

Abstract
Both thyroid hormone (TH) and retinoic acid (RA) induce purified rat oligodendrocyte precursor cells in culture to stop division and differentiate. We show that these responses are blocked by the expression of a dominant-negative form of p53. Moreover, both TH and RA cause a transient, immediate early increase in the same 8 out of 13 mRNAs encoding intracellular cell cycle regulators and gene regulatory proteins, but only if protein synthesis is inhibited. Platelet-derived growth factor (PDGF) withdrawal also induces these cells to differentiate, but we show that the intracellular mechanisms involved are different from those involved in the hormone responses: the changes in cell cycle regulators differ, and the differentiation induced by PDGF withdrawal (or that which occurs spontaneously in the presence of PDGF) is not blocked by the dominant-negative p53. These results suggest that TH and RA activate the same intracellular pathway leading to oligodendrocyte differentiation, and that this pathway depends on a p53 family protein. Differentiation that occurs independently of TH and RA apparently involves a different pathway. It is likely that both pathways operate in vivo.
Keywords: immediate early genes/oligodendrocyte precursors/p53 family/retinoic acid/thyroid hormone
Introduction
In many vertebrate cell lineages, precursor cells divide a limited number of times before they stop and terminally differentiate into post-mitotic cells. It is not known what limits cell proliferation and causes the cells to stop dividing when they do. The stopping mechanisms are important as they influence both the timing of cell differentiation and the number of terminally differentiated cells generated.
We have studied the mechanisms that stop precursor cell division and initiate differentiation in the oligodendrocyte cell lineage in the rodent optic nerve. Oligodendrocyte precursor cells (OPCs) migrate into the developing rat optic nerve from the brain from before birth (Small et al., 1987). After a period of proliferation, they stop dividing and terminally differentiate into oligodendrocytes, which myelinate the axons in the nerve. The first oligodendrocytes appear in the nerve around the day of birth and then increase in number for the next 6 weeks (Skoff et al., 1976; Miller et al., 1985; Barres et al., 1992).
Similar timing of oligodendrocyte development can be reconstituted in cultures of either mixed (Raff et al., 1985, 1988) or purified (Gao et al., 1998) embryonic optic nerve OPCs. When the OPCs are stimulated to proliferate by either astrocytes (Raff et al., 1985) or platelet-derived growth factor (PDGF) (Raff et al., 1988; Gao et al., 1998), oligodendrocytes begin to appear around the equivalent of the day of birth. Clonal analyses of either single (Temple and Raff, 1986) or purified (Barres et al., 1994) OPCs isolated from postnatal day 7–8 (P7–8) rat optic nerve suggest that both a cell-intrinsic programme and extracellular signals can play important parts in determining when OPCs stop dividing and differentiate. In the presence of appropriate signalling molecules, the P7 OPCs divide up to eight times before they stop and differentiate, and the progeny of an individual OPC tend to stop dividing and differentiate at about the same time (Temple and Raff, 1986; Barres et al., 1994). Moreover, when the two daughter cells of an individual OPC are separated and cultured on astrocyte monolayers in separate microwells, they tend to differentiate more or less synchronously (Temple and Raff, 1986). These findings suggest that a cell-intrinsic mechanism helps control when OPCs withdraw from the cell cycle and differentiate, at least in culture (Temple and Raff, 1986). When OPCs are cultured at 33 rather than 37°C, they divide more slowly but differentiate sooner, after fewer cell divisions, suggesting that the intrinsic mechanism does not operate by simply counting cell divisions, but instead may measure elapsed time in some other way (Gao et al., 1997). We therefore refer to this intracellular mechanism as a timer. One component of the timer seems to be the gradual accumulation in the proliferating OPCs of the cyclin-dependent protein kinase inhibitor (CdkI) p27/Kip1 (Casaccia-Bonnefil et al., 1997; Durand et al., 1997, 1998). Another seems to be a progressive decrease in the Hes5 and Id4 helix–loop–helix proteins, either of which can inhibit differentiation when overexpressed (Kondo and Raff, 2000a,b).
Although the timer is cell intrinsic, it is not cell autonomous. It requires signals from other cells to work normally. The mitogen PDGF, for example, is one such required signal (Noble et al., 1988; Raff et al., 1988; Richardson et al., 1988). In the absence of PDGF, cultured OPCs prematurely stop dividing and differentiate into oligodendrocytes within 1–2 days (Noble and Murray, 1984; Temple and Raff, 1985). It seems likely that a lack of sufficient PDGF is responsible for the differentiation of at least some OPCs in vivo, especially early in development. In the embryonic mouse spinal cord, for instance, the availability of PDGF normally limits OPC proliferation: increasing PDGF availability by the expression of PDGF transgenes greatly increases OPC numbers in proportion to the total number of PDGF genes expressed (Calver et al., 1998). It has been proposed that, in normal embryos, the expanding population of OPCs consumes an increasing amount of PDGF, and so the PDGF concentration progressively falls, causing the cell cycle to slow and some OPCs to stop dividing and differentiate (Calver et al., 1998; van Heyningen et al., 2001). It is unclear how PDGF withdrawal induces OPCs to differentiate. There is evidence that cell cycle arrest may not be enough, as the overexpression of CdkI p27/Kip1 arrests the cycle but does not induce cultured OPCs to differentiate in the presence of mitogens (X.M.Tang et al., 1998; Tikoo et al., 1998).
Other extracellular signals are also required for the normal operation of the cell-intrinsic timer in cultured OPCs, including hydrophobic signals such as thyroid hormone (TH) or retinoic acid (RA) (Barres et al., 1994; Ibarrola et al., 1996; Ahlgren et al., 1997; Baas et al., 1997; Gao et al., 1998). When purified P7 OPCs are cultured in a saturating concentration of PDGF in the absence of TH and RA, most of the cells keep dividing and do not differentiate (Barres et al., 1994; Tang et al., 2000, 2001). If TH is added after 8 days, however, most of the cells stop dividing and differentiate within 4 days (Barres et al., 1994). These findings and others (Bögler and Noble, 1994) suggest that the intrinsic timer consists of at least two components: a timing component that measures elapsed time independently of hydrophobic signals, and an effector component that is regulated by hydrophobic signals and that stops cell proliferation and initiates differentiation when the time is up. Thus, TH and RA can induce OPCs to differentiate only when the OPCs have reached a certain stage of maturation (Gao et al., 1998), whereas PDGF withdrawal induces OPCs to differentiate at any stage of maturation, whether TH or RA is present or not (Barres et al., 1994; Ahlgren et al., 1997; Gao et al., 1998).
Although it is unclear whether RA regulates oligodendrocyte development in vivo, it has long been known that TH does. Myelination, for example, is retarded in hypothyroid animals and accelerated in hyperthyroid animals (Walters and Morell, 1981; Legrand, 1986; Dussault and Ruel, 1987). Moreover, there are fewer oligodendrocytes in the developing postnatal optic nerves of hypothyroid mouse (Ibarolla et al., 1996) and rat (Ahlgren et al., 1997) pups than in the optic nerves of euthyroid pups at the same age. Thus, it seems likely that the TH-regulated intrinsic timer is responsible for the differentiation of at least some OPCs in vivo, especially postnatally when TH levels are rising and OPCs are becoming more responsive to TH (Gao et al., 1998). But TH is not required for oligodendrocyte development, as even in its absence some oligodendrocytes develop both in vivo (Ahlgren et al., 1997) and in vitro (Ibarrola et al., 1996; Ahlgren et al., 1997). Because TH influences the timing of differentiation in a number of cell lineages, it is likely that it plays a part in coordinating the timing of differentiation in different tissues during vertebrate development (see for example Knipper et al., 1998).
It remains uncertain how TH or RA triggers the effector component of the timer to initiate OPC differentiation. Both act by binding to nuclear receptors that are members of the same superfamily of ligand-activated gene regulatory proteins (Evans, 1988; Mangelsdorf et al., 1995). We showed previously that they both induce an antigenic change on the surface of purified OPCs within 4 h and that this change is inhibited if RNA synthesis is suppressed (Tokumoto et al., 1999). These findings suggest that both TH and RA stimulate oligodendrocyte differentiation, at least in part, by directly regulating gene transcription. We previously used RT–PCR to study the transcription of some candidate genes, including several immediate early genes and all of the genes that encode known CdkIs. We found that none of these genes was activated 1, 4 or 8 h after treatment of purified OPCs with either TH or RA (Tokumoto et al., 1999).
In the present study, we compare some of the intracellular events induced by TH or RA treatment with those induced by PDGF withdrawal. Our findings suggest that TH and RA activate the same intracellular pathway leading to differentiation, whereas PDGF withdrawal acti vates a different pathway. The first pathway depends on a p53 family protein, while the second pathway does not. These findings suggest that p53 or one of its relatives is an essential part of the effector component of the intrinsic timer that helps control when OPCs differentiate in the presence of PDGF and TH or RA.
Results
In most experiments, we purified OPCs from P7 rat optic nerve by sequential immunopanning (Barres et al., 1992), and studied the cells after 10 days in serum-free culture in the presence of PDGF to stimulate proliferation and in the absence of both TH and RA to inhibit differentiation (Barres et al., 1994). We then either treated the cells with TH or RA or washed them free of PDGF and cultured them without PDGF, TH or RA. These treatments all induce most of the OPCs to stop dividing and differentiate within several days (Barres et al., 1994). After variable periods, we harvested the cells and analysed either the mRNAs by RT–PCR or the proteins by western blotting. In some experiments, we pretreated the cells with protein synthesis inhibitors before treating them with TH or RA. In others, we infected optic nerve cells with retroviral vectors encoding either enhanced green fluorescent protein (GFP) alone or GFP and a dominant-negative form of p53 before purifying the OPCs and studying their responses to TH, RA or PDGF withdrawal. All experiments were repeated at least twice, with similar results.
If protein synthesis is inhibited, TH and RA induce a rapid and transient increase in the same eight mRNAs
To test for direct, immediate early responses to TH and RA, we pretreated purified OPCs with cycloheximide (CHX; 50 µg/ml) for 6 h to inhibit protein synthesis and then added TH or RA for 1 or 2 h. We then harvested the mRNAs and analysed them by RT–PCR, using oligonucleotide primers to detect mRNAs that encode various cell cycle regulatory proteins or immediate early gene regulatory proteins. We found previously that neither TH nor RA directly increased these mRNAs in the absence of CHX pretreatment (Tokumoto et al., 1999). As shown in Figure 1, however, in CHX-pretreated cells both TH and RA induced transient increases in mRNAs encoding the INK4 family CdkIs p15/INK4b (p15) and p19/INK4d (p19), the Cip/Kip family CdkIs p21/Cip1 (p21) and p27/Kip1 (p27), the cell cycle inhibitor p19ARF (Figure 1A), and the immediate early gene regulatory proteins c-Myc, c-Fos and Krox24/NGFI-A (Figure 1B). In all cases, the increase was greater at 1 h than at 2 h. By contrast, neither TH nor RA under the same conditions had a significant effect on the mRNAs encoding the INK4 family CdkI p18/INK4c (p18) (Figure 1A) or the cell cycle promoters cyclins D1, D2 or E (not shown). Although we were able to detect the mRNA encoding the INK4 family CdkI p16/INK4a (p16) in cells that were not pretreated with CHX (see below), we could not detect it in CHX-pretreated cells (Figure 1A), suggesting that the CHX pretreatment shut off p16 transcription.
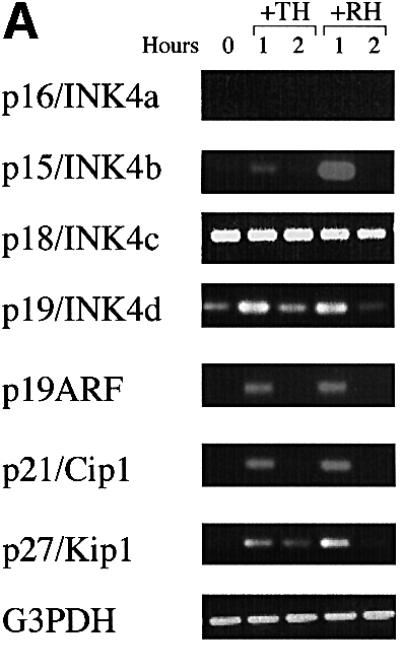
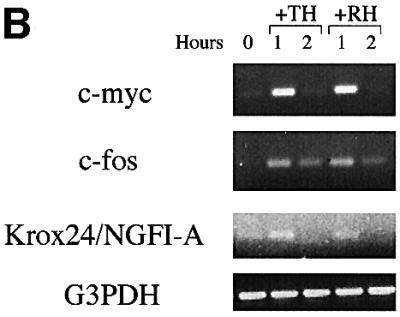
Fig. 1. RT–PCR analysis of immediate early mRNA responses to TH or RA. With the exception of Figure 4, all OPCs were purified and cultured for 10 days in PDGF without TH or RA before they were studied. In this experiment, the cells were then treated with CHX for 6 h before treatment with TH or RA for 1 or 2 h before they were harvested and cDNA was prepared for RT–PCR analysis. The number of PCR cycles was: 40 for p16; 32 for p15, p18, p19 and p27; 35 for p19ARF, p21, c-myc, c-fos and Krox24/NGFI-A; and 25 for G3PDH.
To confirm that the effect of CHX resulted from its ability to inhibit protein synthesis, we pretreated the cells with another protein synthesis inhibitor, emetine (10 µg/ml for 15 min). This had the same effect as pretreatment with CHX (not shown).
The finding that TH and RA treatments lead to an immediate early increase in the same 8 out of 13 mRNAs tested in OPCs is consistent with the possibility that TH and RA act on the same genes to induce differentiation.
TH treatment and PDGF withdrawal have distinct but overlapping effects on cell cycle regulators
Both TH (and RA) treatment and PDGF withdrawal induce purified OPCs to stop dividing and differentiate within several days, but it remains unclear to what extent they use the same intracellular mechanisms. We found previously that neither treatment (without protein synthesis inhibition) influences the levels of mRNAs that encode various cell cycle regulators within the first 8 h of treatment (Tokumoto et al., 1999). We therefore extended these observations to 16, 24 and 48 h after treatment. As shown in Figure 2A, both TH treatment and PDGF withdrawal led to a sustained increase in the mRNAs encoding the negative cell cycle regulators p19, p21, p27 and p19ARF at each of the three time points. TH treatment, but not PDGF withdrawal, led to a transient increase in p16 mRNA at 16 and 24 h, whereas PDGF withdrawal, but not TH treatment, led to a dramatic and sustained decrease in p15 mRNA. Surprisingly, TH treatment led to a sustained increase in cyclin D1 mRNA, but had no effect on cyclin D2 mRNA levels (Figure 2B); by contrast, PDGF withdrawal led to a sustained fall in both cyclin D1 and cyclin D2 mRNAs by 16 h (Figure 2B). Both treatments led to a late fall in cyclin E mRNA at 48 h, which was much more pronounced with PDGF withdrawal (Figure 2A).

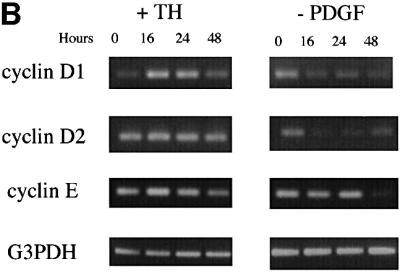
Fig. 2. RT–PCR analysis of late mRNA responses to either TH treatment or PDGF withdrawal. The cells were harvested before treatment (0 h) or at 16, 24 and 48 h after treatment. The number of PCR cycles was: 35 for p16; 33 for p15 and p21; 30 for p18, p19, p19ARF, cyclin D1 (after TH addition), cyclin D2 and cyclin E; 32 for p27 and cyclin D1 (after PDGF withdrawal); and 23 for G3PDH.
To test the influence of these treatments on cell cycle regulatory proteins, we performed western blots on extracts of the cells 24 and 48 h after either TH treatment or PDGF withdrawal. As shown in Figure 3, both treatments led to decreases in cyclin D1 and cyclin D2, and an increase in p27, which was greater after TH treatment than after PDGF withdrawal; TH treatment, but not PDGF withdrawal, led to increases in p18 and p21, whereas the levels of p19 and p19ARF remained largely unchanged after either treatment.
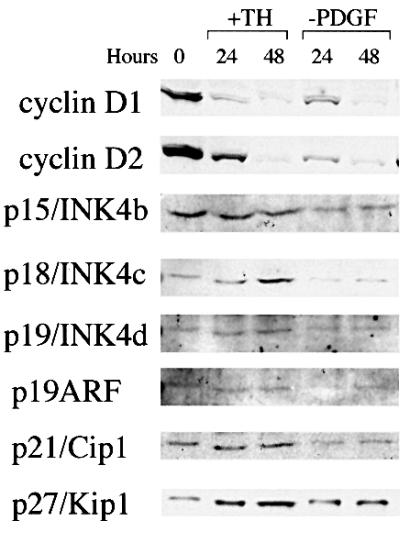
Fig. 3. Western blot analysis of cell cycle regulatory proteins in response to either TH treatment or PDGF withdrawal. The analysis was performed as described previously (D.G.Tang et al., 1998).
The early and late changes in mRNAs and proteins stimulated by TH (or RA) treatment or PDGF withdrawal are summarized in Table I.
Table I. Summary of early and late changes in expression of cell cycle regulators in response to TH (or RA) treatment or PDGF withdrawal.
| Molecules | mRNA (early) |
mRNA (late) |
|
Protein (late) |
|
| |
(TH or RA treatment after CHX Rx) |
TH treatment |
PDGF withdrawal |
TH treatment |
PDGF withdrawal |
| p16/INK4a | – | ↑ | – | ND | ND |
| p15/INK4b | ↑ | → | ↓ | → | ↓ |
| p18/INK4c | → | → | → | ↑ | → |
| p19/INK4d | ↑ | ↑ | ↑ | → | → |
| p19ARF | ↑ | ↑ | ↑ | → | → |
| p21/Cip1 | ↑ | ↑ | ↑ | ↑ | → |
| p27/Kip1 | ↑ | ↑ | ↑ | ↑ | ↑ |
| cyclin D1 | → | ↑ | ↓ | ↓ | ↓ |
| cyclin D2 | → | → | ↓ | ↓ | ↓ |
| cyclin E | → | ↓ | ↓ | ND | ND |
Differentiation induced by TH or RA, but not by PDGF withdrawal, is inhibited by a dominant-negative form of p53
The finding that TH caused a delayed increase in p21 mRNA and protein raised the possibility that TH-induced differentiation depends on the tumour suppressor protein p53, which is a potent activator of p21 transcription (El-Deiry et al., 1993; Brugarolas et al., 1995; Deng et al., 1995; Waldman et al., 1995). To test this possibility, we infected freshly isolated P7 optic nerve cells with retroviral vectors encoding either enhanced GFP alone or GFP and a naturally occurring mutant form of p53 that acts as a dominant-negative inhibitor of wild-type p53 (R175H; Kern et al., 1992). We then purified the OPCs and cultured them at clonal density for 10 days in PDGF, without TH or RA, and then either added TH (or RA) or removed PDGF. After 3 and 5 days, we examined the fluorescent cells in an inverted fluorescence microscope to assess clone size and the proportion of transfected cells that had acquired the characteristic morphology of oligodendrocytes. In some experiments, we assessed differentiation by staining the cells for galactocerebroside (GC), a specific marker for differentiated oligodendrocytes (Raff et al., 1978).
As shown in Figure 4A, whereas ∼70% of the cells expressing GFP alone had acquired oligodendrocyte morphology, <15% of the cells that expressed both GFP and the dominant-negative p53 had done so. Moreover, whereas <5% of either uninfected clones or clones that expressed GFP alone expanded between 3 and 5 days after TH treatment, >95% of clones that expressed the dominant-negative p53 expanded during this time (not shown). Thus, expression of the dominant-negative p53 completely inhibited both cell cycle withdrawal and differentiation induced by TH. It also inhibited RA-induced cell cycle withdrawal and oligodendrocyte differentiation (not shown). By contrast, it had no effect on the proportion of cells that acquired oligodendrocyte morphology in response to PDGF withdrawal or that spontaneously acquired oligodendrocyte morphology in PDGF without TH (Figure 4A). We confirmed these results by staining transfected cells with a monoclonal anti-GC antibody after 2 days of either TH treatment or PDGF withdrawal (Figure 4B).


Fig. 4. Effect of dominant-negative p53 on OPC differentiation. The cells were infected with retroviral vectors encoding either GFP alone (Bird–GFP) or GFP and a dominant-negative p53 (Bird–GFP–DNp53) and were induced to differentiate by either TH treatment or PDGF withdrawal. (A) The cells were assessed by morphology after 3 days of treatment. (B) The cells were stained with monoclonal anti-GC antibody after 2 days of treatment. The results are shown as the mean ± SD of five flasks for each condition, with >200 GFP-positive cells counted in each flask.
Discussion
OPCs are arguably the best understood precursor cells in the vertebrate central nervous system, but the intracellular mechanisms involved in their differentiation remain largely mysterious. In the present study, we have compared some of the intracellular events that are induced by TH, RA or PDGF withdrawal; all these treatments induce purified, postnatal OPCs in culture to stop dividing and differentiate into oligodendrocytes. Our findings suggest that there are at least two independent intracellular pathways leading to OPC cell cycle arrest and differentiation, and that TH and RA activate one of them and PDGF withdrawal activates the other. Whereas the first depends on p53 or one of its relatives, the second does not. As discussed in the Introduction, it is likely that oligodendrocytes develop by both pathways in vivo.
TH and RA increase many of the same mRNAs when protein synthesis is inhibited
One surprising finding of the study is that TH and RA induce an immediate early increase in the same 8 out of 13 mRNAs in OPCs when protein synthesis is inhibited by pretreatment with either CHX or emetine. These mRNAs include those encoding the cell cycle inhibitors p15, p19, p21, p27 and p19ARF, and the gene regulatory proteins c-Myc, c-Fos and Krox 24/NGFI-A. In all these cases, the response is transient, being greater at 1 h than at 2 h. The increases are specific, as protein synthesis inhibition alone does not induce them, and five other mRNAs that we tested do not increase under the same conditions. The simplest interpretation of these findings is that, in OPCs, the receptors for TH and RA, which function as homodimers or heterodimers with RXR proteins and recognize distinct response elements in DNA (Evans, 1988; Mangelsdorf et al., 1995; Glass, 1996), can bind to the regulatory regions of these eight genes and activate their transcription when protein synthesis is inhibited. It is unclear why the responses require inhibition of protein synthesis in order to be seen, but the findings raise the possibility that TH and RA may both induce OPCs to differentiate by directly activating the same genes. It remains to be determined what the relevant primary response genes are. Whatever they are, they seem to depend on a p53 family protein to induce OPC differentiation (discussed below).
TH treatment and PDGF withdrawal decrease D cyclins by different mechanisms
Although TH (or RA) treatment and PDGF withdrawal can both induce OPCs in culture to differentiate, it has been unclear whether they do so by similar intracellular mechanisms. We find that both inducers cause a decrease in cyclin D1 and D2 proteins within 24 h and an almost complete disappearance of these proteins by 48 h. In both cases, it seems likely that the decreases in D cyclins contribute to the cell cycle arrest that accompanies differentiation. The mechanisms involved in these decreases, however, are apparently different in the two cases. Whereas TH treatment causes an increase in cyclin D1 mRNA at 16 and 24 h, and has no effect on cyclin D2 mRNA up to 48 h, PDGF withdrawal causes a sustained fall in both cyclin D1 and D2 mRNAs. Thus, whereas the rapid fall in cyclin D1 and D2 protein levels induced by PDGF withdrawal is likely to involve, at least in part, a decrease in D cyclin transcription and/or mRNA stability, this cannot be the case for the fall induced by TH. The TH-induced, post-transcriptional decrease in cyclin D1 and D2 proteins is specific, in that most of the other proteins we studied did not decrease with TH treatment. It remains to be determined whether the TH-induced decrease in cyclin D1 and D2 proteins is caused by a decrease in the synthesis of these cyclins, an increase in their degradation, or both.
The level of cyclin E mRNA is unchanged 24 h after either TH treatment or PDGF withdrawal, when most of the OPCs would have already permanently stopped dividing (Durand et al., 1997), although in both cases the level is decreased by 48 h. Thus, a decrease in cyclin E mRNA is unlikely to contribute to cell cycle withdrawal after either TH treatment or PDGF deprivation. It remains to be determined whether a fall in cyclin E protein does contribute.
TH treatment causes multiple CdkI proteins to increase
The decrease in cyclin D1 and D2 proteins is unlikely to be the only reason that OPCs withdraw from the cell cycle in response to TH. TH treatment also stimulates increases in several CdkI proteins, including p18, p21 and p27, which are increased at 24 and 48 h after TH addition. These increases may well contribute to the slowing of the cell cycle seen in OPCs after treatment with TH (Barres et al., 1994). The increase in p18 seems to reflect exclusively post-transcriptional regulation, as p18 mRNA levels do not change detectably in response to TH. By contrast, p21 and p27 mRNAs increase in parallel with p21 and p27 proteins, suggesting that at least part of the increase in the proteins reflects an increase in either the transcription of their genes or the stability or their mRNAs. The mRNAs that encode three other cell cycle inhibitors (p16, p19 and p19 ARF) also increase in response to TH treatment, but we cannot detect an increase in the proteins. If the increases in the mRNAs for p16, p19, p21, p27 and p19 ARF reflect increased transcription, this may depend on a change in the activity of gene regulatory proteins other than TH receptors themselves, as the direct stimulation of the expression of these genes by TH is transient and detectable only if protein synthesis is first inhibited (see above). The increase in p21 transcription in response to TH, for example, might depend on p53 or one of its relatives (see below).
In contrast to TH treatment, PDGF withdrawal leads to an increase in p27 protein, but not in any of the other cell cycle inhibitory proteins that we assessed. However, the mRNAs that encode p19, p21 and p19ARF, as well as the mRNA that encodes p27, increase in response to PDGF withdrawal. Surprisingly, p15 mRNA is dramatically decreased 16 h after PDGF withdrawal, and remains undetectable at 24 and 48 h. These findings support the hypothesis that the intracellular mechanisms responsible for cell cycle arrest are different when differentiation is induced by TH treatment than when it is induced by PDGF withdrawal.
A p53 family protein is part of the effector component of the timer
The most compelling evidence for at least two intracellular pathways of oligodendrocyte differentiation comes from our experiments on OPCs infected with a retroviral vector encoding a dominant-negative form of p53. These OPCs fail to stop dividing and differentiate in response to TH or RA, but stop dividing and differentiate normally in response to PDGF withdrawal. They also display a normal rate of spontaneous differentiation in the presence of PDGF and the absence of both TH and RA. These findings suggest that TH- and RA-induced OPC differentiation depends on p53 or one of its relatives [reviewed by Levrero et al. (2000) and Yang and McKeon (2000)], whereas both spontaneous OPC differentiation and PDGF-withdrawal-induced OPC differentiation do not. More importantly, they suggest that a p53 family protein is an essential part of the effector component of the intrinsic timer that helps control OPC differentiation in the presence of PDGF and TH or RA. A role for a p53 family member in TH-induced differentiation could explain why p21 mRNA and protein levels are increased more after TH treatment than after PDGF withdrawal.
Eizenberg et al. (1996) previously reported that p53 protein translocates from the cytoplasm to the nucleus when brain-derived OPCs differentiate in culture. They also found that a dominant-negative p53 inhibited the differentiation of these cells when they were deprived of mitogens present in medium conditioned by B104 cells. Although the latter finding appears to conflict with our finding that dominant-negative p53 does not inhibit differentiation induced by PDGF withdrawal, Eizenberg et al. apparently had TH in the culture medium when they withdrew mitogen. Moreover, it is possible that astrocytes in their cultures produced some PDGF (Noble et al., 1988; Richardson et al., 1988). Thus, it is difficult to dissect the roles of TH and mitogen deprivation in their experiments.
The relevant family member may not be p53
There are several reasons for thinking that the TH- and RA-regulated pathway of OPC differentiation may depend on a p53 homologue such as p63 or p73 [reviewed by Levrero et al. (2000) and Yang and McKeon (2000)], rather than on p53 itself. First, the dominant-negative form of p53 used in our experiments (R175H; Kern et al., 1992) has been shown to bind to and inhibit the activity of most isoforms of p63 and p73 (Gaiddon et al., 2001). Secondly, the main function of p53 seems to be to maintain genome stability and thereby suppress the development of cancer [reviewed by Lane (1992) and Levine (1997)], whereas the main functions of p63 and p73 seem to be in development. Thus, p53-deficient mice generally develop normally, whereas mice deficient in either p63 (Mills et al., 1999) or p73 (Yang et al., 2000) have serious developmental abnormalities, including neural abnormalities in the case of p73 (Yang et al., 2000). Thirdly, RA-induced differentiation of a mouse neuroblastoma line has been shown to depend on p73, and overexpression of p73 in these cells induces their differentiation in the absence of RA (De Laurenzi et al., 2000). Further experiments will be required to determine which p53 family member(s) is required for TH- and RA-regulated differentiation of OPCs.
Materials and methods
Animals and materials
Sprague–Dawley rats were obtained from the breeding colony of University College London (London, UK). All chemicals were from Sigma, unless indicated otherwise. Recombinant human PDGF-AA and neurotrophin-3 (NT-3) were purchased from Peprotech. Monoclonal anti-cyclin D1 (72-13G), anti-cyclin D2 (M-20), anti-p15 (M-20), anti-p19 (E-11), anti-p21 (F-5) and anti-p27 (F-8) were purchased from Santa Cruz Biotechnology. Rabbit anti-rat p19ARF antibodies were a gift from Alison Lloyd. The rabbit anti-p18 antibodies (06-555) were purchased from Upstate Biotechnology. Emetine was a gift from Louis Mahadevan.
Cultures of purified OPCs
Optic nerve cells were prepared from P7 rats, and OPCs were purified by sequential immunopanning, as described previously (Barres et al., 1992). The purified cells were cultured (2000 cells in 3 ml of culture medium) in poly-d-lysine (PDL)-coated 25 cm2 flasks (Falcon) in modified Bottenstein–Sato medium (Bottenstein and Sato, 1979) containing N-acetyl-cysteine (60 µg/ml), forskolin (5 µM), PDGF-AA (10 ng/ml), NT-3 (5 ng/ml), penicillin and streptomycin (Gibco). In some experiments, the purified cells were cultured (1000 cells in 2 ml of culture medium) in PDL-coated Nunc slide flasks.
Preparation of cDNA from purified OPCs
Purified OPCs were removed from the culture flasks with trypsin after 10 days in culture in PDGF without TH or RA. In some cases, the cells were treated with TH (40 ng/ml 3,5,3′-triiodo-l-thyronine, 40 ng/ml l-thyroxine) or RA (all-trans RA; 300 ng/ml) for 1, 2, 16, 24 or 48 h before they were harvested with trypsin. In other cases, the cells were washed well and cultured without PDGF, TH or RA for the same periods before they were harvested. About 6 × 104 cells were harvested from each flask. Poly(A)+ mRNA was prepared using a QuickPrep Micro mRNA Purification kit (Amersham Pharmacia Biotech) according to the supplier’s instructions. The RNA was then dissolved in doubly distilled water and cDNA synthesis was carried out using a SMART PCR cDNA Synthesis kit (Clontech) according to the supplier’s instructions. The PCR was carried out and the amplified total cDNA was used as the template for the RT–PCR. Before performing the PCR using the gene-specific primers, the amplified total cDNA samples were all electrophoresed in the same 1% agarose gel, stained with ethidium bromide (EtBr; 0.4 µg/ml) and normalized according to the intensity of staining.
RT–PCR
The following oligonucleotide DNA primers were synthesized. For p15/INK4b, the 5′ primer was 5′-CCGCTTCGGGAGGCGC-3′ and the 3′ primer was 5′-ACCTCGCAATATCACGGTG-3′. For p16/INK4a, the 5′ primer was 5′-TCACCAAACGCCCCGAAC-3′ and the 3′ primer was 5′-ATCGCGCACATCCAGCC-3′. For p19ARF, the 5′ primer was 5′-TGGGTCGCAGGTTCGTGG-3′ and the 3′ primer was the same as the 3′ primer used for p16/INK4a. For glyceraldehyde-3-phosphate dehydrogenase (G3PDH), the 5′ primer was 5′-ACCACAGTCCATGCC ATCAC-3′ and the 3′ primer was 5′-TCCACCACCCTGTTGCTGTA-3′. The sequences of the other RT–PCR primers used have been described previously (Tokumoto et al., 1999).
All primers were dissolved in TE buffer (10 mM Tris–HCl and 1 mM EDTA pH 8). The RT–PCRs were carried out in 25 µl of reaction mixture, containing 0.2 ng of PCR-amplified total cDNA as template, 0.3 µM 5′ PCR primer, 0.3 µM 3′ PCR primer, 0.2 mM dNTP, 1.25 mM MgCl2, 10 mM Tris–HCl pH 9.0, 50 mM KCl, 0.1% Triton X-100 and 1.25 U of Taq DNA polymerase (Promega). The reaction mixture was denatured for 1 min at 94°C. The PCR reaction was then started, using 94°C for 10 s for the denaturing step, 53°C for 30 s for the annealing step, and 72°C for 1 min for the elongation step. To determine how many PCR cycles to use, we removed 10 µl of each reaction mixture after 30 cycles, electrophoresed it in 2% agarose gel and stained it with EtBr; depending on the intensity of staining, we either stopped the reaction at 30 cycles, continued for several more cycles, or started again and stopped before 30 cycles.
Retroviral vectors and infection
Two retroviral vectors were used (Tang et al., 2001), both based on pBabe-puro (Morgenstern and Land, 1990). One (Bird–GFP) encoded enhanced GFP alone, while the other (Bird–GFP–DNp53) encoded GFP and a dominant-negative form of p53 (R175H; Kern et al., 1992). P7 rat optic nerve cells were cultured in slide flasks in PDGF without TH or RA and infected with vector for 16 h. The OPCs were then purified and cultured at clonal density in slide flasks (1000 cells/flask) in PDGF without TH or RA for 10 days. At this point, the flasks were examined using a Leica DM1RB inverted fluorescence microscope, and between 15 and 40% of the clones were found to be infected, as assessed by GFP expression. The cells were then either treated with TH or RA, or washed and cultured without PDGF for 2–5 days. Clone size and differentiation were assessed by morphology in the inverted fluorescence microscope at 3 and 5 days, or by staining for GC after 2 days (see below).
Immunofluorescence staining
Cells in PDL-coated slide flasks were fixed in 2% paraformaldehyde for 3 min at room temperature. After washing, they were incubated for 15 min in 50% normal goat serum to block non-specific staining and then treated with monoclonal anti-GC antibody (Ranscht et al., 1982). After washing, the cells were incubated with biotin-conjugated goat anti-mouse IgG antibody (Amersham; diluted 1:100), followed by Texas Red-conjugated streptavidin (Amersham; diluted 1:100). All incubations were for 30 min at room temperature. Finally, the coverslips were mounted in Citifluor mounting medium (CitiFluor), sealed with nail varnish, and examined using a Zeiss Axioskop fluorescence microscope.
Acknowledgments
Acknowledgements
We thank N.Billon and L.Mahadevan for helpful discussions, A.Lloyd for antibodies and L.Mahadevan for emetine. Y.M.T. was supported by a research fellowship from the British Council and a Project Grant from the Medical Research Council to M.C.R. Y.M.T. thanks members of the Raff, Mudge and Lloyd laboratories for their support. D.G.T. was a recipient of a Hitchings–Elion Award from Burroughs–Wellcome Fund. M.C.R. and the work described here were supported by a Program Grant from the MRC.
References
- Ahlgren S.C., Wallace,H., Bishop,J., Neophytou,C. and Raff,M.C. (1997) Effect of thyroid hormone on embryonic oligodendrocyte precursor cell development in vivo and in vitro. Mol. Cell. Neurosci., 9, 420–432. [DOI] [PubMed] [Google Scholar]
- Baas D., Bourbeau,D., Sarlieve,L.L., Ittel,M.E., Dussault,J.H. and Puymirat,J. (1997) Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia, 19, 324–332. [DOI] [PubMed] [Google Scholar]
- Barres B.A., Hart,I.K., Coles,H.S.R., Burne,J.F., Voyvodic,J.T., Richardson,W.D. and Raff,M.C. (1992) Cell death and control of cell survival in the oligodendrocyte lineage. Cell, 70, 31–46. [DOI] [PubMed] [Google Scholar]
- Barres B.A., Lazar,M.A. and Raff,M.C. (1994) A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development, 120, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Bögler O. and Noble,M. (1994) Measurement of time in oligodendrocyte-type-2 astrocyte (O-2A) progenitors is a cellular process distinct from differentiation or division. Dev. Biol., 162, 525–538. [DOI] [PubMed] [Google Scholar]
- Bottenstein J.E. and Sato,G.H. (1979) Growth of rat neuroblastoma cell line in serum-free supplemented medium. Proc. Natl Acad. Sci. USA, 76, 514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J., Chandrasekaran,C., Gordon,J.I., Beach,D., Jacks,T. and Hannon,G.J. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature, 377, 552–557. [DOI] [PubMed] [Google Scholar]
- Calver A.R., Hall,A.C., Yu,W.P., Walsh,F.S., Heath,J.K., Betsholtz,C. and Richardson,W.D. (1998) Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron, 20, 869–882. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P., Tikoo,R., Kiyokawa,H., Friedrich,V.,Jr, Chao,M.V. and Koff,A. (1997) Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27/Kip1. Genes Dev., 11, 2335–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laurenzi V. et al. (2000) Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem., 275, 15226–15231. [DOI] [PubMed] [Google Scholar]
- Deng C., Zhang,P., Harper,J.W., Elledge,S.J. and Leder,P. (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell, 82, 675–684. [DOI] [PubMed] [Google Scholar]
- Durand B., Gao,F.B. and Raff,M. (1997) Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J., 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand B., Fero,M.L., Roberts,J.M. and Raff,M.C. (1998) p27/Kip1 alters the response of cells to mitogen and is part of a cell-intrinsic timer that arrests cell division and initiates differentiation. Curr. Biol., 8, 431–440. [DOI] [PubMed] [Google Scholar]
- Dussault J.H. and Ruel,J. (1987) Thyroid hormones and brain development. Annu. Rev. Physiol., 49, 321–334. [DOI] [PubMed] [Google Scholar]
- Eizenberg O., Faber-Elman,A., Gottlieb,E., Oren,M., Rotter,V. and Schwartz,M. (1996) p53 plays a regulatory role in differentiation and apoptosis of central nerve system-associated cells. Mol. Cell. Biol., 16, 5178–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumour suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Evans R.M. (1988) The steroid and thyroid receptor superfamily. Science, 240, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiddon C., Lokshin,M., Ahn,J., Zhang,T. and Prives,C. (2001) A subset of tumour-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol., 21, 1874–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.B., Durand,B. and Raff,M. (1997) Oligodendrocyte precursor cells count time but not cell divisions before differentiation. Curr. Biol., 7, 152–155. [DOI] [PubMed] [Google Scholar]
- Gao F.B., Apperly,J. and Raff,M. (1998) Cell-intrinsic timers and thyroid hormone regulate the probability of cell-cycle withdrawal and differentiation of oligodendrocyte precursor cells. Dev. Biol., 197, 54–66. [DOI] [PubMed] [Google Scholar]
- Glass C.K. (1996) Some new twists in the regulation of gene expression by thyroid hormone and retinoic acid receptors. J. Endocrinol., 150, 349–357. [DOI] [PubMed] [Google Scholar]
- Ibarrola N., Mayer-Pröuschel,M., Rodriguez-Pena,A. and Noble,M. (1996) Evidence for the existence of at least two timing mechanisms that contribute to oligodendrocyte generation in vitro.Dev. Biol., 180, 1–21. [DOI] [PubMed] [Google Scholar]
- Kern S.E., Pietenpol,J.A., Thiagalingam,S., Seymour,A., Kinzler,K.W. and Vogelstein,B. (1992) Oncogenic forms of p53 inhibit p53-regulated gene expression. Science, 256, 827–830. [DOI] [PubMed] [Google Scholar]
- Knipper M., Bandtlow,C., Gestwa,L., Kopschall,I., Rohbock,K., Wiechers,B., Zenner,H.P. and Zimmermann,U. (1998) Thyroid hormone affects Schwann cell and oligodendrocyte gene expression at the glial transition zone of the VIIIth nerve prior to cochlea function. Development, 125, 3709–3718. [DOI] [PubMed] [Google Scholar]
- Kondo T. and Raff,M. (2000a) Basic helix–loop–helix proteins and the timing of oligodendrocyte differentiation. Development, 127, 2989–2998. [DOI] [PubMed] [Google Scholar]
- Kondo T. and Raff,M. (2000b) The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J., 19, 1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane O.P. (1992) p53, guardian of the genome. Nature, 358, 83–86. [DOI] [PubMed] [Google Scholar]
- Legrand J. (1986) Thyroid hormone effects on growth and development. In Hennemann,G. (ed.), Thyroid Hormone Metabolism. Marcel Dekker, Inc., New York, NY, pp. 503–534.
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Levrero M., De Laurenzi,V., Costanzo,A., Sabatini,S., Gong,J., Wang,J.Y.J. and Melino,G. (2000) The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci., 113, 1661–1670. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D.J. et al. (1995) The nuclear receptor superfamily: the second decade. Cell, 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R.H., David,S., Patel,R., Abney,E.R. and Raff,M.C. (1985) A quantitative immunohistochemical study of macroglial cell development in the rat optic nerve: in vivo evidence for two distinct astrocyte lineages. Dev. Biol., 111, 35–41. [DOI] [PubMed] [Google Scholar]
- Mills A.A., Zheng,B., Wang,X.J., Vogel,H., Roop,D.R. and Bradley,A. (1999) p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature, 398, 708–713. [DOI] [PubMed] [Google Scholar]
- Morgenstern J.P. and Land,H. (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res., 18, 3587–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M. and Murray,K. (1984) Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. EMBO J., 3, 2243–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M., Murray,K., Stroobant,P., Waterfield,M.D. and Riddle,P. (1988) Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature, 333, 560–562. [DOI] [PubMed] [Google Scholar]
- Raff M.C., Mirsky,R., Fields,K.L., Lisak,R.P., Dorfman,S.H., Silberberg,D.H., Gregson,N.A., Leibowitz,S. and Kennedy,M.C. (1978) Galactocerebroside is a specific cell-surface antigenic marker for oligodendrocytes in culture. Nature, 274, 813–816. [PubMed] [Google Scholar]
- Raff M.C., Abney,E.R. and Fok-Seang,J. (1985) Reconstitution of a developmental clock in vitro: a critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell, 42, 61–69. [DOI] [PubMed] [Google Scholar]
- Raff M.C., Lillien,L.E., Richardson,W.D., Burne,J.F. and Noble,M.D. (1988) Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature, 333, 562–565. [DOI] [PubMed] [Google Scholar]
- Ranscht B., Clapshaw,P.A., Price,J., Noble,M. and Seifert,W. (1982) Development of oligodendrocytes and Schwann cells studied with a monoclonal antibody against galactocerebroside. Proc. Natl Acad. Sci. USA, 79, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson W.D., Pringle,N., Mosley,M.J., Westermark,B. and Dubois-Dalcq,M. (1988) A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell, 53, 309–319. [DOI] [PubMed] [Google Scholar]
- Skoff R.P., Price,D.L. and Stocks,A. (1976) Electron microscopic autoradiographic studies of gliogenesis in rat optic nerve. II. Time of origin. J. Comp. Neurol., 169, 313–334. [DOI] [PubMed] [Google Scholar]
- Small R.K., Riddle,P. and Noble,M. (1987) Evidence for migration of oligodendrocyte-type-2 astrocyte progenitor cells into the developing rat optic nerve. Nature, 328, 155–157. [DOI] [PubMed] [Google Scholar]
- Tang D.G., Li,L., Chopra,D. and Poter,A.T. (1998) Extended survivability of prostate cancer cells in the absence of trophic factors: Increased proliferation, evasion of apoptosis and the role of apoptosis proteins. Cancer Res., 58, 3466–3479. [PubMed] [Google Scholar]
- Tang D.G., Tokumoto,Y.M. and Raff,M.C. (2000) Long-term culture of purified postnatal oligodendrocyte precursor cells: evidence for an intrinsic maturation program that plays out over months. J. Cell Biol., 148, 971–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D.G., Tokumoto,Y.M., Apperly,J.A., Lloyd,A.C. and Raff,M.C. (2001) Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science, 291, 868–871. [DOI] [PubMed] [Google Scholar]
- Tang X.M., Strocchi,P. and Cambi,F. (1998) Changes in the activity of cdk2 and cdk5 accompany differentiation of rat primary oligodendrocytes. J. Cell. Biochem., 68, 128–137. [DOI] [PubMed] [Google Scholar]
- Temple S. and Raff,M.C. (1985) Differentiation of a bipotential glial progenitor cell in a single cell microculture. Nature, 313, 223–225. [DOI] [PubMed] [Google Scholar]
- Temple S and Raff,M.C. (1986) Clonal analysis of oligodendrocyte development in culture; evidence for developmental clock that counts cell divisions. Cell, 44, 773–779. [DOI] [PubMed] [Google Scholar]
- Tikoo R., Osterhout,D.J., Casaccia-Bonnefil,P., Seth,P., Koff,A. and Chao,M.V. (1998) Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell-cycle growth arrest. J. Neurobiol., 36, 431–440. [PubMed] [Google Scholar]
- Tokumoto M.Y., Durand,M. and Raff,M.C. (1999) An analysis of the early events when oligodendrocyte precursor cells are triggered to differentiate by thyroid hormone, retinoic acid, or PDGF withdrawal. Dev. Biol., 213, 323–339. [DOI] [PubMed] [Google Scholar]
- van Heyningen P., Calver,A.R. and Richardson,W.D. (2001) Control of progenitor cell number by mitogen supply and demand. Curr. Biol., 11, 232–241. [DOI] [PubMed] [Google Scholar]
- Waldman T., Kinzler,K.W. and Vogelstein,B. (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res., 55, 5187–5190. [PubMed] [Google Scholar]
- Walters S.N. and Morell,P. (1981) Effect of altered thyroid states on myelinogenesis. J. Neurochem., 36, 1792–1801. [DOI] [PubMed] [Google Scholar]
- Yang A. and McKeon,F. (2000) p63 and p73: p53 mimics, menaces and more. Nature Rev. Mol. Cell Biol., 1, 199–207. [DOI] [PubMed] [Google Scholar]
- Yang A. et al. (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature, 404, 99–103. [DOI] [PubMed] [Google Scholar]