk-1 is expressed throughout development
To determine the RNA expression pattern of jnk-1 during development, northern blot analysis was carried out using membranes containing total RNA from embryos, L1–L2, L3, L4 and young adult worms. High stringency hybridization, using the complete coding region of the jnk-1β cDNA as a probe, showed a single band of 1.5 kb (Figure 3A). It should be noted that this probe recognizes both jnk-1 isoforms, which differ by only 82 bp and hence cannot be distinguished on a northern blot. The jnk-1 message was most abundant during embryonic (E) and L1/L2 stages, and decreased gradually from early L3 larva to the young adult worm (Figure 3A). The same result was obtained using the 82 bp fragment only present in jnk-1α, as a probe (not shown). In both cases, the transcript size was the same as that predicted by the sizes of the cloned cDNAs, confirming that full-length cDNAs had been isolated. Semi-quantitative RT–PCR using primers that selectively amplify the individual isoforms confirmed that both isoforms had abundant RNA expression during embryonic and L1 stages (Figure 3B). To identify the individual JNK-1 isoforms, we performed western analysis on C.elegans lysates using a monoclonal anti-human JNK1 antibody. The antibody recognized two bands of the expected sizes for JNK-1α (46 kDa) and JNK-1β (38 kDa) (Figure 3C). To provide evidence that the bands represent bona fide isoforms rather than a single form post-translationally modified, we showed that the migration pattern of the endogenous JNKs corresponded to the Flag-tagged versions from COS-7 cells. (The small size difference between endogenous C.elegans isoforms and epitope-tagged proteins is due to 25 amino acids included in the cloning process.) A close correlation between mRNA and protein levels at L1 to adult stages was detected using this antibody (not shown). However, at the embryo stage, low levels of protein were detected, suggesting that post-transcriptional or post-translational mechanisms regulate JNK-1 levels in embryos.

Fig. 3. Expression pattern of jnk-1 during development. (A) Northern blot and (B) RT–PCR analysis using total RNA from different C.elegans developmental stages. Northern blot was probed with jnk-1β full-length cDNA and RT–PCR was performed using a specific set of primers that amplified both isoforms. Ethidium bromide staining was used to control for RNA quality and loading (lower panel). (C) Western blot analysis of 100 µg of total protein from mixed C.elegans stages using a monoclonal anti-human JNK1 antibody (sc-1648, Santa Cruz Biotechnology) shows expression of both JNK-1 proteins. Flag-tagged JNK-1α or -β, isolated by immunoprecipitation from COS-7 lysates using anti-FLAG monoclonal antibody, served as controls. E, embryos; L1–L2, L1 and L2 larvae; L3, L3 larvae; L4, L4 larvae; AD, young adult worms; MS, mixed stages; M, DNA molecular weight marker.
Transiently expressed JNK-1α and JNK-1β are activated in response to stress stimuli
Mammalian JNKs are activated in response to diverse stress stimuli (Derijard et al., 1994, 1995; Galcheva-Gargova et al., 1994; Kyriakis et al., 1994). To investigate activation of JNK-1 isoforms, COS-7 cells were transiently transfected with mammalian expression plasmids encoding Myc-tagged JNK-1α, Flag-tagged JNK-1β or vector alone. After 48 h, cells were left untreated or stimulated with UV light (40 J/m2), hyperosmotic shock (400 mM sorbitol), heat shock (42°C for 20 min), TNF-α (100 ng/ml) or lipopolysaccharide (LPS; 1 µg/ml). Epitope-tagged JNK-1α and JNK-1β were isolated by immunoprecipitation with anti-Myc and anti-FLAG antibodies, respectively, and kinase activity was determined in an immune complex kinase assay using glutathione S-transferase (GST)–c-Jun as substrate. These stimuli similarly activated both JNK-1α (not shown) and JNK-1β (Figure 4A, top panel). Similar results were obtained with γ-radiation (20 Gy) or H2O2 (75 µM) (not shown). In parallel, a control experiment was performed in cells transfected with human hemagglutinin (HA)-tagged JNK1 and yielded quantitatively similar results (not shown). Western blot analysis of COS-7 cells extracts showed that the epitope-tagged proteins were expressed at comparable levels (Figure 4A, lower panel).

Fig. 4. Activation of C.elegans JNK-1 proteins by various stress stimuli and by co-expressed mouse MKK4/SEK1 or C.elegans MEK-1. (A and B) COS-7 cells were transfected with plasmid encoding Flag-tagged JNK-1β. After 48 h, cells were left unstimulated or stimulated for 20 min as indicated. Epitope-tagged proteins were isolated by immunoprecipitation using anti-FLAG monoclonal antibody, and protein kinase activity measured in an immune complex kinase assay using GST–c-Jun (A), or GST–ATF2 or GST–ELK-1 (B), as substrates. (C and D) COS-7 cells were co-transfected with plasmids encoding (C) Flag-tagged JNK-1β and mouse GST-tagged MKK4/SEK1 or dominant-negative GST-tagged MKK4/SEK1(K129R) (Verheij et al., 1996), or (D) Flag-tagged JNK-1α and C.elegans HA-tagged MEK-1 or dominant-negative HA-tagged MEK1(K99R). Flag-JNK-1α or -β were immunoprecipitated, and protein kinase activity was measured as above using GST–c-Jun as a substrate. In all experiments, an aliquot of each sample was analyzed by western blot to confirm equal levels of expression of the epitope-tagged proteins.
The different human JNK isoforms can phosphorylate c-Jun, ATF-2 and ELK-1 transcription factors (Gupta et al., 1995, 1996; Jacobs et al., 1999). In a separate set of studies, both C.elegans JNK-1 isoforms also phosphorylated human recombinant GST–ATF-2 and GST–ELK-1, when supplied as substrates (Figure 4B, not shown for JNK-1α).
Caenorhabditis elegans JNK-1 isoforms are activated by phosphorylation on residues corresponding to Thr183 and Tyr185 of mammalian JNK1
In vertebrates and Drosophila, evidence suggests that JNK is activated by two MAPKKs, MKK7 and MKK4/SEK1, in response to diverse stimuli (Derijard et al., 1995; Tournier et al., 1997). Recently, two MAPKKs, mek-1 (Koga et al., 2000) and jkk-1 (Kawasaki et al., 1999), have been cloned in C.elegans and a third, mkk-4, has been identified (corresponding to cosmid F42G10.2). mek-1 and jkk-1 encode two distinct MKK7 homologs. MEK-1 shows 53.5% amino acid identity in the kinase domain to MKK7, while JKK-1 shows 41.6% identity (Koga et al., 2000). JKK-1 specifically stimulates the kinase activity of JNK-1α but not C.elegans p38 in transfected 293 cells (Kawasaki et al., 1999). These data are consistent with the notion that JKK-1 is a MAPKK specific for JNK.
To investigate whether MKK4/SEK1 might also activate C.elegans JNK-1 isoforms, we transiently transfected COS-7 cells with plasmids encoding Myc-tagged JNK-1α or Flag-tagged JNK-1β either alone, or in combination with mouse GST-tagged MKK4/SEK1 or dominant-negative kinase-inactive GST-tagged MKK4/SEK1(K129R) (Verheij et al., 1996). As shown in Figure 4C, wild-type GST–MKK4/SEK1 activated Flag-JNK-1β in the absence of any stress stimuli, while dominant-negative GST– MKK4/SEK1 was not activating. Similar results were obtained with Myc-tagged JNK-1α (not shown). COS-7 cells were also co-transfected with expression vectors encoding C.elegans HA-tagged MEK-1 and Flag-tagged JNK-1α or -β. Co-expression with HA-MEK-1 resulted in strong Flag-JNK-1α activation (Figure 4D). Consistent with a requirement for the kinase function of MEK-1, expression of kinase-inactive HA-tagged MEK-1(K99R) did not result in Flag-JNK-1α activation. Similar results were obtained with Flag epitope-tagged JNK-1β (not shown).
Mammalian JNK proteins are activated by dual phosphorylation on threonine and tyrosine residues. In particular, JNK1 is phosphorylated on Thr183 and Tyr185, located within its activation loop (kinase subdomain VIII) (Galcheva-Gargova et al., 1994). To investigate whether the corresponding residues are also phosphorylated in C.elegans JNK-1 isoforms in response to stress stimuli, we transiently transfected COS-7 cells with plasmids encoding Flag-tagged JNK-1β or mutant Flag-tagged JNK-1β(AF) lacking the requisite phosphorylation sites. Kinase activity measured in an immune complex assay using GST–c-Jun as substrate showed that substitution of amino acids Thr185 and Tyr187 (corresponding to human JNK1 Thr183 and Tyr185) for alanine and phenylalanine blocked the activation of JNK-1β in response to UV light (Figure 5). Western blot analysis using an anti-human phospho-JNK (Thr183/Tyr185) antibody (New England Biolabs) that also recognizes the phosphorylated form of C.elegans JNK-1 confirmed that phosphorylated isoforms were only present in immunoprecipitates obtained from stimulated cells, or from cells co-expressing GST-tagged MKK4/SEK1 or HA-tagged MEK-1 (not shown). Similar results were obtained using Myc-tagged JNK-1α and mutant Myc-tagged JNK-1α (AF) (not shown).
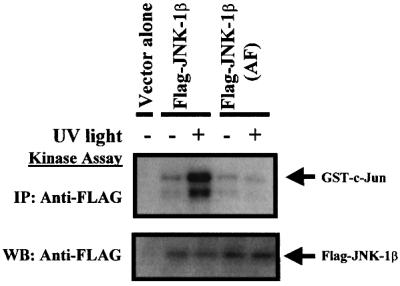
Fig. 5. JNK-1α and JNK-1β are activated by dual threonine and tyrosine phosphorylation within the conserved TPY JNK domain. COS-7 cells were transfected with plasmids encoding wild-type Flag-tagged JNK-1β or mutated Flag-tagged JNK-1β(AF) (where amino acids Thr185 and Tyr187 corresponding to human JNK1 Thr183 and Tyr185 were substituted with alanine and phenylalanine, respectively). After 48 h, cells were left unstimulated (–), or stimulated (+) for 20 min with UV light. Flag-JNK-1β was immunoprecipitated using anti-FLAG antibody, and protein kinase activity measured in an immune complex kinase assay. In all experiments, an aliquot of each sample was analyzed by western blot to confirm equal levels of expression of the epitope-tagged proteins.
Isolation of a jnk-1 loss-of-function allele
To investigate the physiological role of jnk-1, we undertook a reverse genetic approach to isolate loss-of-function mutations in the jnk-1 gene locus. Using a PCR-based screening strategy, we detected and isolated an individual worm with a deletion of 1186 bp (Figure 6A, right panel) involving exons 3–6 (Figure 6A, left panel), corresponding to nucleotides 30 128–31 314 of cosmid B0478. We refer to this deletion allele as jnk-1(gk7). This deletion eliminated sequences encoding amino acids 53–243 of JNK-1α and 1–182 of JNK-1β. In both cases, the deletion includes kinase subdomains I–VIA. RT–PCR using specific primers that amplified each mutant isoform confirmed the existence of two mutant transcripts in jnk-1(gk7), named jnk-1(gk7)α and jnk-1(gk7)β (Figure 6B). However, neither an anti-human monoclonal antibody (generated against the full-length human protein), which recognizes mammalian and C.elegans JNK1 isoforms, nor anti-human phospho-JNK (Thr183/Tyr185) detected any mutant JNK-1 isoforms in jnk-1(gk7) worm extracts. Even if a small amount of truncated JNK-1 were expressed, undetectable by our antibodies, it would probably be inactive as critical elements of the kinase domain are lacking. To address this issue directly, COS-7 cells were transiently transfected with plasmids encoding Myc-tagged JNK-1(gk7)α and JNK-1(gk7)β mutant isoforms and subjected to stress. The mutant Myc-JNK-1(gk7) isoforms failed to display any kinase activity in an immune complex kinase assay [Figure 6C; not shown for Myc-tagged JNK-1(gk7)β], despite adequate protein expression (not shown). We conclude that jnk-1(gk7) is a loss-of-function allele for the jnk-1 gene, resulting in loss of both JNK-1α and JNK-1β kinase activities.

Fig. 6. Structure of the mutant jnk-1(gk7)α and jnk-1(gk7)β transcripts generated from the jnk-1(gk7) allele. (A) Exons are indicated by boxes, while introns are represented as lines. DNA sequencing of the PCR product from jnk-1(gk7) showed an in-frame deletion of 1186 bp involving exons 3–6 (right panel), corresponding to nucleotides 30 128–31 314 of the cosmid B0458 (left panel). Sequences encoding JNK-1α amino acids 53–243 and JNK-1β amino acids 1–182 are deleted, respectively, in the gk7 mutant. (B) RT–PCR using specific primers that amplified each mutant isoform confirms the existence of two mutant transcripts expressed in jnk-1(gk7). (C) COS-7 cells were transfected with the plasmid encoding mutated Myc-tagged JNK-1(gk7)α and, after 48 h, cells were left unstimulated or stress stimulated for 20 min, as indicated. Myc-JNK-1(gk7)α was immunoprecipitated and protein kinase activity measured in an immune complex kinase assay using GST–c-Jun. The positive control is from COS-7 cells expressing wild-type Myc-JNK-1β stimulated with UV light. M, DNA molecular weight marker. The other abbreviations are as in Figure 1.
Loss of jnk-1 function results in body movement and mechanosensory defects
Wild-type C.elegans moves by propagating waves of alternating dorsal and ventral flexions along its body length, producing regular sinusoidal tracks on a bacterial lawn. Similarly to jkk-1(km2) knockout worms (Kawasaki et al., 1999), jnk-1(gk7) mutants left tracks with increased wave amplitude, approximately double that of a wild-type N2 animal (Table I; Figure 7A). The wavelength, however, was unchanged. The locomotion of jnk-1(gk7) animals was slightly sluggish and hence the distance covered during a fixed time (5 min) was reduced substantially. The jnk-1 knockout worms seldom moved in a straight trajectory and instead moved in small circles, while wild-type worms covered straight long distances (Figure 7B). Compatible with the notion that both kinases function in the same pathway, jnk-1(gk7);jkk-1(km2) manifested movement defects equivalent to those of either jnk-1(gk7) or jkk-1(km2).
Table I. Behavioral analysis.
| Locomotion assays | |||
|---|---|---|---|
| Strain | Amplitude | Wavelength | Locomotion |
| Wild type (N2) | 0.10 ± 0.04 (40) | 0.42 ± 0.02 (40) | + |
| jnk-1(gk7) | 0.22 ± 0.02 (40) | 0.48 ± 0.05 (40) | slightly sluggish |
| jnk-1(gk7)[Myc-JNK-1α]a | 0.14 ± 0.02 (20) | 0.43 ± 0.05 (20) | + |
| jnk-1(gk7)[Flag-JNK-1β]b | 0.12 ± 0.03 (20) | 0.44 ± 0.04 (20) | + |
| jnk-1(gk7)[vector alone]c | 0.23 ± 0.03 (10) | 0.44 ± 0.03 (10) | slightly sluggish |
| unc-25(e156) | 0.09 ± 0.04 (20) | 0.36 ± 0.04 (20) | shrinkerd |
| jnk-1(gk7);unc-25(e156) | 0.12 ± 0.02 (20) | 0.48 ± 0.05 (20) | shrinker |
| Sensory responses | |||||
|---|---|---|---|---|---|
| Strain | Osmotic avoidance escape time (min) | Foraging | Defecation (s) | Light nose touch (% responding) | Harsh body touch (% responding) |
| Wild type (N2) | 9 ± 1 (100, 5) | + | 44 ± 2 (10, 10) | 80 ± 5 (20, 10) | 85 ± 2 (20, 10) |
| jnk-1(gk7) | 8 ± 1 (100, 5) | slow | 45 ± 3 (10, 10) | 44 ± 6 (20, 10) | 51 ± 5 (20, 10) |
| jnk-1(gk7)[Myc-JNK-1α]a | ND | + | ND | 75 ± 5 (10, 10) | 80 ± 4 (10, 10) |
| jnk-1(gk7)[Flag-JNK-1β]b | ND | + | ND | 71 ± 5 (10, 10) | 82 ± 2 (10, 10) |
| jnk-1(gk7)[vector alone]c | ND | slow | ND | 47 ± 4 (10, 10) | 58 ± 3 (10, 10) |
| Volatile chemical odorants and salt chemotaxis assays (% responding) | |||||||
|---|---|---|---|---|---|---|---|
| Strain | Chemical odorantse |
Salts |
|||||
| Alcohol (isoamyl alcohol) | Ketone (acetone) | Ester (ethyl acetate) | Aromatic compound (benzonitrile) | Ether (diethy ether) | NH4Cl | NaCl | |
| Wild type (N2) | 95 | 72 | 77 | 69 | 71 | + | + |
| jnk-1(gk7) | 90 | 77 | 70 | 72 | 76 | + | + |
ND, not determined.
Errors indicate SEM. The number and trials per animal are shown in parentheses.
Young adult jnk-1(gk7) harboring as extrachromosomal arrays: apPD49.78-Myc-JNK-1α and pPD49.83-Myc-JNK-1α; bpPD49.78-Flag-JNK-1β and pPD49.83-Flag-JNK-1β; and cpPD49.78 and pPD49.83 as control plasmids, were heat treated for 30 min at 33°C, and individual worms were placed on NGM plates seeded with E.coli at 20°C. Animals were monitored every 4 h to select worms showing a wild-type movement pattern. After 24 h, amplitude, wavelength and locomotion were scored. Amplitude and wavelength were measured in millimeters. Transgenic animals exhibited the same locomotion defects and modest reductions in mechanosensory responses in the absence of heat as the parental strain.
dAn animal was scored as a shrinker if it failed to move backwards and decreased in body length (McIntire et al., 1993a,b).
eChemical odorant and salt chemotaxis studies were performed as described (Culotti and Russell, 1978; Bargmann et al., 1993).

Fig. 7. jnk-1(gk7) mutants worms exhibit defects in coordination of body movement. Single young adult N2 and jnk-1(gk7) worms were spotted in the center of NGM plates and wave forms (A) and trajectory (B) evaluated at 5 min. Tracks inscribed in the bacterial lawn (B) were traced using a black pen.
jnk-1(gk7) worms also displayed modest reductions in some mechanosensory responses: light touch to the nose was 45% reduced and harsh touch to the body 40% reduced when compared with N2 worms (Table I). Consistent with the concept that touch responses through JNK-1 were regulated via a JKK-1 independent mechanism, abnormalities in touch responses were not detected in jkk-1(km2), while jnk-1(gk7);jkk-1(km2) still displayed the modest reductions in mechanosensory responses observed in jnk-1(gk7) (not shown).
In contrast, jnk-1(gk7) worms manifested normal responses to a variety of attractive and repulsive water-soluble and volatile compounds (Table I). Indeed, no changes in the uptake of the lipophilic dye DiO by the amphid and phasmid neurons were detected (not shown), suggesting that the anatomy of these sensory structures is normal in jnk-1 mutants.
Other behaviors including pharyngeal pumping, egg laying, dauer formation, male mating and defecation were also normal in the jnk-1(gk7) strain. Consistent with the proposed loss-of-function nature of the jnk-1(gk7) deletion, we found that jnk-1(gk7)/+ heterozygous animals did not show any of the behavioral defects observed in the homozygous mutant animals (not shown).
Conditional overexpression of JNK-1α or JNK-1β reverses movement and mechanosensory response defects in jnk-1(gk7)
To determine whether the defects observed in jnk-1(gk7) mutants are due to abnormal development or cell function, we generated transgenic animals in the jnk-1(gk7) background harboring, as extrachromosomal arrays, plasmids encoding Myc epitope-tagged JNK-1α or Flag-tagged JNK-1β under control of the heat shock promoters hsp16-2 and hsp16-41. These promoters direct protein expression in the majority of tissues, including neurons (Stringham et al., 1992; Mello and Fire, 1995). Transgenic animals exhibited the same locomotion defects and modest reductions in mechanosensory responses in the absence of heat treatment as the parental strain. When heat treated at the young adult stage, movement defects (wave amplitude, locomotion and trajectory) and mechanosensory responses (light touch to the nose and harsh body touch) were rescued by either isoform within 12–24 h (Table I), demonstrating that these defects are not due to developmental abnormalities, but rather to defects in functions regulated by JNK-1 protein isoforms. It should be noted that we did not detect low levels of jnk-1 cDNA by RT–PCR in the absence of heat (not shown). In contrast, heat treatment of worms carrying empty vector did not rescue the mutant phenotype (Table I).
Increased amplitude phenotype in jnk-1(gk7) mutants requires GABAergic synaptic transmission
Recently, it has been demonstrated that JNK-1, like JKK-1, is expressed in most or all neurons and their processes, including the nerve ring, head ganglion, dorsal and ventral nerve cords and tail ganglion (Kawasaki et al., 1999). Caenorhabditis elegans body wall muscle contraction depends on a balance between inhibitory GABAergic inputs from the DD and VD neurons (McIntire et al., 1993a,b) and excitatory cholinergic inputs (Lewis et al., 1980). The six DD neurons (which innervate the dorsal body wall muscles) and the 13 VD neurons (which innervate the ventral body wall muscles) coordinate muscle relaxation during locomotion. Animals in which the DD and VD neurons have been destroyed by laser ablation, or genetic mutations in the genes unc-25, unc-30, unc-46, unc-47 and unc-49, which regulate GABA synthesis/action (McIntire et al., 1993a; Eastman et al., 1999), manifest simultaneous contraction of antagonistic dorsal and ventral body muscles, causing these animals to ‘shrink’ (White et al., 1976). To investigate the genetic relationship between unc-25 (which encodes glutamic acid decarboxylase), the GABA biosynthetic enzyme (Jin et al., 1999) and jnk-1, an unc-25(e156);jnk-1(gk7) double mutant was constructed. Similarly to results obtained with unc-25(e156);jkk-1(km2) worms (Kawasaki et al., 1999), loss of unc-25 function reversed the jnk-1(gk7) movement phenotype; the unc-25(e156);jnk-1(gk7) double mutant transversed the bacterial lawn in a manner similar to unc-25(e156) single mutants, and the amplitude of the wave of motion was normalized (Table I). In order to investigate if other genes involved in the GABA signaling pathway were also genetically related to jnk-1, we used double-stranded RNA-mediated interference (RNAi) (Fire et al., 1998) to inactivate unc-25, unc-30 (which encodes a homeobox gene that controls the fate of 19 type-D GABAergic neurons) (Eastman et al., 1999), and unc-47 (which encodes a vesicular GABA transporter) (McIntire et al., 1997). unc-25, unc-30 or unc-47 inactivation by RNAi resulted in a shrinker phenotype in both wild-type and jnk-1(gk7) animals, indicating that RNAi was an effective technique for inactivation of these genes (Table II). Further, inactivation of unc-30 or unc-47, like unc-25, reversed the jnk-1(gk7) movement defect, whereas injection of a non-specific dsRNA for green fluorescent protein (GFP), was without effect. These investigations provide additional support for the concept that the locomotion defect resulting from loss of JNK-1 function is mediated through a GABAergic pathway. Unfortunately, unc-49 (which encodes three distinct GABAA receptor-like subunits) (Bamber et al., 1999), like other genes in the nervous system (Tavernarakis et al., 2000), could not be inactivated by RNAi, and thus is not included in this analysis.
Table II. Inactivation of GABAergic regulatory genes in jnk-1(gk7) rescues the locomotion defect.
| RNAi | Strain | Locomotion assays |
Sensory responses |
|||
|---|---|---|---|---|---|---|
| Locomotion | Amplitude | Wavelength | Light nose touch (% responding) | Harsh body touch (% responding) | ||
| Uninjected | wild type (N2) | + | 0.11 ± 0.03 (15) | 0.45 ± 0.04 (15) | 84 ± 4 (15, 10) | 87 ± 4 (15, 10) |
| Uninjected | jnk-1(gk7) | slightly sluggish | 0.24 ± 0.04 (15) | 0.46 ± 0.04 (15) | 42 ± 5 (15, 10) | 53 ± 3 (15, 10) |
| unc-30 | wild type (N2) | shrinkera | 0.12 ± 0.04 (20) | 0.42 ± 0.04 (20) | 85 ± 3 (20, 10) | 84 ± 3 (20, 10) |
| unc-30 | jnk-1(gk7) | shrinkera | 0.14 ± 0.03 (20) | 0.43 ± 0.03 (20) | 47 ± 4 (20, 10) | 53 ± 5 (20, 10) |
| unc-47 | wild type (N2) | shrinkera | 0.09 ± 0.03 (20) | 0.43 ± 0.03 (20) | 82 ± 3 (20, 10) | 85 ± 4 (20, 10) |
| unc-47 | jnk-1(gk7) | shrinkera | 0.12 ± 0.03 (20) | 0.46 ± 0.05 (20) | 48 ± 6 (20, 10) | 55 ± 4 (20, 10) |
| unc-25 | wild type (N2) | shrinkera | 0.10 ± 0.03 (20) | 0.40 ± 0.03 (20) | 86 ± 4 (20, 10) | 85 ± 2 (20, 10) |
| unc-25 | jnk-1(gk7) | shrinkera | 0.15 ± 0.04 (20) | 0.45 ± 0.04 (20) | 48 ± 5 (20, 10) | 56 ± 4 (20, 15) |
| gfp | wild type (N2) | + | 0.10 ± 0.03 (10) | 0.44 ± 0.02 (10) | 83 ± 3 (10, 10) | 86 ± 5 (10, 10) |
| gfp | jnk-1(gk7) | slightly sluggish | 0.23 ± 0.03 (10) | 0.47 ± 0.04 (10) | 43 ± 4 (10, 10) | 53 ± 4 (10, 10) |
Errors indicate SEM. The number and trials per animal are shown in parentheses. Amplitude and wavelength were measured in millimeters.
aAn animal was scored as a shrinker if it failed to move backwards and decreased in body length (McIntire et al., 1993a,b).
The modest mechanosensory deficits reported above, however, could still be observed in the unc-25 (e156);jnk-1(gk7) double mutant (not shown), as well as in jnk-1(gk7) worms subjected to unc-25, unc-30 or unc-47 RNAi (Table II). These results suggest that JNK-1 proteins are also involved in the regulation of functions independent of GABA action in C.elegans.
jnk-1(gk7) are hypersensitive to heavy metal stress
There is little genetic evidence validating the role of the JNK signaling pathway in stress response signaling in whole animals. Recently, it has been shown that the C.elegans MKK7 homolog MEK-1 is involved in some forms of stress response (Koga et al., 2000). Similarly to the mek-1(ks54) null allele, we found that jnk-1(gk7) mutants were also hypersensitive to copper and cadmium ions (Figure 8A and B). N2 animals grew well, becoming adults by 4 days in plates containing low concentrations of copper (Cu; <60 µM) or cadmium (Cd; <10 µM), while jnk-1(gk7) animals grew poorly. In fact most of the animals failed to reach the adult stage within 4 days. At high doses of Cu (100 µM) or Cd (20–30 µM), some N2 animals were viable whereas virtually all jnk-1(gk7) animals were arrested at young larvae stages. Further more, jnk-1(gk7);mek-1(ks54) displayed heavy metal sensitivity identical to that detected in mek-1(ks54) or jnk-1(gk7) (Figure 8A and B), suggesting that these kinases act in a common pathway. jnk-1(gk7); mek-1 (ks54), like jnk-1(gk7);jkk-1(km2), continued to display the modest reductions in touch responses observed in jnk-1(gk7) (not shown).

Fig. 8. jnk-1(gk7) mutant worms show similar hypersensitivity to heavy metals as mek-1(ks54). Effect of copper sulfate (CuSO4) (A) and cadmium chloride (CdCl2) (B) in N2, jnk-1(gk7), mek-1(ks54), jkk-1(km2), jnk-1(gk7);mek-1(ks54) and jnk1(gk7);jkk-1(km2). Toxicity assays were performed as described by Koga et al. (2000). Briefly, four worms were allowed to lay eggs in normal NGMSR plates containing the indicated concentrations of heavy metals. After 6 h, worms were removed, and the number of eggs counted. The percentage (± standard errors) of worms reaching adulthood 4 days after egg laying is compiled from three experiments.
To determine whether restoring JNK-1 activity in jnk-1(gk7) reverts the heavy metal sensitivity, transgenic jnk-1(gk7) animals were generated harboring Flag-JNK-1 isoforms under the control of heat shock promoters. After heat shock induction, copper and cadmium sensitivity in jnk-1(gk7) transgenic worms was significantly, but not completely, rescued (Figure 9A). While only 30–35% of jnk-1(gk7) survived at 60 µM copper, 65% of the transgenic worms survived. Similarly, only 20–25% of jnk-1(gk7) survived at 20 µM cadmium while 55% of the transgenic worms survived (Figure 9A).

Fig. 9. Rescue of jnk-1(gk7) heavy metal hypersensitivity. (A) Transgenic jnk-1(gk7) were generated harboring plasmids expressing Flag-tagged JNK-1 isoforms under the control of heat shock promoters, and pRF4 (dominant rol-6) plasmid as a selection marker. Young adult worms were heat treated for 30 min at 33°C, after 16 h allowed to lay eggs for 6 h onto plates containing cadmium (20 µM) and copper (60 µM), and survival assessed as in Figure 8. (B) Pmek-1::flag-jnk-1α or -β transgenes were co-injected with the plasmid pRF4 into jnk-1(gk7); progeny with a Roller phenotype were selected, and heavy metal sensitivity determined as in (A) (left panel). Expression of Flag-JNK-1α or -β in transgenic worms was confirmed at the end of these experiments by western blot (right panel). 1, jnk-1(gk7); 2, jnk-1(gk7)[pRF4]; 3, jnk-1(gk7)[pRF4; Pmek-1::flag-jnk-1α]; 4, jnk-1(gk7)[pRF4; Pmek-1::flag-jnk-1β].
To investigate whether jnk-1 expression in cells, where mek-1 is normally expressed, rescues the heavy metal sensitivity, Flag-JNK-1α or -β were expressed in jnk-1(gk7) under the control of the mek-1 promoter (Pmek-1). Pmek-1::flag-jnk-1α or -β transgenes were maintained as extrachromosomal arrays. As shown in Figure 9B, the increased sensitivity to copper (60 µM) and cadmium (20 µM) was partially rescued by expression of Flag-JNK-1α and -β proteins driven by the mek-1 promoter. Furthermore, after 4 days treatment with 10 µM cadmium, an increase in phospho-JNK-1 was detected in N2, while mek-1(ks54) showed a phospho-JNK-1 level similar to untreated N2 (not shown).
jnk-1(gk7) reproduces the loss-of-function phenotypes of jkk-1 and mek-1 but not that of mkk-4
Although biochemical evidence supports the model that two MAPK kinases, MKK4 and MKK7, mediate activation of JNK, genetic evidence supporting this model is incomplete. While jkk-1(km2) and mek-1(ks54) null alleles each show isolated defects detected in jnk-1(gk7), inactivation by RNAi of mkk-4, the C.elegans homolog of mammalian MKK4/SEK1, caused an egg-laying defect in N2 worms (Table III). No egg-laying defect was observed in jnk-1(gk7), jkk-1(km2), mek-1(ks54), jnk-1(gk7);jkk-1(km2) or jnk-1(gk7); mek-1(ks54). To investigate if the egg-laying defect observed in N2 worms where mkk-4 was inactivated occurs independently of jnk-1, we inactivated mkk-4 by RNAi in jnk-1(gk7). As in N2, inactivation of mkk-4 in a jnk-1-deficient worm still resulted in the egg-laying defect (Table III) further dissociating MKK-4 from JNK-1 in C.elegans.
Table III. In vivo effects of inactivation of jkk-1, mek-1 and mkk-4 genes by RNAi in wild-type and jnk-1(gk7) worms.
| RNAi | Strain | Development | Locomotion |
Touch responses |
% egg-laying defective |
|
|---|---|---|---|---|---|---|
| Amplitude | Light nose (% responding) | Harsh body (% responding) | ||||
| Uninjected | wild type (N2) | normal | 0.09 ± 0.03 (10) | 85 ± 3 (10, 10) | 84 ± 6 (10, 10) | 0 (150) |
| Uninjected | jnk-1(gk7) | normal | 0.22 ± 0.04 (10) | 46 ± 4 (10, 10) | 49 ± 3 (10, 10) | 0 (150) |
| jkk-1 | wild type (N2) | normal | 0.17 ± 0.02 (15) | 82 ± 6 (15, 10) | 83 ± 5 (15, 10) | 0 (150) |
| jkk-1 | jnk-1(gk7) | normal | 0.21 ± 0.02 (15) | 42 ± 5 (15, 10) | 53 ± 4 (15, 10) | 0 (150) |
| mek-1 | wild type (N2) | normal | 0.11 ± 0.03 (15) | 86 ± 4 (15, 10) | 88 ± 6 (15, 10) | 0 (150) |
| mek-1 | jnk-1(gk7) | normal | 0.21 ± 0.02 (15) | 47 ± 5 (15, 10) | 50 ± 5 (15, 10) | 0 (150) |
| mkk-4 | wild type (N2) | normal | 0.10 ± 0.03 (15) | 82 ± 5 (15, 10) | 85 ± 4 (15, 10) | 32 (48/150) |
| mkk-4 | jnk-1(gk7) | normal | 0.23 ± 0.02 (15) | 44 ± 6 (15, 10) | 51 ± 6 (15, 10) | 28 (42/150) |
Values indicated mean ± SEM. The number of animals and, when indicated, trials per animal are shown in parentheses, respectively. Locomotion and touch responses were performed as in Table I. jkk-1 and mek-1 correspond, respectively, to F35C8.3 and K08A8.1 genes predicted by BLAST. mkk-4, which is highly homologous to mammalian mkk4/sek1, corresponds to the F42G10.2 gene predicted by BLAST. For mkk-4, RNAi was performed with three different RNA fragments described in Materials and methods. All three yielded similar results. Data from mkk-4(1) are shown.
Discussion
In this study, we characterize the recently cloned C.elegans jnk-1 gene (Kawasaki et al., 1999) at the biochemical and genetic level. In mammalian cells, JNK protein kinases are encoded by three genes, which are alternatively spliced to create 10 JNK isoforms (Gupta et al., 1996). In C.elegans, the system is simpler, a single gene, jnk-1, encodes two different protein isoforms generated either by alternative transcription start site selection or the use of alternative SL1 trans-splice sites. The JNK-1α isoform described previously (Kawasaki et al., 1999) has a unique N-terminus of 91 amino acids fused in-frame to the methionine residue that serves as the JNK-1β N-terminus. In contrast, the amino acid sequence of JNK-1β is conserved almost entirely within mammalian JNKs. Similarly to the insect and mammalian JNKs (Galcheva-Gargova et al., 1994), diverse stimuli, murine MKK4/SEK1 as well as C.elegans MKK7 homologs activate both JNK-1 isoforms by dual phosphorylation on threonine and tyrosine residues within a TPY motif. Further, both JNK-1 isoforms appear capable of phosphorylating human c-Jun, ATF-2 and ELK-1 recombinant proteins, consistent with conservation of function. In contrast to knockouts of the conserved genes of the Drosophila JNK pathway and jnk1/jnk2 in mice, a null mutation for C.elegans jnk-1 is viable. Animals harboring the null allele, jnk-1(gk7), did not display obvious developmental abnormalities. However, jnk-1(gk7) exhibited defects in locomotion and hypersensitivity to several heavy metals, phenotypes found in C.elegans mutants null for the mkk7 homologs, jkk-1(km2) (Kawasaki et al., 1999) and mek-1(ks54) (Koga et al., 2000), respectively. jnk-1(gk7) also exhibited some mechanosensory defects that were not mimicked by jkk-1(km2) or mek-1(ks54), suggesting the existence of additional mechanisms of jnk regulation in this species. Inactivation of mkk-4 by RNAi also did not yield these mechanosensory deficits but rather resulted in an egg-laying defect in hermaphrodites. Despite biochemical activation of JNK-1 by murine MKK4/SEK1, this mkk-4-deficient phenotype was not detected in jnk-1(gk7) worms. These results demonstrate that at least two distinct MKK7s regulate functions through JNK-1. Further, they suggest that MKK-4 acts through a kinase other than JNK-1. In this regard, an analysis of the 19 099 predicted proteins for C.elegans demonstrates the existence of 14 MAPKs (Plowman et al., 1999), including three p38 homologs or potential homologs (Kawasaki et al., 1999; A.Villanueva and R.N.Kolesnick, unpublished data).
Recent reports characterizing mice mutants for the JNK signaling pathway genes suggest that differential expression patterns of these genes may influence the role of this pathway in various tissues. In C.elegans, jnk-1 and jkk-1 appear to be co-expressed in most neurons and their processes, including the nerve ring, the head and tail ganglia and the dorsal and ventral nerve cords (Kawasaki et al., 1999). In contrast, mek-1 is expressed preferentially in pharyngeal muscles, uterine and intestine cells, and in some neurons in the nerve ring and ventral and anal ganglia (Koga et al., 2000). These different expression patterns, together with the different phenotypes of jkk-1 and mek-1 mutant worms, suggest that the JNK pathway is regulated through compartmentalization of function in C.elegans.
In C.elegans, body muscle contraction depends on a balance between excitatory cholinergic (Lewis et al., 1980) and inhibitory GABAergic inputs (McIntire et al., 1993a,b). Animals in which DD and VD neurons have been killed by laser ablation (McIntire et al., 1993b) or which carry mutations in several genes in the GABAergic pathway (McIntire et al., 1993a) simultaneously contract the normally antagonistic dorsal and ventral body muscles, causing these animals to ‘shrink’ (White et al., 1976). Recently, Kawasaki and co-workers reported that jkk-1 regulates coordinated movement in D-type motor neurons by acting as a suppressor of GABA action (Kawasaki et al., 1999). The present study shows that loss of JNK-1 function in C.elegans delivers an identical body movement phenotype as loss of JKK-1 function, and shows that the loss of UNC-25 (glutamic acid decarboxylase) activity suppresses this movement defect. Moreover, inactivation of unc-30 or unc-47 by RNAi also suppressed the movement phenotypes. Further, Kawasaki et al. demonstrated that selective jkk-1 expression in the D-type motor neurons using the unc-30 promoter rescues the jkk-1(km2) movement defects. Since the UNC-30 homeodomain protein coordinates transcriptional regulation of unc-25 and unc-47 genes, taken together the results suggest that functional GABAergic synaptic transmission in D-type motor neurons is required for expression of the jnk-1 locomotion defect. One attractive possibility is that normal action of JNK-1 is to negatively regulate GABA action, and the increased wave amplitude observed in jnk-1 mutants is the result of increased GABAergic signaling. For example, the C.elegans JNK signaling pathway could regulate the quantity of GABA generated by the nervous system, the activity of the UNC-25 protein or the timing of GABA release. However, our results do not exclude other, less direct interactions between JNK-1 function and GABAergic transmission.
There is a little genetic evidence in mice or Drosophila regarding the significance of the JNK signaling pathway in stress responses in whole animals. The embryonic lethality of these mutants in different species makes it difficult to determine whether the JNK signaling pathway is involved in stress responses later in life. In mice, disruption of mkk4, mkk7 or jnk1/jnk2 cause early embryonic lethality (Yang et al., 1997a; Kuan et al., 1999; Dong et al., 2000), while the jnk3 knockout displays defects in stress-induced AP-1 transcriptional activity and apoptosis in the hippocampus (Yang et al., 1997b). The non-essential nature of jnk-1, jkk-1 (Kawasaki et al., 1999), mek-1 (Koga et al., 2000) and mkk-4 for embryogenesis allows an evaluation of the role of these proteins in stress responses. So far, we have shown that the animals harboring a null allele for jnk-1, like mek-1 null mutants, are hypersensitive to the heavy metals cadmium and copper. Consistent with the pattern of mek-1 expression in the intestine, Koga et al. suggested possible explanations for the hypersensitivity of mek-1(ks54) mutant worms to heavy metals: (i) animals might fail to arrest pumping in response to these toxic metals and consequently be exposed to higher doses; (ii) animals might take up more metal into the intestine independent of pumping; or (iii) these metals may alter nutritional status unfavorably (Koga et al., 2000). Further studies will be needed to determine the role of MEK-1 and JNK-1 in the normal disposal/metabolism of these heavy metals to gain insight into this issue.
In conclusion, our results confirm the existence of a highly conserved JNK signaling pathway in C.elegans. This pathway is involved in the regulation of body movement coordination and the response to heavy metals, apparently via compartmentalized expression of two MKK7s, JKK-1 and MEK-1, respectively. However, as in mice and Drosophila, we do not find genetic evidence for MKK4 signaling through JNK in C.elegans. Investigations into functional and genetic associations between JNK-1 and other conserved components of this pathway, the relationship to other MAPKs and the delineation of JNK-1 substrates should allow for a more complete understanding of the C.elegans JNK pathway in the near future.
Materials and methods
Growth and synchronization of C.elegans
All strains were maintained as hermaphrodites at 20°C on NGM standard plates fed with the Escherichia coli OP50 bacterial strain, as described (Brenner, 1974). Worms were observed either under a dissecting microscope (Leica MZ8) or using a Zeiss Axioplan with Nomarski optics. Worms from each developmental stage were obtained from synchronized liquid cultures: L1 larvae were collected 7 h after hatching, L2 larvae at 17 h, L3 larvae at 27 h, L4 larvae at 38 h, and adult worms were harvested at 45 h.
RNA preparation, RT–PCR, RACE, northern blot and RT–SSCP analysis
Total RNA was extracted by a single-step method (Chomczynski and Sacchi, 1987).
RT–PCR. Two micrograms of total RNA were reverse transcribed to cDNA using pd(N6) random hexamers (Amersham-Pharmacia Biotech) as described (Villanueva et al., 1998). Full-length transcripts of jnk-1α and jnk-1β were amplified using the 5′ primers 5′-CCCAAGTTT GAGAACCATCA-3′ and 5′-CCCAAGTTTGAGAGAAGCGTG-3′, respectively, and the common 3′ primer 5′-TCAGGAATAAATGTC ATGGGTTCTTG-3′. mek-1 cDNA was amplified using 5′-ATGGAG AGAGACTTCGACCTT-3′ and 5′-TTATCCGCACTCGCCCATCAC ATC-3′ primers. The reactions were performed in a thermal cycler for 30 cycles (94°C, 60 s; 59–66°C, 90 s; 72°C, 120 s) in a final volume of 30 µl. Identities of the amplified fragments were confirmed by sequencing.
RACE. For analysis of the 5′ end, two rounds of RACE were performed using a RACE Kit (Gibco-BRL) following the manufacturer’s directions. Briefly, for the first round, total RNA was reverse transcribed to cDNA using a primer, 5′-TCCTAGTCCACTGATCGATA-3′, derived from sequence within exon 8, and amplified using AAP (Gibco-BRL) and 5′-ACGTATCAGCTCTCCAAATA-3′ primers. Nested PCR amplification subsequently was performed using UAP (Gibco-BRL) and 5′-TTCGAGCCAATCCGAAATCCAA-3′ primers. A second round of RACE was performed using the sequence 5′-AGCACACAAAAGGAGAC-3′ derived from exon 12 for reverse transcription, AAP and 5′-TAGAGATCGAAATGTGACC-3′ primers for amplification, and UAP and 5′-AAAACATGGAATCAGGAAT-3′ primers for nested PCR. For analysis of the 3′ end, total RNA was reverse transcribed to cDNA using a commercial AP primer, and cDNA was amplified using the 5′-TTGGATTTCGGATTGGCTCGAA-3′ and 3′ UAP primers. PCR products were cloned directly into the TOPO system (Invitrogen) and individual clones were selected and sequenced.
Northern blot. Thirty micrograms of total RNA from different developmental stages were electrophoresed in 1.2% agarose gels containing formaldehyde, transferred onto a Hybond N+ Nylon membranes (Amersham-Pharmacia Biotech) following the manufacturer’s instructions, and the RNA was UV-cross-linked to the membrane using a Stratalinker (Stratagene). The blots were pre-hybridized with ExpressHyb hybridization solution (Clontech) for 1 h, and then probed with random-primed (Prime-it II, Stratagene), [α-32P]dCTP (NEN Life Science)-labeled full-length jnk-1β cDNA at 68°C for 1 h. The blots were washed under stringent conditions (65°C and 0.1% SSC/0.1% SDS) and exposed for 48 h to Kodak XAR-5 film at –70°C.
RT–PCR/SSCP. The cDNA region coding for protein kinase subdomains VIII–XI was amplified using 5′-ACTATCGGGCACCGGAAGTTA-3′ and 5′-TGTGACCGTAAAACATGGAATCAGGA-3′ primers, and SSCP was performed as described (Villanueva et al., 1998).
Plasmid construction
Mammalian expression vectors. Epitope-tagged jnk-1 isoforms and mek-1 were generated by PCR. The Flag epitope MDYKDDDK sequence was introduced at the C-terminus of jnk-1β using 5′-GGTACCGCCACCATGGAACCATCCTCTATTCAT-3′ and 5′-CTCGAGTCTACTTGTCATCGTCGTCCTTGTAGTCGGAATAAATGTCATGGGTTCT-3′ primers and cloned into the mammalian expression vector pCEP4 (Invitrogen) to create pCEP4-Flag-JNK-1β. jnk-1α was amplified using 5′-GGATCCATGGAGGAACGATTATCCACA-3′ and 5′-AAGCTTTCAGGAATAAATGTCATGGGT-3′ primers, and cloned into the mammalian expression vector pCMV3B (Stratagene), that contains the c-Myc epitope MEQKLISE sequence in the N-terminus, to create pCMV3B-Myc-JNK-1α. mek-1 was amplified using 5′-GGTACCATGGAGAGAGACTTCGAC-3′ and 5′-GCGGCCGCTTATCCGCACTCGCC-3′ primers and cloned into the mammalian expression vector pHM6 (Boehringer) that contains the HA epitope YPYDVPDYA sequence in the N-terminus to create pHM6-HA-MEK-1. Amino acids Thr185 and Tyr187 of JNK-1 (corresponding to human JNK1 Thr183 and Tyr185) were substituted with alanine and phenylalanine, respectively, using pCMV3B-Myc-JNK-1α and pCEP4-Flag-JNK-1β mammalian expression vectors as templates, by oligonucleotide mutagenesis using 5′-TCGAACTGCGATTGAGGCATTCATGATGGCTCCTTTCGTTGTGACAAGATACTATCGGGC-3′ and 5′-GCCCGATAGTATCTTGTCACAACGAAAGGAGCCATCATGAATGCCTCAATCGCAGTTCGA-3′ primers and the One Site Mutagenesis kit (Stratagene). Similarly, a kinase-inactive form of MEK-1, in which the conserved Lys99 in the ATP-binding domain was substituted with arginine, was generated using pHM6-HA-MEK-1 as a template and the set of primers 5′-GTG ATCATGGCTGTGAATACGATGCCTCGGACG-3′ and 5′-CGTCCG AGGCATCGTATTCACAGCCATGATCAC-3′. Mutant jnk-1 isoforms, jnk-1(gk7)α and jnk-1(gk7)β, were amplified by RT–PCR from the jnk-1(gk7) knockout and cloned into pCMV3B to generate pCMV3B-Myc-JNK-1(gk7)α and Myc-JNK-1(gk7)β.
Escherichia coli expression vectors. The open reading frames of the jnk-1α and jnk-1β isoforms were cloned into the prokaryotic expression vector pTrcHisB (Invitrogen) to create pTRCHis-JNK-1α and pTRCHis-JNK-1β. Expression and purification of His-tagged JNK-1 proteins were carried out using the TALON system (Clontech) according to the manufacturer’s instructions. Purified proteins were used to validate the specificity of antibody binding.
Caenorhabditis elegans expression vectors. flag-jnk-1α and flag-jnk-1β cDNAs were amplified by PCR using mammalian expression vectors as templates and cloned into the heat shock-inducible vectors pPD49.78 and pPD49.83, placing them under control of heat shock promoters. These promoters direct protein expression in the majority of C.elegans tissues (Jones et al., 1986; Stringham et al., 1992). Pmek-1::flag-jnk-1α or -β transgenes, in which flag-jnk-1α or -β cDNAs are under control of the mek-1 promoter (Pmek-1), were constructed using a PCR fusion approach in several steps. Initially, a 4750 bp DNA fragment, corresponding to the putative mek-1 promoter and initial ATG was amplified from N2 genomic DNA using 5′-AAATCTCCGTAGATCTTAAATGC-3′ and 5′-GTCATCCTTGTAATCCATCTTTTCACCTTAAAATTA-3′ primers. The 3′ primer contains the nucleotide sequences of the Flag epitope tag. Next, DNA fragments including the complete flag-jnk-1α or -β cDNA, respectively, were amplified using mammalian expression vectors as templates, and 5′-TGGAGCTCCACCGCGGTGGC-3′ and 5′-TGACCGTAAAACATGGAATCAGGAATAAATGTCATG-3′ primers. A 2100 bp segment of the jnk-1 3′-untranslated region (UTR), which include 1900 bp beyond the polyadenylation site, were amplified using N2 genomic DNA as a template and 5′-CATGAC ATTTATTCCTGATTCCATGTTTTACGGTC-3′ and 5′-CTAGTG CATTCTCACCGAGACTG-3′ primers. Fusion between flag-jnk-1 cDNAs and the jnk-1 3′-UTR was performed by combing 1.5 µl of each PCR using 5′-ATGGATTACAAGGATGACGACGATAAG-3′ and 5′-TGTCCATCAGTTGCTATAATGC-3′ primers. Finally, PCRs products [Pmek-1-flag and flag-jnk-1 3′-UTR] were fused by combing 1.5 µl of each PCR and subsequent amplification with 5′-GGTACCCTTGGTGGTCTTAAGACTGGC-3′ and 5′-GCGGCCGCTTCTCGTGATGTTCCTGATGGATCC-3′ nested primers from the beginning of the mek-1 promoter and at the end of the jnk-1 3′-UTR region, respectively. The fusion product was inserted between the KpnI and NotI sites of TOPO vector (Invitrogen). For all PCRs, a mixture of Taq polymerase and proofreading PwoI polymerase (Boehringer Mannheim) was used to ensure fidelity of the reaction, and the fusion fragment sequences confirmed by automated DNA sequencing.
Reversal of jnk-1(gk7) defects
To generate transgenic animals, young adult jnk-1(gk7) were injected into both gonads with 25–50 ng/µl DNA with or without pRF4 plasmid (dominant rol-6 marker at 80 ng/µl). For analysis of the recovery of normal movement and touch responses, jnk-1(gk7) worms were injected with heat shock plasmids containing flag-jnk-1α or -β cDNAs without a selection marker. After 30 min at 33°C, young adults were monitored every 4 h to select worms showing a wild-type movement pattern (wave amplitude, locomotion and trajectory). For recovery from heavy metal hypersensitivity, the same heat shock constructs were co-injected into jnk-1(gk7) worms with pRF4 plasmid, as a selection marker, and animals with a Roller phenotype were selected, heat treated, and after 16 h allowed to lay eggs for 6 h onto plates containing heavy metals. For evaluating the expression of JNK-1 via the MEK-1 promoter, Pmek-1::flag-jnk-1α or -β transgenes were co-injected with pRF4 into jnk-1(gk7) progeny with the Roller phenotype were selected, and heavy metal sensitivity assayed. All transgenes were maintained as extrachromosomal arrays. The presence of the different plasmids was confirmed in individual worms at the end of each experiment by specific PCR.
Cell culture and transient transfection
COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Transient transfection was performed with lipofectAMINE according to the manufacturer’s recommendations (Life Technologies). After 48 h, transfected cells were treated with UV light (40 J/m2) or γ-radiation (20 Gy) followed by incubation at 37°C for 20 min. Another set of cells was treated with sorbitol (400 mM), TNF-α (100 ng/ml; Promega), LPS (1 µg/ml; Sigma), H2O2 (75 µM) or heat shocked at 42°C for 20 min. Cells were lysed in buffer [20 mM HEPES pH 7.4, 2 mM EGTA, 50 mM β-glycerophosphate, 1 mM dithiothreitol (DTT), 1 mM Na3VO4, 1% Triton X-100, 10% glycerol, 10 µM leupeptin, 25 µg/ml aprotinin, 100 µM phenylmethylsulfonyl fluoride]. Insoluble material was removed by centrifugation at 14 000 g for 10 min at 4°C and total protein measured by the Bradford method using the Bio-Rad protein assay.
Western blot analysis
To verify expression levels of the different epitope-tagged proteins transiently transfected in COS-7 cells, 15 µg of whole-cell lysates were resolved by SDS–PAGE, transferred to nitrocellulose membranes (Amersham-Pharmacia Biotech), probed with antibodies for 1 h and specific binding was detected by the enhanced chemiluminescence method (Amersham Life Science).
Immunoprecipitation
Epitope-tagged proteins were isolated by immunoprecipitation from cell lysates. Aliquots of 0.5–1 mg of whole-cell lysate were incubated for 2 h at 4°C with 1 µg of anti-FLAG M2 (Stratagene), anti-HA 12CA5 or anti-Myc (Roche Molecular Biochemicals) monoclonal antibodies. Immune complexes were incubated further for 1 h at 4°C with protein AG–Sepharose (Santa Cruz), recovered by centrifugation and washed six times in 1 ml of lysis buffer containing 500 mM NaCl. An additional 1 ml wash in kinase buffer was performed when the immune complexes were used for kinase assays.
Protein kinase assays
Immune complex kinase assays were performed in a final volume of 40 µl of kinase buffer (25 mM MOPS pH 7.4, 25 mM β-glycerophosphate, 25 mM MgCl2, 2 mM DTT and 100 µM Na2VO4). Protein kinase reactions were initiated by addition of recombinant substrate proteins [GST–c-Jun (Upstate Biotechnology), GST–ATF-2 or GST–ELK-1 (New England Biolabs)] and kinase buffer containing 250 µM ATP and 5 µCi of [γ-32P]ATP (3000 Ci/mmol; NEN Life Science). Reactions were incubated for 30 min at 30°C and terminated by the addition of an equal volume of Laemmli sample buffer.
Generation of null alleles
Worms carrying a homozygous deletion of jnk-1 [named jnk-1(gk7)] were isolated at the C.elegans Reverse Genetics Core Facility of Vancouver, British Columbia, Canada, by PCR screening of a chemical (formaldehyde) mutagenesis library using the primers 5′-CTCTGCGTCTTCCTTCCATCT-3′ and 5′-GAAGAGAACTTCAAACGGCG-3′. Nested PCR was performed using primers 5′-AAGACATCCAAACGGACAGC-3′ and 5′-TCATACCTTGGCCGATTTTC-3′. Mutant animals were backcrossed five times onto the wild-type Bristol N2 background before phenotypic analysis.
Strain construction
To generate the unc-25(e156);jnk-1(gk7), jnk-1(gk7);jkk-1(km2) and jnk-1(gk7);mek-1(ks54) double mutant strains, jnk-1(gk7) males were mated with CB156 [genotype: unc-25(e156)] (Brenner et al., 1974), KU2 [genotype: jkk-1(km2)] (Kawasaki et al., 1999) or FK171 [genotype: mek-1(ks54 )] (Koga et al., 2000) hermaphrodites, respectively. F2 unc-25(e156)/+;jnk-1(gk7)/jnk-1(gk7) hermaphrodites, selected by PCR, were allowed to self-fertilize to generate the unc-25(e156);jnk-1(gk7) double mutant strain. unc25(e156) mutant alleles carry a point mutation in codon 383 (TGG→TAG) that generates a new XbaI restriction site. This site allows discrimination of the wild-type and unc-25(e156) alleles by restriction fragment length polymorphism (RFLP)/PCR using the 5′ primer 5′-GGCACAATGATGTTTTCAAGCTTTGGTTGATCT-3′ and the 3′ primer 5′-GCTCACCGAAACTCACATTCTAGATAATCAACT-3′. F2 jnk-1(gk7)/jnk-1(gk7);jkk-1(km2)/+ and jnk-1(gk7)/jnk-1(gk7); mek-1(ks54)/+ hermaphrodites were selected by PCR, and allowed to self-fertilize to generate jnk-1(gk7);jkk-1(km2) and jnk-1(gk7); mek-1(ks54), respectively.
Double-stranded RNA-mediated interference (RNAi)
A 2 µg aliquot of total RNA was reverse transcribed to cDNA using pd(N6) random hexamers, as above, and either full-length cDNA for unc-25, unc-30, unc-47 or a common N-terminus region of 717 bp including amino acids 1–239 of all unc-49 isoforms were amplified using the following primers: unc-25, 5′-ATGTCCTCTGCTGCTGCTGAC-3′ and 5′-TTATTCCAAAGACTCTCCAAT-3′; unc-30, 5′-ATGGATGACAATACGGCCACA-3′ and 5′-CTAAAGTGGTCCACTGTACTG-3′; unc-47, 5′-ATGGCGTCGAATAGATTTCAA-3′ and 5′-TTAAGAATCAGCAGAGTTGAT-3′; unc-49, 5′-ATGGCTCGTCCATTCACACTTA-3′ and 5′CATTGGACACGTTGCAGTGAC-3′. mkk-4, which corresponds to the gene F42G10.2 predicted by BLAST, was amplified using the following different set of primers: (i) 5′-CTTTCGACAGGCACATTAA-3′ and 5′-TGCAAGATACGGCTGACA-3′; (ii) 5′-TGTCAGCCGTATCTTGCA-3′ and 5′-CTTAGCCTCTTCAATTTCG-3′; (iii) 5′-ACATAGAGAACAAATCCGC-3′ and 5′-GTCTGGATGTAGAATTGGA-3′. RNAi using the three different mkk-4 primer sets yielded identical data. gfp was used as a negative control, while bir-1 and unc-22 were used as positive controls in the RNAi experiments. PCR products were cloned into the pCRII-TOPO dual promotor vector using the TOPO system (Invitrogen) and the identities of the amplified fragments confirmed by sequencing. RNAs were in vitro transcribed using T7 and SP6 RNA polymerases according to standard protocols. Sense and antisense strands were mixed prior to injection to 500 ng/µl and then annealed. Young adult wild-type (N2) and jnk-1(gk7) hermaphrodites were injected into both gonads. After 12 h, injected animals were transferred to fresh plates (Fire et al., 1998).
Behavioral assays
Assays for osmolarity, volatile repellents, chemoattractants and chemorepellents, light nose touch, hard body touch, dauer formation, defecation reflex and toxicity assays were performed as previously described (Ward, 1973; Culotti and Russell, 1978; Golden and Riddle, 1982; Bargmann et al., 1993; Kaplan and Horvitz, 1993; Hart et al., 1995, 1999; Maricq et al., 1995; Chou et al., 1996; Koga et al., 2000).
Acknowledgments
Acknowledgements
Some strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). We thank Andy Fire for the pPD49.78 and pPD49.83 plasmids, and members of the Hengartner lab for assistance with this project. This work was supported by grants CA-42385 to R.N.K. and GM-52540 to M.O.H. from the National Institutes of Health. A.V. and J.L. are fellows from the Spanish Ministry of Education, A.M. is a MEC-Fulbright fellow from the Spanish Ministry of Education. M.O.H. is a Rita Allen Foundation Scholar.
References
- Bamber B.A., Beg,A.A., Twyman,R.E. and Jorgensen,E.M. (1999) The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J. Neurosci., 19, 5348–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann C.I., Hartwieg,E. and Horvitz,H.R. (1993) Odorant-selective genes and neurons mediate olfaction in C.elegans. Cell, 74, 515–527. [DOI] [PubMed] [Google Scholar]
- Bektesh S., Van Doren,K. and Hirsh,D. (1988) Presence of the Caenorhabditis elegans spliced leader on different mRNAs and in different genera of nematodes. Genes Dev., 2, 1277–1283. [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavigelli M., Dolfi,F., Claret,F.X. and Karin,M. (1995) Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J., 14, 5957–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi,N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Chou J.H. et al. (1996) Olfactory recognition and discrimination in Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol., 61, 157–164. [PubMed] [Google Scholar]
- Constant S.L., Dong,C., Yang,D.D., Wysk,M., Davis,R.J. and Flavell,R.A. (2000) JNK1 is required for T cell-mediated immunity against Leishmania major infection. J. Immunol., 165, 2671–2676. [DOI] [PubMed] [Google Scholar]
- Coso O.A., Chiariello,M., Kalinec,G., Kyriakis,J.M., Woodgett,J. and Gutkind,J.S. (1995) Transforming G protein-coupled receptors potently activate JNK (SAPK). Evidence for a divergence from the tyrosine kinase signaling pathway. J. Biol. Chem., 270, 5620–5624. [DOI] [PubMed] [Google Scholar]
- Culotti J.G. and Russell,R.L. (1978) Osmotic avoidance defective mutants of the nematode Caenorhabditis elegans. Genetics, 90, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.J. (1993) The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem., 268, 14553–14556. [PubMed] [Google Scholar]
- Davis R.J. (2000) Signal transduction by the JNK group of MAP kinases. Cell, 103, 239–252. [DOI] [PubMed] [Google Scholar]
- Derijard B., Hibi,M., Wu,I.H., Barrett,T., Su,B., Deng,T., Karin,M. and Davis,R.J. (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Derijard B., Raingeaud,J., Barrett,T., Wu,I.H., Han,J., Ulevitch,R.J. and Davis,R.J. (1995) Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science, 267, 682–685. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang,D.D., Wysk,M., Whitmarsh,A.J., Davis,R.J. and Flavell,R.A. (1998) Defective T cell differentiation in the absence of Jnk1. Science, 282, 2092–2095. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang,D.D., Tournier,C., Whitmarsh,A.J., Xu,J., Davis,R.J. and Flavell,R.A. (2000) JNK is required for effector T-cell function but not for T-cell activation. Nature, 405, 91–94. [DOI] [PubMed] [Google Scholar]
- Eastman C., Horvitz,H.R. and Jin,Y. (1999) Coordinated transcriptional regulation of the unc-25 glutamic acid decarboxylase and the unc-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J. Neurosci., 19, 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Fuchs S.Y., Adler,V., Pincus,M.R. and Ronai,Z. (1998) MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl Acad. Sci. USA, 95, 10541–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galcheva-Gargova Z., Derijard,B., Wu,I.H. and Davis,R.J. (1994) An osmosensing signal transduction pathway in mammalian cells. Science, 265, 806–808. [DOI] [PubMed] [Google Scholar]
- Ganiatsas S., Kwee,L., Fujiwara,Y., Perkins,A., Ikeda,T., Labow,M.A. and Zon,L.I. (1998) SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc. Natl Acad. Sci. USA, 95, 6881–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glise B., Bourbon,H. and Noselli,S. (1995) hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell, 83, 451–461. [DOI] [PubMed] [Google Scholar]
- Golden J.W. and Riddle,D.L. (1982) A pheromone influences larval development in the nematode Caenorhabditis elegans. Science, 218, 578–580. [DOI] [PubMed] [Google Scholar]
- Gupta S., Campbell,D., Derijard,B. and Davis,R.J. (1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science, 267, 389–393. [DOI] [PubMed] [Google Scholar]
- Gupta S., Barrett,T., Whitmarsh,A.J., Cavanagh,J., Sluss,H.K., Derijard,B. and Davis,R.J. (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J., 15, 2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Han J., Lee,J.D., Bibbs,L. and Ulevitch,R.J. (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science, 265, 808–811. [DOI] [PubMed] [Google Scholar]
- Han Z.S., Enslen,H., Hu,X., Meng,X., Wu,I.H., Barrett,T., Davis,R.J. and Ip,Y.T. (1998) A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol. Cell. Biol., 18, 3527–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart A.C., Sims,S. and Kaplan,J.M. (1995) Synaptic code for sensory modalities revealed by C.elegans GLR-1 glutamate receptor. Nature, 378, 82–85. [DOI] [PubMed] [Google Scholar]
- Hart A.C., Kass,J., Shapiro,J.E. and Kaplan,J.M. (1999) Distinct signaling pathways mediate touch and osmosensory responses in a polymodal sensory neuron. J. Neurosci., 19, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M., Lin,A., Smeal,T., Minden,A. and Karin,M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev., 7, 2135–2148. [DOI] [PubMed] [Google Scholar]
- Jacobs D., Glossip,D., Xing,H., Muslin,A.J. and Kornfeld,K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev., 13, 163–175. [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Jorgensen,E., Hartwieg,E. and Horvitz,H.R. (1999) The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J. Neurosci., 19, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D., Russnak,R.H., Kay,R.J. and Candido,E.P. (1986) Structure, expression and evolution of a heat shock gene locus in Caenorhabditis elegans that is flanked by repetitive elements. J. Biol. Chem., 261, 12006–12015. [PubMed] [Google Scholar]
- Kaplan J.M. and Horvitz,H.R. (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA, 90, 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki M., Hisamoto,N., Iino,Y., Yamamoto,M., Ninomiya-Tsuji,J. and Matsumoto,K. (1999) A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J., 18, 3604–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M., Zwaal,R., Guan,K.L., Avery,L. and Ohshima,Y. (2000) A Caenorhabditis elegans MAP kinase kinase, MEK-1, is involved in stress responses. EMBO J., 19, 5148–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan C.Y., Yang,D.D., Samanta Roy,D.R., Davis,R.J., Rakic,P. and Flavell,R.A. (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron, 22, 667–676. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. and Avruch,J. (1996) Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem., 271, 24313–24316. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M., Banerjee,P., Nikolakaki,E., Dai,T., Rubie,E.A., Ahmad,M.F., Avruch,J. and Woodgett,J.R. (1994) The stress-activated protein kinase subfamily of c-Jun kinases. Nature, 369, 156–160. [DOI] [PubMed] [Google Scholar]
- Lewis J.A., Wu,C.H., Levine,J.H. and Berg,H. (1980) Levamisole-resistant mutants of the nematode Caenorhabditis elegans appear to lack pharmacological acetylcholine receptors. Neuroscience, 5, 967–989. [DOI] [PubMed] [Google Scholar]
- Maricq A.V., Peckol,E., Driscoll,M. and Bargmann,C.I. (1995) Mechanosensory signalling in C.elegans mediated by the GLR-1 glutamate receptor. Nature, 378, 78–81. [DOI] [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- McIntire S.L., Jorgensen,E. and Horvitz,H.R. (1993a) Genes required for GABA function in Caenorhabditis elegans. Nature, 364, 334–337. [DOI] [PubMed] [Google Scholar]
- McIntire S.L., Jorgensen,E., Kaplan,J. and Horvitz,H.R. (1993b) The GABAergic nervous system of Caenorhabditis elegans. Nature, 364, 337–341. [DOI] [PubMed] [Google Scholar]
- McIntire S.L., Reimer,R.J., Schuske,K., Edwards,R.H. and Jorgensen,E.M. (1997) Identification and characterization of the vesicular GABA transporter. Nature, 389, 870–876. [DOI] [PubMed] [Google Scholar]
- Mello C. and Fire,A. (1995) DNA transformation. Methods Cell Biol., 48, 451–482. [PubMed] [Google Scholar]
- Noselli S. (1998) JNK signaling and morphogenesis in Drosophila. Trends Genet., 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Plowman G.D., Sudarsanam,S., Bingham,J., Whyte,D. and Hunter,T. (1999) The protein kinases of Caenorhabditis elegans: a model for signal transduction in multicellular organisms. Proc. Natl Acad. Sci. USA, 96, 13603–13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesgo-Escovar J.R., Jenni,M., Fritz,A. and Hafen,E. (1996) The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev., 10, 2759–2768. [DOI] [PubMed] [Google Scholar]
- Robinson M.J. and Cobb,M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol., 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Sluss H.K., Han,Z., Barrett,T., Davis,R.J. and Ip,Y.T. (1996) A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev., 10, 2745–2758. [DOI] [PubMed] [Google Scholar]
- Stringham E.G., Dixon,D.K., Jones,D. and Candido,E.P. (1992) Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol. Biol. Cell, 3, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernarakis N., Wang,S.L., Dorovkov,M., Ryazanov,A. and Driscoll,M. (2000) Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nature Genet., 24, 180–183. [DOI] [PubMed] [Google Scholar]
- Tournier C., Whitmarsh,A.J., Cavanagh,J., Barrett,T. and Davis,R.J. (1997) Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl Acad. Sci. USA, 94, 7337–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij M. et al. (1996) Requirement for ceramide-initiated SAPK/JNK signalling in stress- induced apoptosis. Nature, 380, 75–79. [DOI] [PubMed] [Google Scholar]
- Villanueva A. et al. (1998) Disruption of the antiproliferative TGF-β signaling pathways in human pancreatic cancer cells. Oncogene, 17, 1969–1978. [DOI] [PubMed] [Google Scholar]
- Ward S. (1973) Chemotaxis by the nematode Caenorhabditis elegans: identification of attractants and analysis of the response by use of mutants. Proc. Natl Acad. Sci. USA, 70, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.G., Southgate,E., Thomson,J.N. and Brenner,S. (1976) The structure of the ventral nerve cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci., 275, 327–348. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Shore,P., Sharrocks,A.D. and Davis,R.J. (1995) Integration of MAP kinase signal transduction pathways at the serum response element. Science, 269, 403–407. [DOI] [PubMed] [Google Scholar]
- Yang D., Tournier,C., Wysk,M., Lu,H.T., Xu,J., Davis,R.J. and Flavell,R.A. (1997a) Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation and defects in AP-1 transcriptional activity. Proc. Natl Acad. Sci. USA, 94, 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.D., Kuan,C.Y., Whitmarsh,A.J., Rincon,M., Zheng,T.S., Davis,R.J., Rakic,P. and Flavell,R.A. (1997b) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature, 389, 865–870. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Conze,D., Whitmarsh,A.J., Barrett,T., Davis,R.J., Rincon,M. and Flavell,R.A. (1998) Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity, 9, 575–585. [DOI] [PubMed] [Google Scholar]