

Abstract
Yeast Rad52 DNA-repair mutants exhibit pronounced radiation sensitivity and a defect in homologous re combination (HR), whereas vertebrate cells lacking Rad52 exhibit a nearly normal phenotype. Bio chemical studies show that both yeast Rad52 and Rad55–57 (Rad51 paralogs) stimulate DNA-strand exchange mediated by Rad51. These findings raise the possibility that Rad51 paralogs may compensate for lack of Rad52 in vertebrate cells, explaining the absence of prominent phenotypes for Rad52-deficient cells. To test this hypothesis, using chicken DT40 cells, we generated conditional mutants deficient in both RAD52 and XRCC3, which is one of the five vertebrate RAD51 paralogs. Surprisingly, the rad52 xrcc3 double-mutant cells were non-viable and exhibited extensive chromosomal breaks, whereas rad52 and xrcc3 single mutants grew well. Our data reveal an overlapping (but non-reciprocal) role for Rad52 and XRCC3 in repairing DNA double-strand breaks. The present study shows that Rad52 can play an important role in HR repair by partially substituting for a Rad51 paralog.
Keywords: DT40/homologous recombination/Rad51 paralogs/Rad52/XRCC3
Introduction
DNA double-strand breaks (DSBs) arise during DNA replication and from exposure to agents such as ionizing radiation (IR). A single DSB may cause cell death if left unrepaired. Non-homologous end joining (NHEJ) and homologous recombination (HR) are major DSB repair pathways, and both are conserved across eukaryotic cells (reviewed in Jeggo, 1998; Haber, 1999; Morrison and Takeda, 2000; Sonoda et al., 2001; Thompson and Schild, 2001). The genes involved in HR in the yeast Saccharo myces cerevisiae form the RAD52 epistasis group of genes (RAD50, RAD51, RAD52, RAD54, RAD55, RAD57, RAD59, MRE11 and XRS2) (reviewed in Paques and Haber, 1999). Mutants of these genes are hypersensitive to IR and exhibit mitotic and meiotic recombination defects. Among the members of the Rad52 epistasis group, Rad51, a structural and functional homolog of Escherichia coli RecA, is conserved to the highest degree, exhibiting 69% amino acids sequence identity between S.cerevisiae and humans (Shinohara and Ogawa, 1995). This high degree of conservation suggests that the function of Rad51 is also conserved among eukaryotes. Defective Rad51 is lethal to higher eukaryotic cells, indicating a critical role for HR in repairing spontaneous DSBs arising during DNA replication (Tsuzuki et al., 1996; Sonoda et al., 1998). In vitro studies have shown that yeast and human Rad51 proteins form multimeric helical nucleoprotein filaments, similar to RecA proteins, which are assembled on single-stranded DNA (ssDNA) or on double-stranded DNA (dsDNA) containing either 5′ or 3′ single-stranded tails (Mazin et al., 2000; Sigurdsson et al., 2001). The nucleoprotein filaments mediate the search for homology, strand pairing and strand exchange (Baumann and West, 1998).
Relatives of the Rad51 gene that probably arose by gene duplication and the evolution of new functions (paralogs) are present in yeast and higher eukaryotes (Thompson and Schild, 2001). These Rad51 paralogs include Rad55 and Rad57 (Johnson and Symington, 1995) in S.cerevisiae, and in vertebrates, XRCC2 (Cartwright et al., 1998b; Liu et al., 1998), XRCC3 (Tebbs et al., 1995; Liu et al., 1998), Rad51B/Rad51L1 (Albala et al., 1997; Rice et al., 1997; Cartwright et al., 1998a), Rad51C/Rad51L2 (Dosanjh et al., 1998) and Rad51D/Rad51L3 (Cartwright et al., 1998b; Kawabata and Saeki, 1998; Pittman et al., 1998). The five human Rad51 paralogs have only 20–30% identity to human Rad51, with each other, and with yeast Rad55 and Rad57 (reviewed in Thacker, 1999). Yeast two-hybrid studies of human Rad51 paralogs have shown that, unlike Rad51, none of them shows self-association while physical interactions occur between human Rad51 and XRCC3, XRCC3 and Rad51C, Rad51B and Rad51C, Rad51C and Rad51D, and Rad51D and XRCC2. Thus, each Rad51 paralog appears to have different interacting partners within the family (reviewed in Thompson and Schild, 2001). In analogy with the S.cerevisiae Rad55 and Rad57 proteins (Sung, 1997b), the vertebrate paralogs may provide Rad51 accessory functions. This notion is in agreement with the data from our previous genetic study in which all of the Rad51 paralog mutants derived from the chicken B lymphocyte DT40 line exhibited remarkably similar phenotypes (Takata et al., 2000, 2001). Thus, the Rad51 paralogs have non-overlapping roles, and they all participate in DNA repair mediated by HR. Moreover, all the Rad51-paralog mutants show defective Rad51 focus formation and partial correction of the sensitivity to DNA damage from overexpression of human Rad51. These observations suggest that one or more complexes involving Rad51 paralogs facilitate the action of Rad51 in HR.
Rad52 appears to be essential for any type of HR during both meiotic and mitotic processes in S.cerevisiae (reviewed in Paques and Haber, 1999). In marked contrast, murine and chicken cells deficient in Rad52 show a minimal-deficiency phenotype with only a moderate decrease in gene targeting efficiency (Rijkers et al., 1998; Yamaguchi-Iwai et al., 1998) and no radiation sensitivity. Biochemical studies imply that Rad52 and Rad51 paralogs participate in HR in a similar way (Sung, 1997a,b; Benson et al., 1998; reviewed in Kanaar and Hoeijmakers, 1998; New et al., 1998; Shinohara and Ogawa, 1998). Purified mammalian and yeast Rad52 proteins facilitate the respective Rad51-mediated DNA-strand exchange in the presence of RPA protein, a factor binding to ssDNA. Similarly, the addition of Rad55– Rad57 heterodimer to Rad51 and RPA enhances Rad51-mediated strand exchange reaction in vitro (Sung, 1997b), whereas the biochemical activities of the five vertebrate Rad51 paralogs have just begun to be characterized (Braybrooke et al., 2000; Kurumizaka et al., 2001). The similar biochemical activities of yeast Rad55–Rad57 and Rad52 led us to investigate whether in vertebrate cells the Rad51 paralogs can substitute for Rad52, which would explain the nearly normal phenotype of Rad52-deficient cells. To test this hypothesis we generated a conditional RAD52–/–XRCC–/– mutant using an inducible Cre site-specific recombinase MerCreMer (Zhang et al., 1996, 1998). Our results show that Rad52 and Rad51 paralogs are indeed complementary in the maintenance of chromosomal integrity, as well as in HR-mediated repair of IR-induced DSBs in vertebrate cells.
Results
Experimental strategy
To generate cells deficient in both XRCC3 and Rad52, we employed the tamoxifen (TAM)-inducible Cre–loxP system (Zhang et al., 1996, 1998). We generated a transgene containing the human XRCC3 (hXRCC3) and green fluorescent protein (GFP) genes flanked by loxP sequences on both sides (Figure 2A) and introduced this transgene, together with a gene encoding the chimeric Cre recombinase MerCreMer, into RAD52–/–XRCC3+/– cells by random integration to produce RAD52–/–XRCC3+/– hXRCC3+ cells. The intact XRCC3 allele in the transfected cells was subsequently disrupted by gene targeting to generate rad52 xrcc3 hXRCC3+ cells (Figure 1A). (Hereafter we abbreviate the knockout nomenclature for simplification, e.g. rad52 = RAD52–/–.) MerCreMer carries two mutated hormone-binding domains of the murine estrogen receptor (Zhang et al., 1996, 1998), which binds the antagonist 4-hydroxytamoxifen (OH-TAM). Upon the addition of OH-TAM to the culture media, the chimeric Cre recombinase is transported into the nucleus, where it recognizes loxP sites and deletes both human hXRCC3 and GFP transgenes. The Cre-mediated recombination worked efficiently in rad52 xrcc3 hXRCC3+ DT40 cells. The human XRCC3 and GFP transgenes were deleted in virtually all of the cells within 24 h after the addition of OH-TAM, as verified by genomic PCR (Figure 2B) and flow cytometric analysis of GFP fluorescence (data not shown).
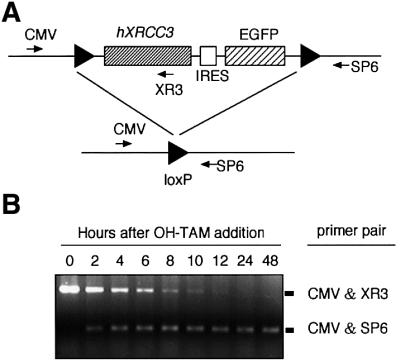
Fig. 2. Time course of MerCreMer-mediated deletion of the hXRCC3 transgene. (A) Schematic representation of the human XRCC3 transgene containing two LoxP recognition sequences (triangles), promoter sequences derived from cytomegalovirus (CMV), internal ribosomal entry site (IRES) and the EGFP gene encoding the enhanced GFP. Exposure of the cells to OH-TAM activates the chimeric Cre recombinase through nuclear localization, causing deletion of both XRCC3 and EGFP. The locations of the three PCR primers are indicated by arrows. (B) The extent of Cre-mediated deletion in the human XRCC3 transgene in rad52 xrcc3 hXRCC3+ hRAD51+ cells was determined by PCR using the indicated primer pairs shown in (A).
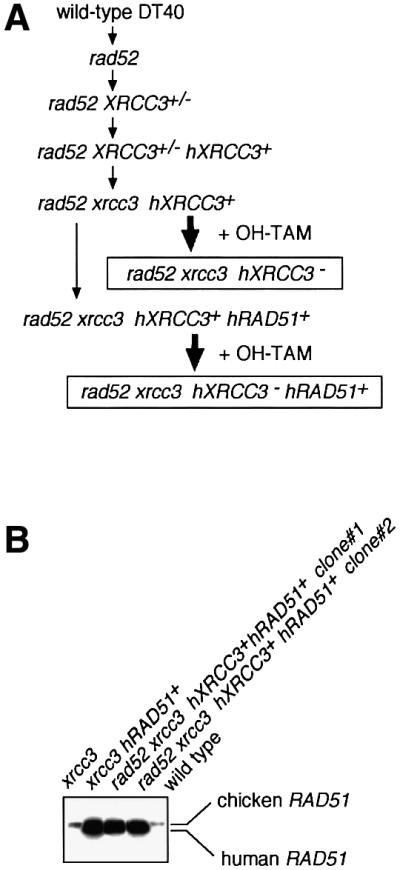
Fig. 1. Experimental strategy. (A) The functional analysis of Rad52 by comparing wild type and rad52, xrcc3 and rad52 xrcc3, and xrcc3 hRAD51+ and rad52 xrcc3 hRAD51+. (B) Western blot analysis of expression of endogenous chicken Rad51 and the human Rad51 transgene.
Figure 1A summarizes the preparation of the DT40 clones employed in the present study. Each gene-targeting event was verified by Southern blotting as previously shown (Takata et al., 2001). The expression levels of human Rad51 transgene that is randomly integrated in xrcc3 hRAD51+ and rad52 xrcc3 hXRCC3+ hRAD51+ clones were measured by western blot analysis (Figure 1B). Two clones of each genotype, which expressed an ∼20-fold higher level of human Rad51 compared with that of endogenous Rad51, were used for subsequent analysis.
Lethality of cells deficient in both XRCC3 and Rad52
The proliferative properties of clones were monitored by growth curves and plating efficiency. As previously observed (Yamaguchi-Iwai et al., 1998; Takata et al., 2001), wild-type and rad52 clones were indistinguishable in their growth properties, whereas xrcc3 cells proliferated significantly more slowly than the wild type (Figure 3A). Similarly, the plating efficiencies of cells in methylcellulose plates were 100% for wild-type and rad52 clones, whereas only ∼70% for xrcc3 cells (Figure 4). We showed previously that higher proportions of dead cells in xrcc3 cultures caused the slower growth rates as well as lower plating efficiencies (Takata et al., 2001).

Fig. 3. Defective proliferation of rad52 xrcc3 hXRCC3– cells. (A and C) Growth curves of the indicated cell cultures in the absence and presence of TAM. The data shown are the average results from two separate clones of each genotype. Standard errors are given by error bars. (B) Cell viability was assessed by flow cytometric analysis of PI uptake and forward scatter (FSC) representing the cell size. A fixed number of plastic beads was added before flow cytometric analysis to calibrate cell number. Cells falling in the R1 and R2 gates identify dead and viable cells, respectively, and numbers given show their percentages. The R3 gate was for the plastic beads, which are used as a reference to measure the cell number.
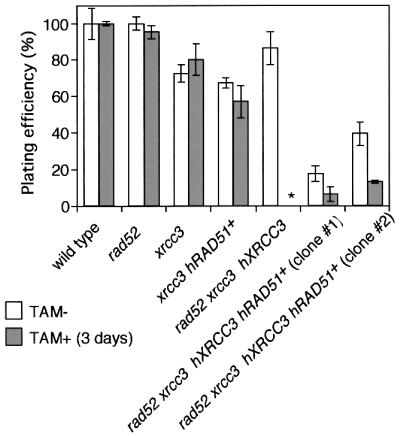
Fig. 4. No detectable colony formation of rad52 xrcc3 hXRCC3– cells. The histogram shows the percentage of metabolically viable cells that gave rise to colonies. The number of viable cells was measured by flow cytometry as shown in Figure 3B before plating cells in methylcellulose plates. Histograms of TAM+ (3 days) show the plating efficiency of cells that were treated with OH-TAM in liquid media for 3 days. No colonies was obtained from the wells plated with 10 000 cells of rad52 xrcc3 hXRCC3– (asterisk).
As OH-TAM covalently binds to DNA (White, 1999), we examined proliferative properties of cells that were exposed to OH-TAM. However, addition of OH-TAM did not affect the proliferation of wild-type and rad52 cells (growth curve in Figure 3A and cloning efficiency in Figure 4). Similarly, exposure of xrcc3 cells to OH-TAM for 3 days did not significantly reduce their plating efficiency. Thus, we used this condition to conduct the following experiments.
While rad52 xrcc3 hXRCC3+ cells multiplied with the same kinetics as wild-type and rad52 cells (Figure 3A, left panel), 3 days after the addition of OH-TAM, rad52 xrcc3 hXRCC3– cells stopped proliferating (Figure 3A, right panel) and began to die (Figure 3B, right panel). These findings were in agreement with the absence of any surviving rad52 xrcc3 hXRCC3– cells after exposure to OH-TAM for 3 days (Figure 4). These observations are in marked contrast to the survival of rad52 and xrcc3 cell populations even after continuous exposure of the cells to OH-TAM (data not shown). From these results, we conclude that cells deficient in both Rad52 and XRCC3 are unable to proliferate.
To investigate the cause of cell death, we analyzed chromosomal breaks in mitotic cells. In agreement with previous findings (Yamaguchi-Iwai et al., 1998; Takata et al., 2001), we found ∼0.1 aberrations per cell in the xrcc3 culture, whereas wild-type and rad52 cells showed few chromosomal aberrations (Figure 5A; Table I). These values were not dependent on OH-TAM. Remarkably, rad52 xrcc3 hXRCC3– cells had 0.08 and 0.38 aberrations per cell at days 2 and 3, respectively, after adding OH-TAM (Tables I and II). We previously showed that the level of spontaneous chromosomal breaks of various HR-deficient DT40 clones is closely correlated with the rate of cell death during the cell cycle (reviewed in Morrison and Takeda, 2000). These observations indicate that the increased chromosomal breaks may account for the massive cell death of rad52 xrcc3 hXRCC3– cells. It should be noted that cells growing with normal kinetics occasionally appeared at <10–5 frequency (Figure 3C). Such clones retained the GFP and XRCC3 transgenes in the continuous presence of OH-TAMs.

Fig. 5. Spontaneous and IR-induced chromosomal aberrations. (A) Spontaneously occurring chromosomal breaks are shown. OH-TAM+ indicates cells that were exposed to OH-TAM for 3 days. One hundred mitotic cells were analyzed in each case. (B) IR-induced chromosomal breaks were determined by subtracting spontaneously occurring breaks from the number of breaks after IR treatment. OH-TAM+ indicates cells that were exposed to OH-TAM for 2 days. The numbers of spontaneous chromosomal breaks are shown in Table I, while IR-induced breaks are shown in Table II.
Table I. Spontaneous chromosomal breaks.
| Cell clone | TAMa | Chromatid type |
Chromosome type |
Total aberrations (per cell ±SE)c | ||
|---|---|---|---|---|---|---|
| Breaksb | Gapsb | Breaksb | Gapsb | |||
| Wild type | – | 0 | 0 | 0 | 1 | 0.01 ± 0.010 |
| + | 0 | 1 | 0 | 0 | 0.01 ± 0.010 | |
| rad52 | – | 1 | 1 | 1 | 0 | 0.03 ± 0.017 |
| + | 0 | 0 | 2 | 2 | 0.04 ± 0.020 | |
| xrcc3 | – | 1 | 1 | 6 | 4 | 0.12 ± 0.035 |
| + | 0 | 3 | 3 | 7 | 0.13 ± 0.036 | |
| rad52 xrcc3 hXRCC3 | – | 2 | 2 | 0 | 2 | 0.06 ± 0.024 |
| + | 7 | 2 | 25 | 4 | 0.38 ± 0.062 | |
| xrcc3 hRAD51+ | – | 0 | 1 | 0 | 1 | 0.02 ± 0.014 |
| + | 0 | 0 | 0 | 2 | 0.02 ± 0.014 | |
| rad52 xrcc3 hXRCC3 hRAD51+ | – | 0 | 0 | 0 | 1 | 0.01 ± 0.010 |
| + | 1 | 4 | 2 | 13 | 0.20 ± 0.045 | |
aCells were treated with colcemid after 3 days of culture in the presence or absence of OH-TAM.
bData are presented as macrochromosomal (1–5 and Z) aberrations per 100 metaphase spreads.
cIf the numbers of cells analyzed and total chromosomal aberrations are defined as N and x, respectively, the number of total aberrations per cell ±SE is calculated as x/N ± √x/N, based on the Poisson distribution of spontaneous chromosomal aberrations we observed previously (Sonoda et al., 1998).
Table II. IR-induced chromosomal breaks.
| Cell clone | IR (Gy)a | Chromatid type | Chromosome type | Total aberrations (per cell ±SE) | ||
|---|---|---|---|---|---|---|
| Breaksb | Gapsb | Breaksb | Gapsb | |||
| Wild type | 0 | 0 | 0 | 0 | 1 | 0.01 ± 0.010 |
| 0.3 | 2 | 0 | 1 | 2 | 0.05 ± 0.022 | |
| 2.0 | 5 | 2 | 5 | 8 | 0.20 ± 0.045 | |
| rad52 | 0 | 1 | 1 | 1 | 0 | 0.03 ± 0.017 |
| 0.3 | 2 | 0 | 4 | 1 | 0.07 ± 0.026 | |
| 2.0 | 7 | 3 | 4 | 2 | 0.16 ± 0.040 | |
| xrcc3 | 0 | 3 | 0 | 3 | 6 | 0.12 ± 0.035 |
| 0.3 | 8 | 3 | 7 | 12 | 0.30 ± 0.055 | |
| 2.0 | 28 | 5 | 4 | 29 | 0.66 ± 0.081 | |
| rad52 xrcc3 hXRCC3+ | 0 | 0 | 2 | 0 | 2 | 0.04 ± 0.020 |
| 0.3 | 1 | 1 | 4 | 0 | 0.06 ± 0.024 | |
| 2.0 | 7 | 4 | 0 | 13 | 0.24 ± 0.049 | |
| rad52 xrcc3 hXRCC3– | 0 | 0 | 0 | 4 | 4 | 0.08 ± 0.028 |
| 0.3 | 16 | 1 | 4 | 13 | 0.34 ± 0.058 | |
| 2.0 | 26 | 19 | 11 | 24 | 0.80 ± 0.089 | |
| rad52 xrcc3 hXRCC3– hRAD51+ | 0 | 0 | 2 | 1 | 7 | 0.10 ± 0.032 |
| 0.3 | 2 | 6 | 0 | 16 | 0.24 ± 0.049 | |
| 2.0 | 0 | 2 | 2 | 52 | 0.56 ± 0.075 | |
Data were calculated and are presented as described for Table I.
aCells were treated with colcemid for 3 h after γ-irradiation. Cells were exposed to OH-TAM for 2 days. At least 100 cells were analyzed.
bThe number of aberrations per 100 cells is presented.
Importance of Rad52 in DSB repair in XRCC3-deficient cells
In order to assess the role of Rad52 in DSB repair, we tried to rescue rad52 xrcc3 cells by expressing the hRad51 cDNA transgene. We previously found that overexpression of hRad51 partially suppressed mutant phenotypes of all Rad51-paralog mutants (Takata et al., 2001). hRad51 overexpression rescued rad52 xrcc3 hXRCC3– cells, although rad52 xrcc3 hXRCC3– hRAD51+ cells proliferated at a significantly slower rate than wild type (Figure 3C). Likewise, only 5–10% of the cells that were exposed to OH-TAM for 3 days gave rise to colonies in methylcellulose plates (Figure 4). The overexpression of hRad51 appears to enhance the repair of spontaneously-occurring DNA damage in rad52 xrcc3 hXRCC3– hRAD51+ cells because their chromosomal breaks were significantly reduced when compared with rad52 xrcc3 hXRCC3– cells (Figure 5A). While this observation is in agreement with the suppression of a mutant phenotype of xrcc3 cells by overexpression of hRad51 (Takata et al., 2001), it is not clear whether this overproduction also substitutes for the lack of Rad52. Rad51 overexpression reduced the plating efficiency of rad52 xrcc3 hXRCC3+ cells but not that of wild-type (data not shown) or xrcc3 cells, implying that Rad51 overexpression is rather toxic in the absence of Rad52.
To analyze the involvement of Rad52 in IR-induced DSB repair, we measured chromosomal breaks following IR by comparing the genotypes of wild type and rad52, and of xrcc3 and rad52 xrcc3. Because of the massive cell death in rad52 xrcc3 cells at day 3 after addition of OH-TAM, we examined the effect of IR at day 2 (Figure 5B; Table II). To evaluate HR-mediated DSB repair capability, we measured chromosomal aberrations in cells that were irradiated in the late S to G2 phases when HR is preferentially used for DSB repair over NHEJ in DT40 cells (Takata et al., 1998). As most cells irradiated in the late S to G2 phases are expected to reach the M phase within 3 h after IR, we measured chromosomal breaks in cells entering mitosis between 0 and 3 h. As expected, rad52 xrcc3 hXRCC3– exhibited greater levels of IR-induced chromosomal aberrations in comparison with xrcc3 (Figure 5B). These results indicate the involvement of Rad52 in repairing IR-induced DSBs in the absence of the XRCC3 gene. This conclusion led us to analyze whether or not introduction of a RAD52 transgene into a xrcc3 clone can suppress its mutant phenotype. However, Rad52 overexpression appears to be toxic to the transfectants because their cloning efficiencies were consistently decreased to 15 to 31% from ∼70% of the parental xrcc3 clone. Furthermore, these Rad52 overexpressing clones were more sensitive to cisplatin than xrcc3 cells by colony formation assay (data not shown). In contrast, Rad51 overexpression in rad52 xrcc3 (Figure 5B) as well as xrcc3 clone (Takata et al., 2001) suppressed their elevated sensitivities to γ-rays.
Discussion
Rad52 and the Rad51 paralogs are complementary in repairing DSB
This is the first genetic study that clearly shows a role of Rad52 in DSB repair in vertebrates. Our results are in agreement with the important role of Rad52 in yeast strains, as well as with co-localization of Rad52 with Rad51 following IR in mammalian cells (Liu and Maizels, 2000). These observations are in marked contrast with no obvious phenotype of rad52 DT40 cells (Yamaguchi-Iwai et al., 1998), murine ES cells and mice (Rijkers et al., 1998). Thus, Rad52 may play an important role in repairing DSBs in XRCC3-deficient cells but not in the wild-type cells. XRCC3 almost fully substitutes for lack of Rad52 while Rad52 can only partially substitute for lack of XRCC3. As cells deficient in the other four Rad51 paralogs have similar phenotypes as XRCC3-deficient cells (Takata et al., 2001), it seems reasonable to expect that the other Rad51 paralogs and Rad52 may also have complementary functions in maintaining chromosomal integrity and repairing IR-induced DSBs.
Although Rad52 is required for virtually every mitotic recombination event in S.cerevisiae (Paques and Haber, 1999), its ortholog in vertebrates appears to be dispensable. There are four possible explanations for this species difference. First, Rad52 may not be involved in conventional HR but in other DNA metabolism pathways in vertebrate cells. However, this possibility is unlikely because immunocytochemical experiments suggest a coordinated response of mammalian Rad52 and Rad51 to DNA damage (Liu and Maizels, 2000). Moreover, both the yeast and human Rad52 proteins form ring structures (Van Dyck et al., 1999; Stasiak et al., 2000) and stimulate DNA-strand exchange promoted by Rad51 protein in vitro (Benson et al., 1998; New et al., 1998; Shinohara and Ogawa, 1998), further emphasizing the conservation of the roles of human and yeast Rad52 in HR. Secondly, there might be an as yet undescribed Rad52 homolog in vertebrates (Kanaar and Hoeijmakers, 1998), although the human genome does not seem to contain other Rad52-like genes (Wood et al., 2001). Thirdly, the relative importance of HR and end joining differs between vertebrates and yeast, such that during evolution the Ku proteins in vertebrate cells may have assumed a portion of the function of Rad52 in yeast DSB repair. The end-joining pathway plays a much more important role in vertebrate cells compared with yeast (Milne et al., 1996; reviewed in Jeggo, 1998; Essers et al., 2000). Biochemical studies suggest that Ku proteins protect DSB ends from exonuclease activity, as does the Rad52 protein (Van Dyck et al., 1999). These results have led to the proposal that recognition of DSB by either Rad52 or Ku protein directs repair by Rad52-dependent HR or Ku-dependent NHEJ pathways, respectively (Van Dyck et al., 1999). To investigate the possibility that the Ku proteins substitute for Rad52 in DSB repair, we generated a DT40 mutant deficient in both Ku70 and Rad52 (rad52 ku70). This double mutant exhibited radiosensitivity very similar to that of ku70 cells (data not shown), although rad54 ku70 cells exhibited much higher sensitivity than either single mutant (Takata et al., 1998). Thus, while the HR and end-joining pathways are complementary to each other in DSB repair, the nearly normal phenotype of Rad52-deficient cells is not explained by an overlapping, compensatory function of the Ku proteins. Fourthly, the precise mechanism of HR likely differs between vertebrates and yeasts so that analogous molecules may compensate for the lack of Rad52 in vertebrate cells. The present data show that a Rad51 paralog (XRCC3) indeed complements defective Rad52 in DSB repair in DT40 cells. This genetic study, combined with biochemical studies (Sung, 1997a,b; Benson et al., 1998; New et al., 1998; Shinohara and Ogawa, 1998) showing that mammalian and yeast Rad52 proteins, as well as yeast Rad55–57, facilitate Rad51-mediated DNA-strand exchange, suggests that Rad52 and the Rad51 paralogs share overlapping roles as co-factors of Rad51 in HR repair.
The Rad51 paralogs and Rad52 may facilitate the action of Rad51 in HR
Among the proteins of the Rad52 epistasis group, Rad51 is the most highly conserved (95% amino acid identity) between humans and chickens (Sonoda et al., 1998), whereas the Rad52 homologs show only 56% identity (Yamaguchi-Iwai et al., 1998). Additionally, human and chicken amino acid sequences of the five Rad51 paralogs showed only modest identities, up to 77% for Rad51B (Takata et al., 2001). Thus, Rad51’s high conservation suggests that it plays a more critical role than Rad52 or the Rad51 paralogs in vertebrates. This notion is supported by the following reverse genetic studies. Upon the depletion of Rad51, a vast majority of the cells were no longer capable of entering the next round of the cell cycle due to massive chromosomal breaks (Sonoda et al., 1998). In contrast, rad52 xrcc3 hRAD51+ DT40 cells exhibited a less severe phenotype, with some cells able to divide several times after deletion of the human XRCC3 transgene. These observations suggest that Rad51 plays a central role in HR repair in vertebrate cells, while both Rad52 and the Rad51 paralogs may act as co-factors of Rad51.
The Rad51 paralogs may have taken over important roles of Rad52 in HR during evolution
Embryonic lethality of mice deficient in XRCC2 (Deans et al., 2000), Rad51B or Rad51D (reviewed in Thompson and Schild, 2001) implies a more essential role for the Rad51 paralogs than Rad52 in mammals. There are two possible reasons. First, single-strand annealing (SSA) requires only Rad52 of the RAD52 epistasis group of genes in yeast, indicating that Rad52 alone is able to facilitate pairing of short stretches of homologous sequences (Mortensen et al., 1996; Sugawara et al., 2000). Active SSA along with other modes of efficient homologous pairing mediated by Rad52 might be useful in yeast, even if such repair events are associated with ectopic recombination and deletion. On the other hand, a higher fidelity of DNA repair may be more important in vertebrates than in yeast to prevent events leading to tumorigenesis. Therefore, homologous pairing mediated by Rad52 should be rather suppressed in vertebrates. Secondly, vertebrates have evolved more proteins that regulate the assembly and activity of Rad51, including the five Rad51 paralogs (Takata et al., 2001; Thompson and Schild, 2001) and the Brca2 cancer susceptibility protein (Davies et al., 2001; Moynahan et al., 2001). Indeed, although HR can operate in DSB repair throughout the cell cycle in yeast, IR-induced Rad51 focus formation in response to DNA damage is absent in the G1 phase in mammalian cells (Bishop et al., 1998), suggesting that HR does not occur under these conditions. The acquisition of such cell cycle specificity, which would avoid heteroallelic recombination leading to the loss of heterozygosity (Takata et al., 1998; Johnson and Jasin, 2000), may have contributed to a diminished role for Rad52 compared with the Rad51 paralogs.
Materials and methods
Plasmid constructs
Two XRCC3 disruption constructs, XRCC3-puro and XRCC3-hygro, were generated from genomic PCR products (Takata et al., 2001). We constructed an expression vector pCR3-loxP-hXRCC3/IRES-EGFP-loxP, in which human XRCC3 and GFP (EGFP) genes are flanked by the loxP sequences, as follows. A DNA fragment containing the loxP sequences and multiple cloning region was excised from pBS246 (Life Technologies, Gland Island, NY) as a 215-bp EcoRI–NotI fragment and subsequently blunt-ended and inserted between the BamHI and NotI sites of the pCR3 expression vector (Invitrogen, Carlsbad, CA) to generate pCR3-loxP-multiple cloning sites (MCS)-loxP. This expression vector was digested at the BamHI site in MCS, and ligated with a 1.3-kb BglII–NotI fragment of pIRES2-EGFP (Clontech, Palo Alto, CA), containing the IRES and EGFP sequences. The resulting plasmid was inserted between the EcoRI and SalI sites with a human XRCC3 cDNA (Liu et al., 1998) tagged with the HA epitope at the C-terminus. To express Rad52 together with GFP, chicken Rad52 cDNA (Yamaguchi-Iwai et al., 1998) was inserted between the EcoRI and SalI sites of the same plasmid. The human RAD51 (Shinohara et al., 1993) was cloned into an expression vector containing the chicken β-actin promoter and the zeocin resistance gene (pA-zeo-hRad51). pAzeo was kindly provided by Tomohiro Kurosaki (Yasuda et al., 2000).
Cell culture, DNA transfection and γ-irradiation
Cells were maintained in RPMI-1640 medium supplemented with 10–5 M β-mercaptoethanol, 10% fetal calf serum (FCS) and 1% chicken serum (Sigma, St Louis, MO) at 39.5°C in the absence of OH-TAM. DNA transfections and selection were performed as described previously (Buerstedde and Takeda, 1991). γ-irradiation was performed using 137Cs (0.02 Gy/s; Gammacell 40, Nordion, Kanata, Ontario, Canada).
Generation of rad52 xrcc3 hXRCC3+ and rad52 xrcc3 hXRCC3+ hRAD51+ cells
The XRCC3-puro targeting construct was transfected into a rad52 null clone (Yamaguchi-Iwai et al., 1998). Double-mutant rad52 XRCC3+/– clones were identified by Southern blot analysis. A rad52 XRCC3+/– clone was co-transfected with both pANMerCreMer-neo (Zhang et al., 1996, 1998; Verrou et al., 1999) and pCR3-loxP-hXRCC3/IRES-EGFP-loxP expression vectors, followed by selection with G418 (2 mg/ml). Among stable transfectants, clones that expressed larger amounts of GFP were identified by FACScaliber (Becton Dickinson, Mountain View, CA) and were isolated and exposed to OH-TAM as previously described. Deletion of the human XRCC3 transgene was examined by FACScaliber and confirmed by Southern blot analysis using a human XRCC3 cDNA as probe. Such rad52 XRCC3+/– hXRCC3+ clones were transfected with the XRCC3-hygro targeting construct to obtain rad52 xrcc3 hXRCC3+ clones. A human Rad51 expression vector, pA-zeo-hRad51, was transfected into rad52 xrcc3 hXRCC3+ as well as xrcc3 cells. Clones expressing hRad51 were identified using western blot analysis using anti-hRad51 antiserum (Figure 1B) (Sonoda et al., 1998). Clones that expressed similar levels of human Rad51 (∼20-fold higher than endogeneous Rad51) were used for subsequent studies.
Monitoring of Cre-mediated recombination using PCR
Genomic DNA was prepared from rad52 xrcc3 hXRCC3– cells at the indicated times following the addition of TAM. PCR was performed with an upstream primer, CMV (5′-CACTGCTTACTGGCTTATCG-3′), and downstream primers, XR3 and SP6 (5′-AGTCTCTTCAAGTCTGGTCC-3′ and 5′-TTTAGGTGACACTATAGAATAG-3′), indicated in Figure 2A.
Flow cytometry and measurement of cloning efficiency
The proliferative properties of clones in the presence and absence of TAM were monitored by FACScalibur using propidium iodide (PI) staining and a fixed number of plastic beads (3 × 105/ml), as previously described (Takata et al., 1998). The number of viable cells was calculated based on the ratio of cell numbers falling in the R2 gate shown in Figure 3B to the number of plastic beads (3 × 105/ml) falling in the R3 gate in flow cytometric analysis. Clonogenic survival of each genotype was monitored by colony formation assay as previously described (Takata et al., 1998). Briefly, appropriate numbers of cells measured by FACScalibur were plated into 6-well cluster plates containing complete medium supplemented with 1.5% methylcellulose (Aldrich, Milwaukee, WI). The effect of OH-TAM on cells was examined in the following manner. Cells were incubated with OH-TAM-containing medium for 3 days, washed with medium once, and plated on OH-TAM-free methylcellulose plates. Colony numbers were counted after 7–10 days. The cloning efficiency was determined as the number of colonies per viable cell (based on the flow cytometric analysis following exposure to OH-TAM; Figure 3B).
Analysis of chromosome breaks
Karyotype analysis was performed as previously described (Sonoda et al., 1998). A 0.1 µg/ml aliquot of colcemid was added for the last 3 h of incubation before harvest. To measure IR-induced chromosomal aberrations, colcemid was added immediately after IR.
Acknowledgments
Acknowledgements
We would like to thank Y.Sato, M.Nagao and M.Fujii for their technical assistance and Prof. Tomohiro Kurosaki (Insititute for Hepatic Research, Kansai Medical School) for kindly providing us with pAzeo plasmid. We also thank Drs Ashok Venkitaraman (University of Cambridge), Kendall L.Knight (University of Massachusetts Medical School) and Edward Egelman (University of Virginia) for critical reading and discussion. Financial support was provided in part by CREST, JST (Saitama, Japan) and Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (M.T. and S.T.) and by grants from The Uehara Memorial Foundation, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, and The Naito Foundation. P.K.D. is a postdoctoral fellow of CREST, JST. A portion of this work was carried out at Lawrence Livermore National Laboratory under the auspices of the US Department of Energy under contract No. W-7405-ENG-48.
References
- Albala J.S., Thelen,M.P., Prange,C., Fan,W., Christensen,M., Thompson,L.H. and Lennon,G.G. (1997) Identification of a novel human RAD51 homolog, RAD51B. Genomics, 46, 476–479. [DOI] [PubMed] [Google Scholar]
- Baumann P. and West,S.C. (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci., 23, 247–251. [DOI] [PubMed] [Google Scholar]
- Benson F.E., Baumann,P. and West,S.C. (1998) Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature, 391, 401–404. [DOI] [PubMed] [Google Scholar]
- Bishop D.K., Ear,U., Bhattacharyya,A., Calderone,C., Beckett,M., Weichselbaum,R.R. and Shinohara,A. (1998) Xrcc3 is required for assembly of Rad51 complexes in vivo. J. Biol. Chem., 273, 21482–21488. [DOI] [PubMed] [Google Scholar]
- Braybrooke J.P., Spink,K.G., Thacker,J. and Hickson,I.D. (2000) The RAD51 family member, RAD51L3, is a DNA-stimulated ATPase that forms a complex with XRCC2. J. Biol. Chem., 275, 29100–29106. [DOI] [PubMed] [Google Scholar]
- Buerstedde J.M. and Takeda,S. (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell, 67, 179–188. [DOI] [PubMed] [Google Scholar]
- Cartwright R., Dunn,A.M., Simpson,P.J., Tambini,C.E. and Thacker,J. (1998a) Isolation of novel human and mouse genes of the recA/RAD51 recombination-repair gene family. Nucleic Acids Res., 26, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright R., Tambini,C.E., Simpson,P.J. and Thacker,J. (1998b) The XRCC2 DNA repair gene from human and mouse encodes a novel member of the recA/RAD51 family. Nucleic Acids Res., 26, 3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A.A., Masson,J., McIlwraith,M.J., Stasiak,A.Z., Stasiak,A., Venkitaraman,A.R. and West,S.C. (2001) Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell, 7, 273–282. [DOI] [PubMed] [Google Scholar]
- Deans B., Griffin,C.S., Maconochie,M. and Thacker,J. (2000) Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. EMBO J., 19, 6675–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosanjh M.K., Collins,D.W., Fan,W., Lennon,G.G., Albala,J.S., Shen,Z. and Schild,D. (1998) Isolation and characterization of RAD51C, a new human member of the RAD51 family of related genes. Nucleic Acids Res., 26, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J., van Steeg,H., de Wit,J., Swagemakers,S.M., Vermeij,M., Hoeijmakers,J.H. and Kanaar,R. (2000) Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J., 19, 1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. (1999) DNA recombination: the replication connection. Trends Biochem. Sci., 24, 271–275. [DOI] [PubMed] [Google Scholar]
- Jeggo P.A. (1998) Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat. Res., 150, 80–91. [PubMed] [Google Scholar]
- Johnson R.D. and Jasin,M. (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J., 19, 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.D. and Symington,L.S. (1995) Functional differences and interactions among the putative RecA homologs Rad51, Rad55 and Rad57. Mol. Cell. Biol., 15, 4843–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaar R. and Hoeijmakers,J.H. (1998) Genetic recombination. From competition to collaboration. Nature, 391, 337–338. [DOI] [PubMed] [Google Scholar]
- Kawabata M. and Saeki,K. (1998) Sequence analysis and expression of a novel mouse homolog of Escherichia coli recA gene. Biochim. Biophys. Acta, 1398, 353–358. [DOI] [PubMed] [Google Scholar]
- Kurumizaka H., Ikawa,S., Nakada,M., Eda,K., Kagawa,W., Takata,M., Takeda,S., Yokoyama,S. and Shibata,T. (2001) Homologous-pairing activity of the human DNA-repair proteins Xrcc3.Rad51C. Proc. Natl Acad. Sci. USA, 98, 5538–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N. et al. (1998) XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA crosslinks and other damages. Mol. Cell, 1, 783–793. [DOI] [PubMed] [Google Scholar]
- Liu Y. and Maizels,N. (2000) Coordinated response of mammalian Rad51 and Rad52 to DNA damage. EMBO Rep., 1, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin A.V., Zaitseva,E., Sung,P. and Kowalczykowski,S.C. (2000) Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. EMBO J., 19, 1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne G.T., Jin,S., Shannon,K.B. and Weaver,D.T. (1996) Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C. and Takeda,S. (2000) Genetic analysis of homologous DNA recombination in vertebrate somatic cells. Int. J. Biochem. Cell Biol., 32, 817–831. [DOI] [PubMed] [Google Scholar]
- Mortensen U.H., Bendixen,C., Sunjevaric,I. and Rothstein,R. (1996) DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl Acad. Sci. USA, 93, 10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Pierce,A.J. and Jasin,M. (2001) BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell, 7, 263–272. [DOI] [PubMed] [Google Scholar]
- New J.H., Sugiyama,T., Zaitseva,E. and Kowalczykowski,S.C. (1998) Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature, 391, 407–410. [DOI] [PubMed] [Google Scholar]
- Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman D.L., Weinberg,L.R. and Schimenti,J.C. (1998) Identification, characterization and genetic mapping of Rad51d, a new mouse and human RAD51/RecA-related gene. Genomics, 49, 103–111. [DOI] [PubMed] [Google Scholar]
- Rice M.C., Smith,S.T., Bullrich,F., Havre,P. and Kmiec,E.B. (1997) Isolation of human and mouse genes based on homology to REC2, a recombinational repair gene from the fungus Ustilago maydis. Proc. Natl Acad. Sci. USA, 94, 7417–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers T., van den Ouweland,J., Morolli,B., Rolink,A.G., Baarends,W.M., Van Sloun,P.P.H., Lohman,P.H.M. and Pastink,A. (1998) Targeted inactivation of MmRAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol., 18, 6423–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A. and Ogawa,T. (1995) Homologous recombination and the roles of double-strand breaks. Trends Biochem. Sci., 20, 387–391. [DOI] [PubMed] [Google Scholar]
- Shinohara A. and Ogawa,T. (1998) Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature, 391, 404–407. [DOI] [PubMed] [Google Scholar]
- Shinohara A., Ogawa,H., Matsuda,Y., Ushio,N., Ikeo,K. and Ogawa,T. (1993) Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nature Genet., 4, 239–243. [DOI] [PubMed] [Google Scholar]
- Sigurdsson S., Trujillo,K., Song,B., Stratton,S. and Sung,P. (2001) Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J. Biol. Chem., 276, 8798–8806. [DOI] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,M.S., Buerstedde,J.-M., Bezzubova,O., Shinohara,A., Ogawa,H., Takata,M., Yamaguchi-Iwai,Y. and Takeda,S. (1998) Rad51 deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J., 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E., Takata,M., Yamashita,Y.M., Morrison,C. and Takeda,S. (2001) Homologous DNA recombination in vertebrate cells. Proc. Natl Acad. Sci. USA, 98, 8388–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasiak A.Z., Larquet,E., Stasiak,A., Muller,S., Engel,A., Van Dyck,E., West,S.C. and Egelman,E.H. (2000) The human Rad52 protein exists as a heptameric ring. Curr. Biol., 10, 337–340. [DOI] [PubMed] [Google Scholar]
- Sugawara N., Ira,G. and Haber,J.E. (2000) DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell. Biol., 20, 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. (1997a) Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem., 272, 28194–28197. [DOI] [PubMed] [Google Scholar]
- Sung P. (1997b) Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev., 11, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y., Shinohara,A. and Takeda,S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M. et al. (2000) The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol., 20, 6476–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M., Sasaki,M.S., Tachiiri,S., Fukushima,T., Sonoda,E., Schild,D., Thompson,L.H. and Takeda,S. (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol., 21, 2858–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs R.S., Zhao,Y., Tucker,J.D., Scheerer,J.B., Siciliano,M.J., Hwang,M., Liu,N., Legerski,R.J. and Thompson,L.H. (1995) Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned cDNA of the XRCC3 DNA repair gene. Proc. Natl Acad. Sci. USA, 92, 6354–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J. (1999) A surfeit of RAD51-like genes? Trends Genet., 15, 166–168. [DOI] [PubMed] [Google Scholar]
- Thompson L.H. and Schild,D. (2001) Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res., 477, 131–153. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T., Fujii,Y., Sakumi,K., Tominaga,Y., Nakao,K., Sekiguchi,M., Matsushiro,A., Yoshimura,Y. and Morita,T. (1996) Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl Acad. Sci. USA, 93, 6236–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E., Stasiak,A.Z., Stasiak,A. and West,S.C. (1999) Binding of double-strand breaks in DNA by human Rad52 protein. Nature, 398, 728–731. [DOI] [PubMed] [Google Scholar]
- Verrou C., Zhang,Y., Zurn,C., Schamel,W.W. and Reth,M. (1999) Comparison of the tamoxifen regulated chimeric Cre recombinases MerCreMer and CreMer. J. Biol. Chem., 380, 1435–1438. [DOI] [PubMed] [Google Scholar]
- White I.N. (1999) The tamoxifen dilemma. Carcinogenesis, 20, 1153–1160. [DOI] [PubMed] [Google Scholar]
- Wood R.D., Mitchell,M., Sgouros,J. and Lindahl,T. (2001) Human DNA repair genes. Science, 291, 1284–1289. [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y., Sonoda,E., Buerstedde,J.-M., Bezzubova,O., Morrison,C., Takata,M., Shinohara,A. and Takeda,S. (1998) Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol., 18, 6430–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T., Maeda,A., Kurosaki,M., Tezuka,T., Hironaka,K., Yamamoto,T. and Kurosaki,T. (2000) Cbl suppresses B cell receptor-mediated phospholipase C (PLC)-γ2 activation by regulating B cell linker protein-PLC-γ2 binding. J. Exp. Med., 191, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Riesterer,C., Ayrall,A.M., Sablitzky,F., Littlewood,T.D. and Reth,M. (1996) Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res., 24, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wienands,J., Zurn,C. and Reth,M. (1998) Induction of the antigen receptor expression on B lymphocytes results in rapid competence for signaling of SLP-65 and Syk. EMBO J., 17, 7304–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]