

Abstract
Adenovirus type 9 (Ad9) is distinct among human adenoviruses because it elicits solely mammary tumors in animals and its primary oncogenic determinant is the E4 region-encoded ORF1 (E4-ORF1) protein. We report here that the PDZ domain-containing protein ZO-2, which is a candidate tumor suppressor protein, is a cellular target for tumorigenic Ad9 E4-ORF1 but not for non-tumorigenic wild-type E4-ORF1 proteins encoded by adenovirus types 5 and 12. Complex formation was mediated by the C-terminal PDZ domain-binding motif of Ad9 E4- ORF1 and the first PDZ domain of ZO-2, and in cells this interaction resulted in aberrant sequestration of ZO-2 within the cytoplasm. Furthermore, transformation-defective Ad9 E4-ORF1 mutants exhibited impaired binding to and sequestration of ZO-2 in cells, and overexpression of wild-type ZO-2, but not mutant ZO-2 lacking the second and third PDZ domains, interfered with Ad9 E4-ORF1-induced focus formation. Our results suggest that the select capacity to complex with the candidate tumor suppressor protein ZO-2 is key to defining the unique transforming and tumorigenic properties of the Ad9 E4-ORF1 oncoprotein.
Keywords: adenovirus/E4-ORF1/PDZ/tumor suppressor/ZO-2
Introduction
The 51 different serotypes of human adenovirus are distributed within six subgroups (A–F) based on physical and hemagglutinating properties of virions. In people, these agents are primarily associated with respiratory, gastrointestinal and ocular infections. Under experimental conditions in rodents, however, viruses comprising subgroups A and B, as well as two viruses from subgroup D, are tumorigenic (Shenk, 1996). Such viruses can be further subdivided into two categories based on the types of tumor they elicit in animals and the viral genes responsible for their oncogenic potential. Adenoviruses from subgroups A and B generate undifferentiated sarcomas, and the oncogenic determinants of these viruses are their E1A and E1B genes (Graham, 1984). In contrast, human adenovirus type 9 (Ad9) from subgroup D elicits only estrogen-dependent mammary tumors (Javier et al., 1991), and its oncogenic determinant is the E4 region-encoded ORF1 (E4-ORF1) gene (Javier, 1994; Thomas et al., 1999, 2001). Whereas the oncogenic potential of the E1A and E1B proteins stem largely from their capacity to inactivate the crucial cellular pRb and p53 tumor suppressors (Shenk, 1996), respectively, the mechanisms underlying tumorigenesis by the Ad9 E4-ORF1 protein are still unknown.
The Ad9 E4-ORF1 gene encodes a 125 amino acid residue polypeptide which, following expression in the rat embryo fibroblast cell line CREF, induces a multitude of transformed properties, including morphological changes, focus formation, anchorage-independent growth and increased saturation densities (Weiss et al., 1996). With the exception of adenovirus types 40 and 41 comprising subgroup F, all human adenoviruses code for an E4-ORF1 protein. Representative E4-ORF1 polypeptides from different viral subgroups display sequence similarities ranging from 45% to 51% identity and, in addition, these viral proteins share the ability to induce anchorage-independent growth in human TE85 cells (Weiss et al., 1997b), revealing a common transforming activity in vitro. Nevertheless, among this family of viral proteins, only Ad9 E4-ORF1 possesses the capacities to promote tumors in animals and to transform CREF cells (Javier, 1994; Weiss et al., 1997b; Thomas et al., 2001). An interesting possible explanation for the unique properties of Ad9 E4-ORF1 would be that it possesses an undetermined activity absent from other adenovirus E4-ORF1 proteins.
The results of mutational analyses demonstrate that transformation of CREF cells by Ad9 E4-ORF1 is dependent on three separate protein regions (Weiss et al., 1997a). One region located at the extreme C-terminus of Ad9 E4-ORF1 has been found to represent a PDZ domain-binding motif that mediates interactions with a specific group of cellular PDZ domain-containing proteins (Lee et al., 1997; Weiss and Javier, 1997). Three of these PDZ proteins were recently identified as the multi-PDZ protein MUPP1 and the Membrane-Associated GUanylate Kinase (MAGUK) homology proteins DLG and MAGI-1 (Lee et al., 1997, 2000; Glaunsinger et al., 2000). These interactions result from the ability of the Ad9 E4-ORF1 C-terminal motif to bind specific PDZ domains within each cellular factor. Also pertinent is that these three PDZ proteins represent common cellular targets for the subgroup C adenovirus type 5 (Ad5) and subgroup A adenovirus type 12 (Ad12) E4-ORF1 proteins, both of which fail to promote tumors in animals and to transform CREF cells (Lee et al., 1997, 2000; Glaunsinger et al., 2000). Thus, despite making predicted important contributions to the limited transforming potential common to all adenovirus E4-ORF1 proteins, interactions with MUPP1, MAGI-1 and DLG would not account for the additional, unique properties of Ad9 E4-ORF1.
PDZ domains are protein–protein interaction modules present primarily within cellular factors that function in signal transduction (Craven and Bredt, 1998). A distinguishing feature of these domains is that their recognition motifs are typically located at the extreme C-terminus of target proteins (Songyang et al., 1997). Several different types of PDZ domain-binding motifs are known (Fanning and Anderson, 1999) and, at their C-termini, adenovirus E4-ORF1 proteins possess a type I motif having the consensus sequence -(S/T)-X-(V/I/L)-COOH (where X is any amino acid residue). With respect to known functions for PDZ proteins, these cellular factors generally act as scaffolding proteins, which organize membrane receptors and cytosolic proteins into large signaling complexes and localize these large complexes to specialized membrane sites of cell–cell contact (Craven and Bredt, 1998; Fanning and Anderson, 1999).
While the precise signaling functions of the Ad9 E4-ORF1-associated PDZ proteins have not been determined, it seems pertinent that DLG is a mammalian homolog of the Drosophila discs large (dlg) tumor suppressor protein (Lue et al., 1994; Muller et al., 1995). In addition, overexpression of DLG has been shown to block progression of NIH 3T3 fibroblasts from G0/G1 to S phase of the cell cycle (Ishidate et al., 2000). Such findings have led us to propose the two hypotheses that DLG, and perhaps other Ad9 E4-ORF1-associated PDZ proteins, function to suppress abnormal cellular proliferation and that Ad9 E4-ORF1 targets these cellular factors for inactivation. Our results showing that Ad9 E4-ORF1 aberrantly sequesters MUPP1 and MAGI-1 in cells are consistent with these ideas (Glaunsinger et al., 2000; Lee et al., 2000).
We reported previously that, in addition to binding MUPP1, MAGI-1 and DLG, Ad9 E4-ORF1 likewise complexes with two unidentified cellular PDZ proteins designated p155 and p160 (Weiss and Javier, 1997). Contrary to other Ad9 E4-ORF1-associated PDZ proteins, however, p160 fails to interact with non-tumorigenic Ad5 and Ad12 E4-ORF1 (Weiss and Javier, 1997). The goal of the present study was to identify this uniquely Ad9 E4-ORF1-specific binding protein. We demonstrate here that p160 is the cellular MAGUK protein ZO-2 (Jesaitis and Goodenough, 1994; Beatch et al., 1996), which was recently identified as a candidate tumor suppressor protein (Chlenski et al., 1999a,b, 2000). Regarding the latter assertion, ZO-2 expression was either lost or significantly decreased in 80% (4/5) of breast cancer lines examined and in 83% (5/6) of primary breast adenocarcinomas examined, although this effect was rarely seen in colon cancers or prostate adenocarcinomas (Chlenski et al., 2000). Moreover, the ZO-2 gene utilizes two alternative promoters, giving rise to two ZO-2 isoforms that differ at their N-terminus by 23 amino acid residues. Whereas both ZO-2 isoforms were detected in normal pancreatic duct epithelial cells, the longer isoform was absent in 90% (9/10) of pancreatic duct carcinoma lines examined and in 100% (4/4) of primary pancreatic adenocarcinomas examined (Chlenski et al., 1999a,b). In accordance with these observations, we show that the transforming potential of Ad9 E4-ORF1 in CREF cells is associated with its ability to bind and aberrantly sequester ZO-2 and that overexpression of ZO-2 in these cells inhibits Ad9 E4-ORF1-induced transformation. Additional results also confirm the expected failure of ZO-2 to bind non-tumorigenic adenovirus E4-ORF1 proteins. In light of these findings, we propose that the exclusive interaction between ZO-2 and Ad9 E4-ORF1 bestows distinct transforming and tumorigenic properties on this viral oncoprotein.
Results
Ad9 E4-ORF1 uniquely complexes with ZO-2 in cells
Ad9 E4-ORF1 is set apart from other adenovirus E4-ORF1 polypeptides by its abilities to promote tumors in animals and to transform CREF cells. With respect to a specific Ad9 E4-ORF1 activity that may account for these unique properties, we previously detected an unidentified cellular factor, p160, which binds to tumorigenic Ad9 E4-ORF1 but not to non-tumorigenic Ad12 and Ad5 E4-ORF1 (Weiss and Javier, 1997). Given that disruption of the PDZ domain-binding motif of Ad9 E4-ORF1 abolishes its interaction with p160 (Weiss and Javier, 1997), we reasoned that p160 is a cellular PDZ domain-containing protein. A search for known 160 kDa cellular PDZ proteins led to ZO-2 (Beatch et al., 1996), a cell junction-associated MAGUK protein related to the Ad9 E4-ORF1-associated proteins DLG and MAGI-1. In addition, recent reports indicate that ZO-2 is a candidate tumor suppressor protein (Chlenski et al., 1999a,b, 2000). These observations prompted studies to assess whether p160 is ZO-2.
In GST pulldown assays, wild-type ZO-2 tagged at its N-terminus with an HA epitope (HA–ZO-2) and expressed in COS-7 cells complexed with the wild-type Ad9 E4-ORF1 GST fusion protein but not with the GST protein control (Figure 1A). This interaction is specific because, in similar assays, we have shown that Ad9 E4-ORF1 fails to bind other cellular PDZ proteins, including the closely related MAGUK proteins ZO-1 and ZO-3, the multi-PDZ proteins FAP-1 and hINADL, and the Ras effector protein AF-6 (Glaunsinger et al., 2000). Transformation-defective Ad9 E4-ORF1 mutants having altered PDZ domain-binding motifs were also tested in the GST pulldown assays. Mutant IIIA lacks a functional PDZ domain-binding motif and detectable transforming activity in CREF cells, whereas mutants IIIC and IIID have less disruptive PDZ domain-binding motif mutations and retain weak transforming activity in these cells (Weiss et al., 1997a). In pulldown assays, mutants IIIA and IIIC failed to interact with HA–ZO-2, while mutant IIID displayed some binding activity, albeit substantially less than that of the wild-type viral protein (Figure 1A). Moreover, the results of co-immunoprecipitation assays performed with COS-7 cells transiently co-expressing HA–ZO-2 and either wild-type or mutant Ad9 E4-ORF1 agreed with those of the GST pulldown assays (Figure 1B). Particularly notable was the failure of Ad5 and Ad12 E4-ORF1 to complex with HA–ZO-2 in GST pulldown assays (Figure 1C), despite the presence of a functional type I PDZ domain-binding motif at the C-terminus of these viral proteins (Lee et al., 1997, 2000; Glaunsinger et al., 2000). We likewise did not detect binding of HA–ZO-2 to non-tumorigenic subgroup B adenovirus type 3 E4-ORF1, or the HTLV-1 Tax and high-risk HPV E6 oncoproteins (our unpublished data) (Glaunsinger et al., 2000). Therefore, binding of ZO-2 to Ad9 E4-ORF1 is highly selective and dependent on the PDZ domain-binding motif of this viral protein.
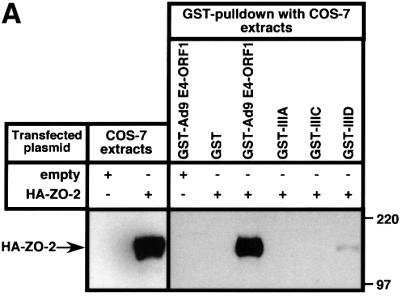

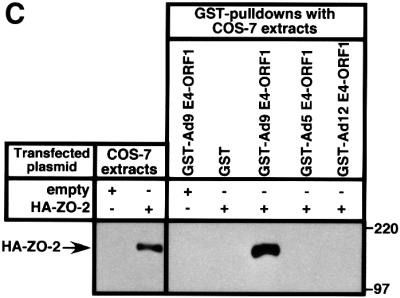
Fig. 1. Binding of wild-type but not mutant Ad9 E4-ORF1 to ZO-2 in GST pulldown assays. (A) ZO-2 binds to wild-type Ad9 E4-ORF1 in GST pulldown assays. Protein (100 µg) from RIPA buffer-lysed COS-7 cells transfected with 5 µg of either empty GW1 plasmid or GW1 plasmid expressing HA–ZO-2 was subjected to GST pulldown assays with the fusion protein indicated. Recovered proteins were separated by SDS–PAGE and immunoblotted with anti-HA antibodies. As a control, one-tenth the amount of protein used in GST pulldown assays was directly immunoblotted with the same antibodies. (B) Wild-type Ad9 E4-ORF1 co-immunoprecipitates with HA–ZO-2. Protein (150 µg) from RIPA buffer-lysed COS-7 cells co-transfected with 5 µg of GW1 plasmid expressing HA–ZO-2 and 5 µg of either empty GW1 plasmid or GW1 plasmid expressing wild-type or the indicated mutant Ad9 E4-ORF1 was immunoprecipitated with Ad9 E4-ORF1 antibodies. Recovered proteins were separated by SDS–PAGE and immunoblotted with either anti-HA or anti-Ad9 E4-ORF1 antibodies. As a control, one-tenth the amount of protein used in the immunoprecipitation reactions was directly immunoblotted with the same antibodies. (C) Binding of ZO-2 to Ad9 E4-ORF1 but not to Ad5 and Ad12 E4-ORF1 in GST pulldown assays. Experiments were performed as described above in (A).
We were next interested in determining whether Ad9 E4-ORF1 also complexes with ZO-2 endogenously expressed in cells. For specific detection of ZO-2, we raised rabbit polyclonal antisera to a unique 100 amino acid region between ZO-2 PDZ2 and PDZ3 (amino acids 394–495). The ZO-2 antisera, but not the matched pre-immmune sera (our unpublished data), recognized an expected 160 kDa polypeptide in CREF cells and several other cell lines, including Madine-Darby canine kidney (MDCK) cells, human 293 and TE85 cells, and murine 3T3 fibroblasts (Figure 2A). The size of the protein detected in these lines was identical to that of authentic wild-type ZO-2 exogenously expressed in COS-7 cells. We also demonstrated that ZO-2 and Ad9 E4-ORF1-associated protein p160 co-migrate in a protein gel (Figure 2B). This finding, coupled with results showing that these two proteins likewise display identical binding profiles to Ad9 E4-ORF1 mutants and fail to interact with Ad5 and Ad12 E4-ORF1 (see Figure 1C) (Weiss and Javier, 1997), argue strongly that ZO-2 and p160 are the same protein.
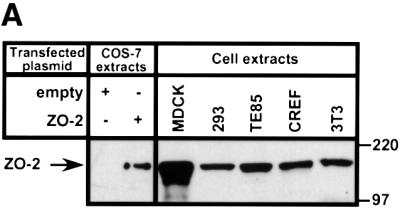
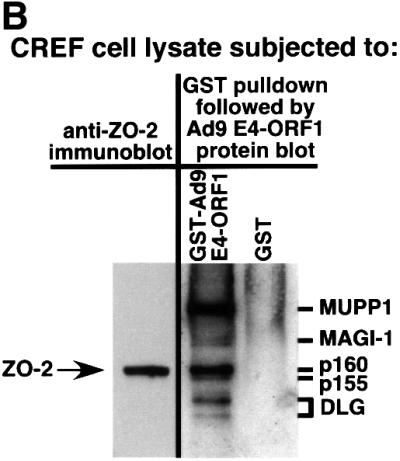
Fig. 2. Co-migration of ZO-2 and Ad9 E4-ORF1-associated protein p160. (A) Gel mobilities of ZO-2 proteins expressed in various cell lines. Protein (100 µg) from the indicated RIPA buffer-lysed cells was separated by SDS–PAGE and immunoblotted with ZO-2 antibodies. As controls, 2.5 µg of protein from RIPA buffer-lysed COS-7 cells transfected with 4 µg of either empty GW1 plasmid or GW1 plasmid expressing wild-type ZO-2 was run on the same protein gel. (B) Ad9 E4-ORF1-associated cellular protein p160 co-migrates with endogenous ZO-2 of CREF cells. Protein (50 µg) from RIPA buffer-lysed normal CREF cells was separated by SDS–PAGE and then immunoblotted with ZO-2 antibodies (left panel). Alternatively, 4 mg of protein from RIPA buffer-lysed normal CREF cells was subjected to GST pulldown assays with the fusion proteins indicated. Recovered proteins were separated by SDS–PAGE and blotted with a radiolabeled Ad9 E4-ORF1 protein probe (right panel). Samples in both panels were run on the same gel to allow comparison of protein mobilities.
To examine whether Ad9 E4-ORF1 also complexes with endogenous ZO-2 in CREF cells, we employed the ZO-2 antiserum in co-immunoprecipitation assays. The results, shown in Figure 3, indicated that wild-type Ad9 E4-ORF1, but none of the Ad9 E4-ORF1 mutants, co-immunoprecipitates with endogenous ZO-2 from lysates of CREF cells stably expressing these viral proteins. These results in CREF cells differed slightly from those in COS-7 cells (see Figure 1A and B), where viral mutant IIID showed substantially diminished yet detectable binding to overexpressed ZO-2. This discrepancy likely reflects enhanced detection of weak binding between mutant IIID and ZO-2 when high levels of this cellular protein are expressed in COS-7 cells. Nonetheless, these findings are important in suggesting that the interaction between ZO-2 and Ad9 E4-ORF1 is required for Ad9 E4-ORF1-induced transformation of CREF cells.

Fig. 3. Binding of wild-type but not mutant Ad9 E4-ORF1 to endogenous ZO-2 of CREF cells. One milligram of protein from RIPA buffer-lysed normal CREF cells or CREF cells stably expressing wild-type or mutant Ad9 E4-ORF1 were immunoprecipitated with ZO-2 antiserum or the matched pre-immune serum (pre). Recovered proteins were separated by SDS–PAGE and immunoblotted with either ZO-2 or Ad9 E4-ORF1 antiserum (top panel). As a control, 100 µg of protein from RIPA buffer-lysed normal CREF cells was also directly immunoblotted with the same antisera (bottom panel). Sample 1 of the top panel was not analyzed in the bottom panel.
Ad9 E4-ORF1 binds ZO-2 PDZ1
To initially identify the ZO-2 PDZ domain(s) that mediates binding to Ad9 E4-ORF1, we blotted membrane-immobilized fragments of ZO-2 with a radiolabeled Ad9 E4-ORF1 protein probe. In these assays, Ad9 E4-ORF1 interacted with ZO-2 PDZ1 but not with a ZO-2 fragment containing both PDZ2 and PDZ3 (PDZ2+3) (Figure 4A and B). This interaction required the Ad9 E4-ORF1 PDZ domain-binding motif because a mutant IIIA protein probe failed to react with either of these ZO-2 peptides (our unpublished data). Consistent with results of the protein blotting assays, wild-type ZO-2 and mutant ZO-2 missing both PDZ2 and PDZ3 (HA–ZO-2ΔPDZ2+3) co-immunoprecipitated with Ad9 E4-ORF1 from lysates of COS-7 cells, whereas mutant ZO-2 missing PDZ1 (HA–ZO-2ΔPDZ1) did not (Figure 5A and C). Therefore, ZO-2 PDZ1 is both necessary and sufficient for mediating ZO-2 binding to Ad9 E4-ORF1 in cells.

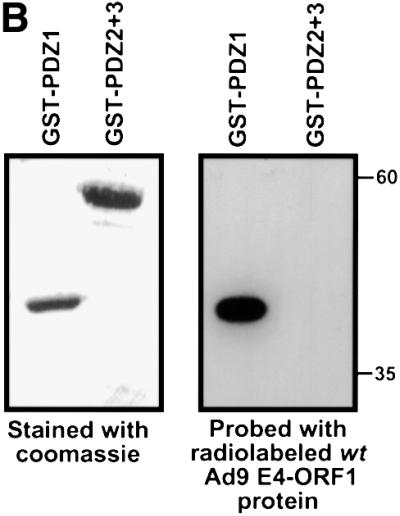

Fig. 4. ZO-2 PDZ1 mediates binding of ZO-2 to Ad9 E4-ORF1. (A) Illustration of ZO-2 protein fragments and ZO-2 deletion mutants. (B) Specific binding of Ad9 E4-ORF1 to ZO-2 PDZ1 in protein blotting assays. Approximately 5 µg of the indicated ZO-2 GST fusion protein, separated by SDS–PAGE and immobilized on a membrane, was either stained with Coomassie Blue dye to verify the presence of equivalent amounts of protein (left panel) or probed with a radiolabeled wild-type Ad9 E4-ORF1 protein probe (right panel). (C) Requirement of ZO-2 PDZ1 for ZO-2 to co-immunoprecipitate with Ad9 E4-ORF1 from cell extracts. Protein (400 µg) from RIPA buffer-lysed COS-7 cells co-transfected with 5 µg of either empty GW1 plasmid or GW1 plasmid expressing Ad9 E4-ORF1 and 5 µg of GW1 plasmid expressing wild-type HA–ZO-2 or mutant HA–ZO-2 lacking either PDZ1 (HA–ZO-2ΔPDZ1) or both PDZ2 and PDZ3 (HA–ZO-2ΔPDZ2+3) was immunoprecipitated with Ad9 E4-ORF1 antibodies. Recovered proteins were separated by SDS–PAGE and immunoblotted with anti-HA or anti-Ad9 E4-ORF1 antibodies. As a control, one-fifteenth the amount of protein used in the immuno precipitation reactions was also directly immunoblotted with the same antibodies.



Fig. 5. Ad9 E4-ORF1 aberrantly sequesters ZO-2 in the cytoplasm of CREF cells. (A) Distribution of ZO-2 in normal CREF cells or CREF cells stably expressing either wild-type or the indicated mutant Ad9 E4-ORF1. IF assays were performed with either affinity-purified ZO-2 antibodies (a, c–f) or normal rabbit IgG (b) and visualized by fluorescence microscopy. (B) ZO-2 and Ad9 E4-ORF1 co-localize within cytoplasmic punctate bodies in CREF cells. Double-label IF assays were performed using both affinity-purified ZO-2 and anti-HA antibodies in CREF cells stably expressing HA–Ad9 E4-ORF1. Each of the three panels represents the same field of five cells stained for ZO-2 (left panel), HA–Ad9 E4-ORF1 (center panel) or the merged images (right panel). (C) Ad9 E4-ORF1 aberrantly sequesters ZO-2 within detergent-insoluble complexes in CREF cells. Normal CREF cells or CREF cells stably expressing either wild-type or the indicated mutant Ad9 E4-ORF1 were lysed in RIPA buffer, and extracts were centrifuged to produce RIPA buffer-soluble (S) supernatant and RIPA buffer-insoluble (I) pellet fractions. Protein (100 µg) from S fractions or an equivalent amount from I fractions (see Materials and methods) was separated by SDS–PAGE and immunoblotted with either ZO-2 or Ad9 E4-ORF1 antiserum.
Ad9 E4-ORF1 aberrantly sequesters ZO-2 in the cytoplasm of CREF cells
We have shown previously that association of Ad9 E4-ORF1 with the cellular PDZ proteins MUPP1 and MAGI-1 results in their aberrant sequestration within punctate bodies in the cytoplasm of CREF cells (Glaunsinger et al., 2000; Lee et al., 2000). It was therefore of interest to examine whether ZO-2 is similarly affected by Ad9 E4-ORF1 in these cells. ZO-2 has been reported to localize primarily at tight junctions in polarized epithelial cells (Jesaitis and Goodenough, 1994), or at adherens junctions in non-epithelial cells such as mouse 3T3 and rat 3Y1 fibroblasts that lack tight junctions (Itoh et al., 1999b). Consistent with these observations, immunofluorescence (IF) assays performed with our antiserum to ZO-2 revealed prominent cell–cell contact staining for this cellular protein in MDCK epithelial cells (our unpublished data). In IF assays using affinity-purified ZO-2 antibodies with normal CREF fibroblasts, however, we found that ZO-2 was located primarily in the cytoplasm with a diffuse staining pattern (Figure 5A). In contrast, the majority of ZO-2 in CREF cells stably expressing wild-type Ad9 E4-ORF1 was instead aberrantly sequestered within cytoplasmic punctate bodies (Figure 5A). Double-labeling IF assays further showed that ZO-2 and wild-type Ad9 E4-ORF1 co-localize in these structures (Figure 5B). This effect is linked to Ad9 E4-ORF1-induced transformation in that transformation-defective mutants IIIA and IIIC failed, or mutant IIID showed a substantially reduced capacity, to aberrantly sequester ZO-2 in these cells (Figure 5A).
The results of cell fractionation experiments confirmed the aberrant sequestration of ZO-2 by Ad9 E4-ORF1 in CREF cells. In these experiments, normal CREF cells or CREF cells stably expressing either wild-type or mutant Ad9 E4-ORF1 were lysed in RIPA buffer, and the cell extracts were separated by centrifugation into soluble (S) supernatant and insoluble (I) pellet fractions. Immunoblot analyses with ZO-2 antiserum were carried out to determine the relative amount of ZO-2 present in each fraction. In normal CREF cells, ZO-2 was present primarily in the soluble fraction whereas, in CREF cells stably expressing wild-type Ad9 E4-ORF1, the majority of ZO-2 was redistributed into the insoluble fraction (Figure 5C). Concordant with results of IF assays (see Figure 5A), we also found that ZO-2 from CREF lines expressing Ad9 E4-ORF1 mutants exhibited a fractionation profile similar to that of normal CREF cells (Figure 5C). In addition, the ZO-2 protein present in the insoluble fraction of wild-type Ad9 E4-ORF1-expressing CREF cells displayed a reduced gel mobility compared with that of normal CREF cells or CREF cells expressing Ad9 E4-ORF1 mutants (Figure 5C; see also Figure 3A). This finding suggests that, in addition to aberrantly sequestering ZO-2, wild-type Ad9 E4-ORF1 also promotes an unknown post-translational modification(s) to this cellular factor.
ZO-2 blocks Ad9 E4-ORF1-mediated transformation of CREF cells
Recent reports showing that ZO-2 expression is lost or reduced in certain human cancers (Chlenski et al., 1999b, 2000) may indicate that ZO-2 suppresses the neoplastic growth of cells. This idea prompted experiments to determine whether overexpression of ZO-2 can inhibit transformation by Ad9 E4-ORF1. In accordance with our previous findings (Weiss et al., 1996), transfection of a wild-type Ad9 E4-ORF1 expression plasmid into CREF cells resulted in numerous transformed foci (Figure 6A). In similar assays, however, inclusion of a plasmid expressing either wild-type HA–ZO-2 or mutant HA–ZO-2ΔPDZ1 significantly decreased the number of Ad9 E4-ORF1-induced foci (2.9- and 2.5-fold reductions, respectively), whereas inclusion of a plasmid expressing mutant HA–ZO-2ΔPDZ2+3 did not (1.1-fold reduction) (Figure 6A). The latter defect of HA–ZO-2ΔPDZ2+3 does not result from an expression deficiency, as this mutant achieved steady-state protein levels comparable to those of wild-type HA–ZO-2 in both COS-7 cells (see Figure 4C) and CREF cells (our unpublished data). These findings are important in providing direct evidence that ZO-2 possesses a transformation-repressive activity and in specifically localizing this activity to the protein sequences deleted from HA–ZO-2ΔPDZ2+3.


Fig. 6. ZO-2 inhibits oncogene-induced focus formation in CREF cells. (A) ZO-2 interferes with Ad9 E4-ORF1-induced focus formation. CREF cells were transfected either alone with 8 µg of empty GW1 plasmid or GW1 plasmid expressing the indicated wild-type or mutant HA–ZO-2 protein (lanes 1–4) or together with 2 µg of pJ4Ω plasmid expressing wild-type Ad9 E4-ORF1 (lanes 5–8). At 3 weeks post-transfection, transformed foci were counted. Numbers of transformed foci are presented as percent relative to the Ad9 E4-ORF1 plasmid alone (control), which was normalized to 100%. Data are compiled from three independent assays, each performed in duplicate. (B) ZO-2 also blocks focus formation by the RasV12 and polyomavirus middle T oncoproteins. CREF cells were transfected either alone with 9 µg of empty GW1 plasmid or GW1 plasmid expressing wild-type HA–ZO-2 protein (lanes 1–2) or together with 3 µg of either pJ4Ω plasmid expressing wild-type Ad9 E4-ORF1 (lanes 3–4), GW1 plasmid expressing RasV12 (lanes 5–6) or pPyMT1 plasmid expressing polyomavirus middle T (PyMT) (lanes 7–8). Assays were scored as indicated above in (A). Numbers of transformed foci are presented as percent relative to the respective Ad9 E4-ORF1, RasV12 or PyMT plasmid alone (control), each of which was normalized to 100%. Data are compiled from two independent assays, each performed in duplicate.
Nevertheless, our results with mutants HA–ZO- 2ΔPDZ1 and HA–ZO-2ΔPDZ2+3, showing that suppression of Ad9 E4-ORF1-induced focus formation by overexpressed ZO-2 was independent of the interaction between this cellular protein and Ad9 E4-ORF1, were somewhat unexpected (Figure 6A). One possible explanation for this observation is provided by a study examining the interaction between the adenovirus E1A oncoprotein and the cellular p300 transcriptional co-activator (Eckner et al., 1994). Although E1A must bind to and sequester p300 to transform cells, it was found that p300 overexpression can overcome E1A-mediated transcriptional repression of the SV40 promoter in either an interaction-independent manner when E1A levels are low or an interaction-dependent manner when E1A levels are high. Hence, we postulate that, for ZO-2 suppression of Ad9 E4-ORF1-induced focus formation, the levels of ZO-2 relative to Ad9 E4-ORF1 stably expressed in the CREF cells are above those needed to reveal the expected interaction dependence of this effect.
As our results imply that overexpression of ZO-2 suppresses transformation by Ad9 E4-ORF1 by elevating the levels of free, active transformation-inhibitory ZO-2 in cells, we predicted that ZO-2 would similarly be able to suppress transformation by other viral or cellular oncoproteins. Consistent with this idea, we showed that overexpression of HA–ZO-2 in CREF cells likewise significantly diminishes focus formation by the activated RasV12 and the polyomavirus middle T proteins (3.7- and 2.9-fold reduction, respectively) (Figure 6B), which lack any recognizable PDZ domain-binding motifs.
Discussion
Among human adenovirus E4-ORF1 proteins, Ad9 E4-ORF1 is unique in its abilities to promote tumors in animals and to transform CREF cells. One possible explanation for these striking distinctions would be that Ad9 E4-ORF1 possesses a crucial oncogenic activity lacking from non-tumorigenic adenovirus E4-ORF1 proteins. Consistent with this notion, we have detected previously a 160 kDa cellular polypeptide that complexes with Ad9 E4-ORF1, yet fails to interact with non-tumorigenic Ad5 and Ad12 E4-ORF1 (Weiss and Javier, 1997). Here, we identified this specific Ad9 E4-ORF1-associated 160 kDa polypeptide as the cellular MAGUK protein ZO-2. Additional results in CREF cells demonstrated that transformation-defective Ad9 E4-ORF1 mutants either failed or showed a substantially reduced capacity to bind ZO-2 and that overexpression of this cellular protein blocked Ad9 E4-ORF1-induced focus formation. These findings are significant in suggesting that this highly specific interaction with ZO-2 is responsible for the unique tumorigenic properties of the Ad9 E4-ORF1 oncoprotein.
It is also important to note that Ad9 E4-ORF1 interacts with additional cellular PDZ proteins, including MUPP1, MAGI-1 and DLG, and that transformation-defective Ad9 E4-ORF1 mutants likewise display impaired binding to these cellular factors (Lee et al., 1997, 2000; Weiss and Javier, 1997; Glaunsinger et al., 2000). These three PDZ proteins also represent common cellular targets for Ad5 and Ad12 E4-ORF1 which, despite lacking tumorigenic potential, are similar to Ad9 E4-ORF1 in having the ability to transform anchorage-dependent human TE85 cells to grow in soft agar (Lee et al., 1997, 2000; Weiss et al., 1997b; Glaunsinger et al., 2000). We therefore favor a model whereby the combined interactions of multiple cellular PDZ proteins with Ad9 E4-ORF1 are required for manifestation of its full transforming potential. Never theless, the fact that ZO-2 binds to tumorigenic Ad9 E4-ORF1, but not to non-tumorigenic Ad5 and Ad12 E4-ORF1, suggests that this particular interaction plays a central role in defining the unique tumorigenic properties of Ad9 E4-ORF1. Moreover, whereas Ad9 E4-ORF1 efficiently transforms CREF cells, we have been unsuccessful in our attempts to transform CREF cells with Ad5 and Ad12 E4-ORF1 or to establish CREF lines stably expressing these proteins (Weiss et al., 1997b), even though they can be transiently expressed to levels comparable to those of Ad9 E4-ORF1 in these cells (S.S.Lee, unpublished data). The latter observations may indicate that the ZO-2 interaction also serves to broaden the range of cell types susceptible to transformation by Ad9 E4-ORF1. This idea is attractive, as our findings argue that Ad9 E4-ORF1 plays an important role in targeting tumorigenesis by Ad9 exclusively to cells of the rat mammary gland (Thomas et al., 2001).
ZO-2 was originally identified through its association with the closely related MAGUK protein ZO-1 at tight junctions (Jesaitis and Goodenough, 1994), specialized cell–cell contact sites forming a belt-like region that separates the apical from the lateral plasma membrane in polarized epithelial cells. ZO-2 has subsequently been found to associate with a number of other cellular proteins, including tight junction transmembrane proteins occludin and claudins (Itoh et al., 1999a; Wittchen et al., 1999), tight junction submembranous proteins ZO-3 and cingulin (Haskins et al., 1998; Cordenonsi et al., 1999), adherens junction protein α-catenin (Itoh et al., 1999b), actin-associated non-erythroid protein 4.1R (Mattagajasingh et al., 2000) and F-actin (Wittchen et al., 1999). Therefore, it is believed that ZO-2 acts as a bridge linking transmembrane proteins with the actin cytoskeleton at specialized membrane regions of cell–cell contact, as well as a regulator of signals emanating from these sites (Gonzalez-Mariscal et al., 2000). Such processes have a clear association with carcinogenesis because the hallmark properties of transformed cells, including morphological changes, anchorage-independent growth, and loss of contact inhibition, are often traced both to deficiencies in processing signals transmitted from neighboring cells and to disruption of the cytoskeleton (Ben-Ze’ev, 1997).
Although ZO-2 has been found to localize at adherens junction membrane sites in non-epithelial cells (Jesaitis and Goodenough, 1994; Itoh et al., 1999b), we found it present primarily in the cytoplasm of CREF fibroblasts. This observation is consistent with our previous results showing that MUPP1 and MAGI-1 likewise localize predominantly within the cytoplasm of CREF cells. Nevertheless, MAGUK proteins are likely to function at multiple sites in cells. For example, in confluent cells, ZO-1 accumulates at the plasma membrane where it complexes with the Y-box transcription factor ZONAB to modulate expression of the ErbB-2 proto-oncogene product (Balda and Matter, 2000), yet, in subconfluent cells, this cellular protein is found in the nucleus (Gottardi et al., 1996). Because ZO-2 contains both putative nuclear localization and export signals and likewise accumulates in the nucleus of subconfluent cells (Gonzalez-Mariscal et al., 2000), it seems probable that this cellular factor also performs distinct functions in several different cellular compartments.
Of particular significance to the present study is that ZO-2 has recently been identified as a candidate tumor suppressor protein. This observation suggests that ZO-2 functions to inhibit inappropriate cellular proliferation and, consequently, that it would be functionally inactivated by Ad9 E4-ORF1. Supporting both of these notions, we demonstrated in CREF cells that overexpression of ZO-2 blocks focus formation by Ad9 E4-ORF1 and other oncoproteins and that Ad9 E4-ORF1 sequesters ZO-2, perhaps post-translationally modified, within detergent-insoluble cytoplasmic complexes. Ad9 E4-ORF1 similarly sequesters and promotes an unknown post-translational modification of MUPP1 and MAGI-1 (Glaunsinger et al., 2000; Lee et al., 2000), but not DLG (S.S.Lee, unpublished data). We have suggested that Ad9 E4-ORF1-induced sequestration may functionally inactivate PDZ proteins by preventing their proper localization in cells and/or their interactions with critical cellular factors (Glaunsinger et al., 2000; Lee et al., 2000).
Our results showing that overexpression of ZO-2 blocks Ad9 E4-ORF1-induced focus formation in CREF cells warrant further consideration. Because binding of overexpressed ZO-2 to Ad9 E4-ORF1 was neither necessary nor sufficient to produce this effect, the transformation-repressive activity of ZO-2 is not due to it titrating Ad9 E4-ORF1 away from other critical cellular targets. Our demonstration that overexpressed ZO-2 likewise interferes with transformation by unrelated oncoproteins is consistent with this conclusion. Instead, we propose that ZO-2 has an intrinsic transformation-repressive activity associated with a specific protein region that includes both PDZ2 and PDZ3. Based on this hypothesis and others discussed earlier, we have formulated a model whereby, in the context of normal cells expressing low physiological levels of ZO-2, Ad9 E4-ORF1 is able to sequester the majority of this cellular factor in an inactive form, thereby neutralizing the transformation-repressive activity associated with PDZ2 and/or PDZ3. With respect to this model, it is interesting that ZO-2 PDZ2 has been shown to mediate binding to ZO-1, which also represents a candidate tumor suppressor protein. Supporting the latter claim, expression of ZO-1 was lost or significantly decreased in 78% (14/18) of human breast adenocarcinoma lines examined (Sommers et al., 1994) and in 69% (33/48) of primary breast carcinomas examined (Hoover et al., 1998). Loss of ZO-1 expression also correlates with increased in vitro invasiveness and decreased differentiation of cancer cells (Sommers et al., 1994) and, in addition, the N-terminal region of ZO-1 can transform epithelial cells to a mesenchymal morphology (Reichert et al., 2000; Ryeom et al., 2000). Thus, an intriguing possible scenario is that ZO-1 and ZO-2 form functional tumor-suppressor complexes, which can be targeted for inactivation by Ad9 E4-ORF1 through its ability to bind and sequester ZO-2. While additional work is needed to determine whether this idea may be correct, we anticipate that future studies of the interaction between Ad9 E4-ORF1 and ZO-2 will aid in revealing molecular mechanisms whereby this candidate tumor suppressor inhibits the neoplastic transformation of cells.
Materials and methods
Cells, transfections, and extracts
Cell lines were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 20 µg/ml gentamicin and 6 or 10% fetal calf serum (FCS). Transfections were carried out with Fugene 6 (Roche Molecular Biochemicals), Lipofectamine or Lipofectamine Plus (Life Technologies), as recommended by the manufacturers. Preparation of cell extracts in RIPA buffer [50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS] was carried out as described previously (Glaunsinger et al., 2000). For cell fractionation experiments, the pellet recovered after centrifugation of cell extracts was suspended in a volume of 1 × sample buffer equivalent to the volume of RIPA buffer originally used for cell lysis (Glaunsinger et al., 2000). Protein concentrations were determined by the Bradford assay (Bradford, 1976).
Plasmids
Plasmids GW1-9ORF1wt, GW1-9ORF1IIIA, GW1-9ORF1IIIC, GW1- 9ORF1IIID and pJ4Ω-9ORF1 have been described previously (Weiss et al., 1997a; Lee et al., 2000). Canine ZO-2, RasV12 and polyomavirus middle T plasmids were kindly provided by Bruce Stevenson (University of Alberta), Julian Downward (ICRF) and Janet Butel (Baylor College of Medicine), respectively. cDNAs coding for RasV12, N-terminal HA-tagged wild-type ZO-2 (HA–ZO-2), HA–ZO-2 missing either the majority of ZO-2 PDZ1 (amino acids 11–66) or a ZO-2 region containing both PDZ2 and PDZ3 (amino acids 288–531) were introduced into the CMV expression plasmid GW1 (British Biotechnology) to generate GW1-RasV12, GW1-HA–ZO-2, GW1-HA–ZO-2ΔPDZ1 or GW1-HA– ZO-2ΔPDZ2+3, respectively. ZO-2 DNA sequences coding for PDZ1 (amino acids 4–113), PDZ2 and PDZ3 (PDZ2+3) (amino acids 290– 585) or unique sequences located between PDZ2 and PDZ3 (US2/3) (amino acids 394–495) were introduced into pGEX-2T to generate plasmids pGEX-PDZ1, pGEX-PDZ2+3 and pGEX-US2/3, respectively. Plasmids pGEX-2TK-9ORF1, pGEX-2T-9ORF1, pGEX-2T-9ORF1IIIA, pGEX-2T-9ORF1IIIC, pGEX-2T-9ORF1IIID, pGEX-2T-5ORF1 and pGEX-2T-12ORF1 have been described previously (Weiss and Javier, 1997). All plasmids were purified on CsCl density gradients and verified by restriction enzyme and limited sequence analyses.
Antisera and antibodies
Rabbit polyclonal antisera to Ad9 E4-ORF1 have been described previously (Javier, 1994). Rabbit polyclonal antisera to ZO-2 were raised against a purified, bacterially expressed GST–US2/3 fusion protein by standard methods (Harlow and Lane, 1988). IgG was purified from ZO-2 antisera or matched pre-immune sera using protein A-coated Sepharose beads (Amersham Pharmacia Biotech). ZO-2 antibodies were affinity purified using the immunizing peptide coupled to an activated Affi-Gel 10 immunoaffinity support (Bio-Rad) (Harlow and Lane, 1988). Com mercially available antibodies to the HA epitope (16B12; Covance), as well as normal rabbit IgG and peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG (Southern Biotechnology Associates), FITC- conjugated goat anti-rabbit IgG (Gibco BRL) and Texas Red-conjugated goat anti-mouse IgG (Molecular Probe) were used.
GST pulldown, immunoprecipitation, immunoblot, and protein blotting assays
GST pulldown and immunoprecipitation assays were performed with cell extracts in RIPA buffer as described previously (Lee et al., 1997). That an equivalent amount of each GST fusion protein (5 mg) was utilized in GST pulldown assays was verified by Coomassie Blue staining of protein gels. Immunoblot assays were carried out with primary antibodies to either Ad9 E4-ORF1 (1:5000), HA (1.2 µg/ml) or ZO-2 (1:5000) and with secondary antibodies to either horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (1:5000). Membranes were developed by enhanced chemiluminescence methods (Pierce). Methods for preparing 32P-labeled GST–Ad9 E4-ORF1 protein probes and performing protein blotting assays have been described previously (Lee et al., 1997).
Immunofluorescence microscopy assays
IF assays, in which cells are fixed in 4% paraformaldehyde, permeablized with 0.1% Triton X-100 and incubated with affinity-purified antibodies, were carried out by standard methods (Harlow and Lane, 1988). Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (5 µg/ml) to visualize nuclei and examined with a Zeiss Axiophot fluorescent microscope.
Focus assays
Forty-eight hours post-transfection, 106 CREF cells were passaged 1:3 and maintained in culture medium supplemented with 3% filtered FCS. At ∼3 weeks post-transfection, cell monolayers were fixed in methanol and stained with Giemsa to quantify numbers of transformed foci (Javier, 1994).
Acknowledgments
Acknowledgements
We thank Andy Rice and Richard Sutton for comments on the manuscript and Isabel Latorre, Kristopher Frese and Darby Thomas for helpful discussions. B.A.G. was the recipient of a predoctoral fellowship from the Molecular Virology Training Grant (T32 AI07471). R.S.W. was the recipient of a predoctoral fellowship from the National Science Foundation. R.S.W. and S.S.L. were recipients of predoctoral fellowships from the US Army Breast Cancer Training Grant (DAMD17-94-J4204). This work was supported by grants to R.T.J. from the National Institutes of Health (RO1 CA58541), the American Cancer Society (RPG-97-668-01-VM) and the US Army (DAMD17-97-1-7082).
References
- Balda M.S. and Matter,K. (2000) The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J., 19, 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatch M., Jesaitis,L.A., Gallin,W.J., Goodenough,D.A. and Stevenson,B.R. (1996) The tight junction protein ZO-2 contains three PDZ (PSD-95/Discs-Large/ZO-1) domains and an alternatively spliced region. J. Biol. Chem., 271, 25723–25726. [DOI] [PubMed] [Google Scholar]
- Ben-Ze’ev A. (1997) Cytoskeletal and adhesion proteins as tumor suppressors. Curr. Opin. Cell Biol., 9, 99–108. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chlenski A., Ketels,K.V., Engeriser,J.L., Talamonti,M.S., Tsao,M.S., Koutnikova,H., Oyasu,R. and Scarpelli,D.G. (1999a) ZO-2 gene alternative promoters in normal and neoplastic human pancreatic duct cells. Int. J. Cancer, 83, 349–358. [DOI] [PubMed] [Google Scholar]
- Chlenski A., Ketels,K.V., Tsao,M.S., Talamonti,M.S., Anderson,M.R., Oyasu,R. and Scarpelli,D.G. (1999b) Tight junction protein ZO-2 is differentially expressed in normal pancreatic ducts compared to human pancreatic adenocarcinoma. Int. J. Cancer, 82, 137–144. [DOI] [PubMed] [Google Scholar]
- Chlenski A., Ketels,K.V., Korovaitseva,G.I., Talamonti,M.S., Oyasu,R. and Scarpelli,D.G. (2000) Organization and expression of the human ZO-2 gene (tjp-2) in normal and neoplastic tissues. Biochim. Biophys. Acta, 1493, 319–324. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M., D’Atri,F., Hammar,E., Parry,D.A., Kendrick-Jones,J., Shore,D. and Citi,S. (1999) Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3 and myosin. J. Cell Biol., 147, 1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven S.E. and Bredt,D.S. (1998) PDZ proteins organize synaptic signaling pathways. Cell, 93, 495–498. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Fanning A.S. and Anderson,J.M. (1999) PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Invest., 103, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaunsinger B.A., Lee,S.S., Thomas,M., Banks,L. and Javier,R. (2000) Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene, 19, 5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L., Betanzos,A. and Avila-Flores,A. (2000) MAGUK proteins: structure and role in the tight junction. Semin. Cell Dev. Biol., 11, 315–324. [DOI] [PubMed] [Google Scholar]
- Gottardi C.J., Arpin,M., Fanning,A.S. and Louvard,D. (1996) The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell–cell contacts. Proc. Natl Acad. Sci. USA, 93, 10779–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F.L. (1984) Transformation by and oncogenicity of human adenoviruses. In Ginsberg,H.S. (ed.), The Adenoviruses. Plenum Press, New York, pp. 339–398.
- Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Haskins J., Gu,L., Wittchen,E.S., Hibbard,J. and Stevenson,B.R. (1998) ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol., 141, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover K.B., Liao,S.Y. and Bryant,P.J. (1998) Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am. J. Pathol., 153, 1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishidate T., Matsumine,A., Toyoshima,K. and Akiyama,T. (2000) The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene, 19, 365–372. [DOI] [PubMed] [Google Scholar]
- Itoh M., Furuse,M., Morita,K., Kubota,K., Saitou,M. and Tsukita,S. (1999a) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2 and ZO-3, with the COOH termini of claudins. J. Cell Biol., 147, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M., Morita,K. and Tsukita,S. (1999b) Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J. Biol. Chem., 274, 5981–5986. [DOI] [PubMed] [Google Scholar]
- Javier R.T. (1994) Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J. Virol., 68, 3917–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javier R., Raska,K.,Jr, Macdonald,G.J. and Shenk,T. (1991) Human adenovirus type 9-induced rat mammary tumors. J. Virol., 65, 3192–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis L.A. and Goodenough,D.A. (1994) Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J. Cell Biol., 124, 949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.S., Weiss,R.S. and Javier,R.T. (1997) Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl Acad. Sci. USA, 94, 6670–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.S., Glaunsinger,B., Mantovani,F., Banks,L. and Javier,R.T. (2000) Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol., 74, 9680–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue R.A., Marfatia,S.M., Branton,D. and Chishti,A.H. (1994) Cloning and charcterization of hdlg: the human homologue of the Drosophila discs large tumor suppressor binds to protein 4.1. Proc. Natl Acad. Sci. USA, 91, 9818–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattagajasingh S.N., Huang,S.C., Hartenstein,J.S. and Benz,E.J. (2000) Characterization of the interaction between protein 4.1R and ZO-2. A possible link between the tight junction and the actin cytoskeleton. J. Biol. Chem., 275, 30573–30585. [DOI] [PubMed] [Google Scholar]
- Muller B.M., Kistner,U., Veh,R.W., Cases-Langhoff,C., Becker,B., Gundelfinger,E.D. and Garner,C.C. (1995) Molecular characteriza tion and spatial distribution of SAP97, a novel presynaptic protein homologous to SAP90 and the Drosophila discs-large tumor suppressor protein. J. Neurosci., 15, 2354–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert M., Muller,T. and Hunziker,W. (2000) The PDZ domains of zonula occludens-1 induce an epithelial to mesenchymal transition of Madin-Darby canine kidney I cells. Evidence for a role of beta-catenin/Tcf/Lef signaling. J. Biol. Chem., 275, 9492–9500. [DOI] [PubMed] [Google Scholar]
- Ryeom S.W., Paul,D. and Goodenough,D.A. (2000) Truncation mutants of the tight junction protein ZO-1 disrupt corneal epithelial cell morphology. Mol. Biol. Cell, 11, 1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenk T. (1996) Adenoviridae: the viruses and their replication. In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields’ Virology. Lippincott-Raven, Philadelphia, PA, Vol. 2, pp. 2111–2148.
- Sommers C.L., Byers,S.W., Thompson,E.W., Torri,J.A. and Gelmann,E.P. (1994) Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res. Treat., 31, 325–335. [DOI] [PubMed] [Google Scholar]
- Songyang Z., Fanning,A.S., Fu,C., Xu,J., Marfatia,S.M., Chishti,A.H., Crompton,A., Chan,A.C., Anderson,J.M. and Cantley,L.C. (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science, 275, 73–77. [DOI] [PubMed] [Google Scholar]
- Thomas D.L., Shin,S., Jiang,B.H., Vogel,H., Ross,M.A., Kaplitt,M., Shenk,T.E. and Javier,R.T. (1999) Early region 1 transforming functions are dispensable for mammary tumorigenesis by human adenovirus type 9. J. Virol., 73, 3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D.L., Schaack,J., Vogel,H. and Javier,R.T. (2001) Several E4 region functions influence mammary tumorigenesis by human adenovirus type 9. J. Virol., 75, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.S. and Javier,R.T. (1997) A carboxy-terminal region required by the adenovirus type 9 E4 ORF1 oncoprotein for transformation mediates direct binding to cellular polypeptides. J. Virol., 71, 7873–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.S., McArthur,M.J. and Javier,R.T. (1996) Human adenovirus type 9 E4 open reading frame 1 encodes a cytoplasmic transforming protein capable of increasing the oncogenicity of CREF cells. J. Virol., 70, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.S., Gold,M.O., Vogel,H. and Javier,R.T. (1997a) Mutant adenovirus type 9 E4 ORF1 genes define three protein regions required for transformation of CREF cells. J. Virol., 71, 4385–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.S., Lee,S.S., Prasad,B.V.V. and Javier,R.T. (1997b) Human adenovirus early region 4 open reading frame 1 genes encode growth-transforming proteins that may be distantly related to dUTP pyrophosphatase enzymes. J. Virol., 71, 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen E.S., Haskins,J. and Stevenson,B.R. (1999) Protein interactions at the tight junction. Actin has multiple binding partners and ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem., 274, 35179–35185. [DOI] [PubMed] [Google Scholar]