

Abstract
Mutations in Bruton’s tyrosine kinase (Btk) result in X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (xid) in mice. While targeted disruption of the protein kinase C-β (PKCβ) gene in mice results in an immunodeficiency similar to xid, the overall tyrosine phosphorylation of Btk is significantly enhanced in PKCβ-deficient B cells. We provide direct evidence that PKCβ acts as a feedback loop inhibitor of Btk activation. Inhibition of PKCβ results in a dramatic increase in B-cell receptor (BCR)-mediated Ca2+ signaling. We identified a highly conserved PKCβ serine phosphorylation site in a short linker within the Tec homology domain of Btk. Mutation of this phosphorylation site led to enhanced tyrosine phosphorylation and membrane association of Btk, and augmented BCR and FcεRI-mediated signaling in B and mast cells, respectively. These findings provide a novel mechanism whereby reversible translocation of Btk/Tec kinases regulates the threshold for immunoreceptor signaling and thereby modulates lymphocyte activation.
Keywords: BCR signaling/Bruton’s tyrosine kinase/calcium signaling/membrane localization/protein kinase C
Introduction
Aggregation of antigen receptors on B cells leads to the concerted activation of non-receptor tyrosine kinases, including Src, Syk and Tec family kinases, and lipid kinases including phosphoinositide 3-kinase (PI3K) (Kurosaki, 1999; Rawlings, 1999; Yang et al., 2000). These events initiate the formation of a multimeric signaling complex termed the signalosome. The signalosome promotes enhanced catalytic activity of phospholipase C-γ2 (PLCγ2) and the generation of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). These second messengers lead to intracellular calcium mobilization and protein kinase C (PKC) activation, respectively (Fruman et al., 2000). Synergistic signaling by PKC isoforms and calcium is important in the activation of transcription factors leading to diverse biological responses in activated B cells (Dolmetsch et al., 1997; Healy et al., 1997). Notably, the signalosome complex integrates both positive and negative signal transducers to produce a graded response that modulates and, ultimately, terminates B-cell receptor (BCR) signaling (Cyster and Goodnow, 1997). While signalosome activation events have been relatively well characterized, the events important for negative regulation of BCR signaling remain elusive.
Bruton’s tyrosine kinase (Btk) plays a pivotal role in the regulation of pre-B and mature BCR signaling, and is a major component of the BCR signalosome (Fruman et al., 2000; Guo et al., 2000). Mutation of Btk results in X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (xid) in mice. These diseases are associated with impaired maturation of B cells, diminished immunoglobulin production, compromised T-independent immune response and marked attenuation of the sustained calcium signal upon BCR stimulation (Rawlings, 1999). Btk contains an N-terminal Pleckstrin homology (PH) domain, followed by Tec homology (TH), SH3, SH2 and kinase domains (Yang et al., 2000). While mutations within each of the domains can lead to immunodeficiency, the best characterized is a missense mutation, R28C, within the PH domain responsible for xid (Rawlings et al., 1993). Biochemical and crystallographic studies demonstrate that this mutation interferes with the capacity of the Btk PH domain to bind membrane phospholipids including phosphatidylinositol-3,4,5-trisphosphate (PIP3), thereby impairing its ability to translocate to the plasma membrane. Conversely, another PH domain mutation, E41K, enhances PIP3 binding avidity, augmenting Btk membrane translocation (Li et al., 1995; Baraldi et al., 1999). Interestingly, mice expressing this activated allele exhibit an immunodeficiency that is similar to but more severe than xid (Dingjan et al., 1998). This observation highlights the critical role of the Btk PH–phospholipid interaction in the regulation of optimal threshold and amplitude for BCR signaling.
Membrane recruitment leads to Btk activation and is associated with rapid, but transient, phosphorylation of two major regulatory tyrosine residues. Upon BCR stimulation, Btk is transphosphorylated by Src family kinases at Y551 in the activation loop of the kinase domain. This promotes Btk catalytic activity and subsequent autophosphorylation at Y223 in the SH3 domain (Park et al., 1996; Rawlings et al., 1996). The sequential tyrosine phosphorylation of Btk following BCR engagement is rapidly attenuated, demonstrating the presence of negative regulatory signals that oppose these activating events (Wahl et al., 1997; Nisitani et al., 1999).
Accumulating evidence suggests that PKC serine/threonine isoforms may function to negatively regulate protein tyrosine kinases (Sidorenko et al., 1996; Bagowski et al., 1999). The PKC family is subdivided into classical, novel and atypical isoforms on the basis of the relative requirements for calcium and/or DAG for catalytic activity. The PKC-related isoform PKCµ (also referred to as PKD) is recruited to the BCR and can negatively modulate Syk kinase activity in vitro (Sidorenko et al., 1996). Mice deficient for the classical PKC isoform, PKCβ, display a developmental phenotype similar to xid (Leitges et al., 1996). This observation has suggested a functional link between Btk and PKCβ. Paradoxically, BCR-mediated Btk tyrosine phosphorylation is increased and prolonged in PKCβ-deficient B cells. This complex phenotype suggests that PKCβ exerts a dual function as both a positive and negative regulator of the strength and duration of Btk activation (Tarakhovsky, 1997). The precise biochemical events responsible for the inhibitory function of PKCβ and other PKC isoforms on protein tyrosine kinases remain unknown.
In this report we demonstrate that PKCβ is a potent inhibitor of Btk-mediated calcium signaling. To elucidate the underlying mechanism, we mapped the PKCβ phosphorylation site on Btk. A non-phosphorylatable mimetic of Btk displayed a marked increase in phosphotyrosine content, augmented capacity to support BCR-induced calcium mobilization and enhanced high affinity IgE receptor (FcεRI)-dependent c-Jun N-terminal kinase (JNK) activation. In addition, we provide direct evidence that PKCβ negatively regulates Btk by altering its membrane localization. Taken together, these data demonstrate that PKCβ utilizes a unique regulatory mechanism to modulate the strength and duration of Btk activation. Conservation of the major PKCβ phosphorylation site in nearly all members of the Tec kinase family suggests that this mechanism operates to down-regulate the activity of multiple cell surface receptors over a broad range of immune and hematopoietic cell lineages.
Results
Pharmacological inhibition of PKC results in enhanced BCR-induced Ca2+ signaling, increased Btk membrane translocation and PLCγ2 tyrosine phosphorylation
The overlapping phenotype of Btk and PKCβ-deficient mice suggests that PKCβ is required for peripheral B-cell development and function (Tarakhovsky, 1997). Paradoxically, engagement of receptors in PKCβ-deficient B cells (Leitges et al., 1996) or PKC-depleted mast cells (Yao et al., 1994) results in enhanced and prolonged Btk tyrosine phosphorylation, suggesting that PKCβ may negatively modulate Btk function. To test this possibility directly, we determined whether Btk-mediated downstream signals were modulated when PKCβ activity was inhibited.
Murine (A20) and human (Ramos) B-cell lines were pretreated with PKC inhibitors and analyzed for their Ca2+ responses upon BCR cross-linking. Ligation of the BCR resulted in a typical biphasic calcium response: a transient peak increase in the intracellular Ca2+ concentration followed by low, but sustained levels of Ca2+ (Scharenberg and Kinet, 1998). Strikingly, when cells were pre-treated with Ro318425 (a general PKC inhibitor), the BCR-mediated Ca2+ signal was significantly enhanced in a concentration-dependent manner (Figure 1A). The effect of PKC inhibition is most pronounced during the sustained phase Ca2+ signal versus the initial peak response. Moreover, addition of the PKCβ-specific inhibitor LY379196 also led to a significantly enhanced response, suggesting a direct role for PKCβ in negative feedback regulation of the BCR-induced Ca2+ signal (Figure 1A).

Fig. 1. Pharmacological inhibition of PKC isoforms results in enhanced BCR-induced Ca2+ signaling. A20 and Ramos B cells were pretreated with either general (Ro318425) or PKCβ-specific inhibitor (LY379196) at doses indicated for 5 min, then activated with anti-Ig cross-linking. (A) In A20 and Ramos B cells, Ca2+ mobilization was monitored by spectrofluorimetry after a 30 s baseline measurement. (B) Ramos cells are stimulated with anti-IgM for 2 min or unstimulated after pretreatment with 5 µM Ro318425 or DMSO. Cells were fractionated by hypotonic lysis, and membrane and cytosolic fractions were analyzed by western blot analysis. Membrane enrichment of Btk was detected by immunoblotting with anti-Btk antibody (left). Fractionation efficiency was evaluated by immunoblotting with anti-transferrin receptor (TrfR). Total cell lysates (TCL) were immunoblotted with anti-phosphotyrosine antibody (middle). Enhancement of pp130 signal was observed in both membrane and cytosolic fractions by anti-phosphotyrosine (PY) blotting (right) and the same membrane was stripped and reblotted with anti-PLCγ2 antibody. (C) Ramos cells were treated as in (B), and lysed in BBS following BCR stimulation. Syk (left) or Lyn (right) was immunoprecipitated and immunoblotted with anti-PY antibody and the same membrane was stripped and reblotted with anti-Syk or anti-Lyn antibodies.
The sustained phase of Ca2+ signal is associated with Btk activation, as measured by increased phosphotyrosine content, Btk membrane targeting and increased PLCγ2 activation (Fluckiger et al., 1998; Scharenberg et al., 1998). Accordingly, we considered the possible connection between Btk and PKC-mediated inhibition of Ca2+ signaling. To address the role of PKC in inhibiting Btk membrane targeting, Ramos cells were pretreated with Ro318425 and activated with anti-IgM. Following stimulation, cellular components were fractionated and the distribution of BCR-activated signaling molecules was examined. Constitutively membrane-localized transferrin receptor (TrfR) was used to demonstrate effective partitioning of membrane and cytosolic fractions. While no change in the distribution of TrfR was observed, levels of Btk in the membrane fraction were significantly increased (∼2-fold) in Ro318425-treated cells. No significant changes in Btk distribution were observed in the cytosolic fraction. This is consistent with a previous observation that the membrane-associated fraction constitutes a relatively small component of the overall pool of Btk (Wahl et al., 1997). These data demonstrate that enhancement of BCR-induced Ca2+ signal by PKC inhibition correlates with Btk membrane recruitment.
We sought to determine whether PKC inhibitors affect the activity of other key signal transducers. First, we evaluated the global pattern of BCR-induced tyrosine phosphorylation in the presence and absence of PKC inhibitors using anti-phosphotyrosine antibody 4G10 (Figure 1B, second panel). Although the pattern was for the most part unchanged by PKC inhibition, one protein with approximate molecular weight of 130 kDa (pp130) exhibited enhanced tyrosine phosphorylation upon pretreatment with Ro318425 (Figure 1B, second panel). Based upon its apparent molecular weight, we hypothesized that this protein might be PLCγ2, a likely candidate molecule for a functional link between Btk activation and Ca2+ signal (Fluckiger et al., 1998; Scharenberg et al., 1998). Indeed, when the membrane from Figure 1B (first two panels) was stripped and reblotted with anti-PLCγ2 antibody, pp130 co-migrated with PLCγ2 (Figure 1B, panel 3). Identical results were obtained when PLCγ2 was immunoprecipitated and examined for phosphotyrosine content (data not shown). Notably, we observed a significant level of tyrosine phosphorylated PLCγ2 in the cytosolic fraction upon inhibition of PKC. The exact kinetics and sites for tyrosine phosphorylation on PLCγ2 are currently unknown. However, the presence of tyrosine-phosphorylated PLCγ2 in the cytosolic fraction suggests that PLCγ2 may translocate between the membrane and cytosol during its activation/inactivation process.
In addition, we evaluated the phosphotyrosine content of Syk and Lyn, two critical non-receptor tyrosine kinases that regulate Btk and/or Ca2+ signaling (Kurosaki, 1999). Neither tyrosine phosphorylation nor in vitro kinase activity (data not shown) was altered by PKC inhibitors (Figure 1C). Together, these results suggested that inhibition of PKC leads specifically to increased membrane targeting of Btk, enhanced phosphorylation of PLCγ2 and augmented BCR-mediated Ca2+ signaling.
PKCβ co-expression down-modulates both Btk transphosphorylation and autophosphorylation
We utilized a fibroblast expression system to define directly the functional interaction between Btk and PKC isoforms. To study the effect of PKC co-expression on Lyn-mediated Btk activation, Btk, Lyn and PKCβ proteins were coordinately expressed in NIH 3T3 cells using recombinant vaccinia virus. Btk was immunoprecipitated and its tyrosine phosphorylation content was measured by immunoblotting (Figure 2). Btk tyrosine phosphorylation significantly increased with Lyn co-expression (as described previously by Rawlings et al., 1996). An increasing dosage of PKCβ led to a progressive diminution of Btk phosphorylation, returning to basal levels at the highest dosage (Figure 2A). Identical results were obtained through expression of either PKCβI or PKCβII, isoforms differing only by a short alternatively spliced C-terminus. Therefore, PKCβI was used for all subsequent experiments.

Fig. 2. PKCβ specifically down-modulates both Btk transphosphorylation and autophosphorylation. (A) Btk, Lyn and PKCβ proteins were coordinately expressed in NIH 3T3 cells using recombinant vaccinia virus. Btk was immunoprecipitated and the total tyrosine phosphorylation content was measured by immunoblotting. Btk loading was measure by anti-Btk antibody. Relative tyrosine phosphorylation levels were quantified by densitometric analysis. (B) Left panel: Btk, Lyn and Akt proteins were co-expressed and Btk tyrosine phosphorylation was analyzed as in (A). Right panel: Lyn was immunoprecipitated with anti-Lyn antibody and in vitro kinase assay (IVK) was performed. Bottom: Btk and Lyn were co-expressed with increasing dosage of PKCµ. Btk phosphorylation was analyzed as in (A). (C) Btk-Wt and Btk-E41K were co-expressed with Lyn and increasing dosage of PKCβ as in (A). Btk protein was immunoprecipitated and sequentially immunoblotted with anti-PY, anti-PY551, anti-PY223 and anti-Btk specific antibodies. (D) Btk and Lyn were co-expressed with high dosage PKCβ and cells were treated with increasing doses of Ro318425 for 30 min. Btk was immunoprecipitated and analyzed as in (A).
In contrast to PKCβ, co-expression of an alternative serine/threonine kinase, Akt, had no significant effect on Btk phosphorylation (Figure 2B, left panel). In addition, we tested the possibility that PKC co-expression might indirectly affect Btk activation by altering Lyn activity (Figure 2B, right panel). PKCβ expression, however, did not significantly affect the in vitro kinase activity of Lyn under these conditions. Finally, we also tested whether PKCµ, previously implicated as a negative regulator of BCR signaling, could functionally substitute for PKCβ (Sidorenko et al., 1996). In our co-expression system, PKCµ had no significant effect on Btk (Figure 2B, bottom panel). Taken together, these data indicate that modulation of Btk by PKCβ is most likely a direct and specific effect.
Btk activation requires sequential phosphorylation of two regulatory tyrosines (Y551 and Y223). The phosphorylation level of Btk Y551 is a relatively direct measure of Btk transphosphorylation by Src family kinases (Rawlings et al., 1996; Wahl et al., 1997). Phosphorylation of Btk Y223 represents subsequent autocatalytic activity following Btk activation (Park et al., 1996). Accordingly, we sought to determine the precise steps at which PKC intervenes to regulate Btk activity. Monoclonal antibodies specific for either phosphorylated Y551 or Y223 were used to measure the effect of PKCβ expression on Lyn-mediated Btk activation. In a similar experimental setup to Figure 2A, Btk was immunoprecipitated and sequentially immunoblotted with antibodies to phosphotyrosine, phospho-Y551, phospho-Y223 and Btk. As before, PKCβ dramatically decreased total Btk tyrosine phosphorylation. Interestingly, PKCβ down-modulated phosphorylation of Y551 and Y223, suggesting that both transphosphorylation and autophosphorylation events were attenuated by PKC-mediated inhibition (Figure 2C). We next repeated the experiment using the activated Btk mutant, Btk-E41K. PKCβ also effectively down-modulated both transphosphorylation and autophosphorylation of Btk-E41K (Figure 2C). This result further demonstrates that PKCβ is a potent negative regulator of Btk. The capacity of PKCβ to alter the enhanced membrane targeting phenotype of Btk-E41K is consistent with the pharmacological inhibitor data, suggesting that PKC may regulate Btk membrane targeting.
Finally, we tested whether PKC kinase activity is required for its inhibitory effect. Btk, Lyn and a high dosage of PKCβ were co-expressed in the presence of increasing concentrations of the PKC inhibitor Ro318425. While Lyn-induced Btk tyrosine phosphorylation was significantly down-modulated with PKCβ co-expression, prior treatment with the PKC inhibitor abrogated the inhibitory effect of PKCβ (Figure 2D). Thus, PKCβ potently down-modulates Btk in a kinase activity-dependent manner. Moreover, PKCβ inhibits both transphosphorylation and autophosphorylation of Btk, most likely by affecting the events controlling Btk localization and Lyn-mediated activation of Btk.
PKCβ phosphorylates Btk on a single serine residue in the Tec homology domain
To elucidate the molecular basis of PKCβ-dependent inhibition of Btk activity, we utilized phosphopeptide mapping to identify potential PKCβ phosphorylation sites. NIH 3T3 cells were infected with vaccinia expressing either Btk alone or Btk and PKCβI. Following in vivo [32P]orthophosphate labeling, Btk was immunoprecipitated and analyzed by two-dimensional tryptic phosphopeptide mapping. Since the pattern of PKCβ-induced Btk phosphopeptides was identical with either wild-type Btk or kinase inactive Btk (Btk-K430R), this mutant was used to minimize the complexity of the phosphopeptide maps (Figure 4A and data not shown).

Fig. 4. PKCβ phosphorylates S180 in the Tec-linker of Btk. (A) Phosphopeptide mapping analysis was performed on Btk-Wt and Btk-S180A, with or without the co-expression of PKCβ. As shown in Figure 3A, Btk-Wt displays two predominant phospho-tryptic fragments (P1, P2), and P1 is increased with PKCβ co-expression. The putative PKCβ phosphorylation site mutant Btk-S180A fails to induce P1, while P2 is still intact (panel 4). (B) Sequence alignment of murine Tec family kinases and Raja eglanteria Btk ortholog, skate PTK. Alignment of the conserved PKC consensus sequence is shown. (C) PKCβ-mediated negative regulation of Tec was examined in NIH 3T3 cells coordinately expressing Tec, Lyn and PKCβ. Tec was immunoprecipitated and blotted with anti-PY and reblotted with anti-Tec antibodies.
Expression of Btk-K430R alone led to generation of two major tryptic spots (P1, P2), each with low but equal intensity. Co-expression of PKCβ resulted in a marked enhancement of P1 (Figure 3A). Phospho-amino acid analysis indicated that the peptide extracted from P1 contained only phosphoserine (data not shown). The presence of P1 in the absence of PKCβ suggests that endogenous PKC isoforms may basally phosphorylate this peptide. The presence of the second peptide, P2, which also contained only phosphoserine (data not shown), suggests that additional serine/threonine kinases may phosphorylate Btk on alternative peptides.

Fig. 3. Phosphopeptide mapping of the PKCβ-induced phosphorylation site on Btk. (A) Kinase inactive Btk (KI; Btk-K430R) was expressed in NIH 3T3 cells using recombinant vaccinia virus in the presence or absence of co-expressed PKCβ. Cells (1 × 107) were labeled with [32P]orthophosphate and Btk was immunoprecipitated, digested with trypsin, and tryptic peptides were separated by thin-layer electro phoresis at pH 1.9 followed by chromatography (see Materials and methods). (B) Identification of the domain phosphorylated by PKCβ. Upper panel: schematic representation of IgA cleavage sites within Btk. Bottom panel: partially purified Btk from (A) was incubated with IgA protease. The digested fragments were resolved by SDS–PAGE, and visualized by autoradiography (first panel) and western blot analysis using antibodies against N-terminal (middle) and C-terminal (third panel) regions of Btk.
To map the domain that is phosphorylated by PKCβ, IgA protease analysis was utilized. IgA protease cleaves Btk within the proline-rich region (Park et al., 1996), generating two major fragments consisting of the N-terminal PH/TH domain and a larger C-terminal fragment (Figure 3B). Autoradiography of the resulting nitrocellulose membrane showed predominant incorporation of [32P]orthophosphate in the 30 kDa (pp30) fragment corresponding to the N-terminal fragment of Btk (Figure 3B). In the presence of PKCβI, 32P incorporation in pp30 increased ∼5-fold, consistent with enhanced Btk phosphorylation (Figure 3B, bottom panel). The identity of pp30 was confirmed by immunoblotting with antibodies specific to either the N-terminal or the C-terminal regions of Btk. Taken together, these results reveal a predominant, PKCβ phosphorylation site within the N-terminal region of Btk (Figure 3B).
The specific site of PKCβ-dependent Btk phosphorylation was determined by automated Edman degradation analysis of the phospho-tryptic peptide P1. Analysis revealed a major radioactive peak on the ninth cycle, suggesting that the ninth residue of the tryptic fragment was the site of phosphorylation (data not shown). Our analysis of the Btk/TH tryptic map indicated only one candidate site that clearly met these criteria. This deductive analysis indicated that PKCβ phosphorylates S180 in the region bisecting the Btk motif (BM) and the PRR of the TH domain.
To demonstrate definitively that the correct site had been identified, we mutated the S180 to alanine (Btk-S180A), and repeated the phosphopeptide mapping. While Btk-Wt maintained the expected phosphopeptide profile, the S180A mutation resulted in a complete loss of basal and induced phosphorylation at P1 (Figure 4A, bottom panels). Because PKC isoforms frequently phosphorylate their substrates on multiple sites, we made additional mutations at the serine residues in the vicinity of S180, including S174 and S179. The phosphopeptide map of the triple mutant (Btk-S174, 179, 180A) was indistinguishable from that of the Btk-S180A mutant (data not shown). In addition, we have mutated several other potential sites to identify or eliminate additional phosphorylation events on Btk. Phosphopeptide mapping of S14A, S21A, S51A, S158A and S174A revealed an intact PKCβ phosphorylation profile (data not shown). These data clearly demonstrate that S180 represents the major phosphorylation site likely responsible for the negative regulation of Btk by PKCβ.
Ser180 residue in the Btk Tec-linker represents an evolutionarily conserved PKC phosphorylation consensus site
The TH domain consists of the Btk motif and the proline-rich region present in all Tec kinases (Vihinen et al., 1994). S180 is located in a surface exposed linker that connects these two subdomains. We have designated this region as the Tec-linker (amino acids 166–184; Figure 4B). The Tec-linker region shares minimal sequence homology with other Tec kinases. However, optimal sequence alignment indicates that S180 is conserved among nearly all Tec family members (Figure 4B). Moreover, these conserved serine residues each share a PKC phosphorylation consensus sequence, (S/T)–X–(K/R). Interestingly, a similarity search using the murine/human Tec-linker identified skate PTK as having the highest sequence homology (Figure 4B). Skate PTK is a Btk orthologue in the cartilaginous fish Raja eglanteria, a phylogenetically distant member of the jawed vertebrates. This observation suggests that PKC-mediated regulation of Tec kinases is likely to be highly evolutionarily conserved (Haire et al., 1997).
Based on these results, it seemed likely that other Tec kinases with a conserved PKC site in the Tec-linker would also be targets of PKC-mediated regulation. Accordingly, we tested whether Tec can be similarly modulated by PKCβ. Using the fibroblast expression system, we analyzed the effect of PKCβ co-expression on Tec tyrosine phosphorylation. As predicted, co-expression of PKCβ significantly down-modulated tyrosine phoshorylation of Tec (Figure 4C) and pharmacological inhibition of PKCβ abrogated this effect (data not shown). The effect of PKCβ on other Tec family members is currently being investigated.
The Btk S180A phosphorylation site mutant is a hyperactive Btk allele
If S180 were the target phosphorylation site for PKCβ, then the absence of this target residue would be predicted to result in a protein that is ‘resistant’ to PKC-mediated negative regulation. To test this possibility, we compared Btk-Wt and Btk-S180A for their sensitivity to PKCβ. Indeed, Btk-S180A was resistant and maintained an elevated tyrosine phosphorylation content in the presence of PKCβ (Figure 5A). These findings suggested that mutation of Btk residue S180 was not only sufficient to abrogate PKCβ-mediated down-modulation of Btk activation, but also appeared to confer a hyperactive phenotype.

Fig. 5. Btk-S180A is hyperphosphorylated and exhibits enhanced BCR-induced Ca2+ signaling and FcεRI-induced JNK activation. (A) Btk-Wt or Btk-S180A was coordinately expressed as indicated. Btk is partially purified by immunoprecipitation, and its phosphotyrosine content was evaluated by anti-PY blotting. Duplicate membranes were immunoblotted with anti-Btk antibody to demonstrate equal loading. (B) BCR-induced Ca2+ flux was analyzed from A20 cells expressing Btk-Wt, S180A, or E41K. A20 cells were infected with vaccinia viruses for 8 h, followed by loading with Indo-1. BCR was cross-linked with anti-IgG [10 µg/ml F(ab)2] at the time indicated (arrow), and Ca2+ mobilization was monitored by spectrofluorimetry. Btk expression was evaluated by western blot analysis. The data shown are a representative of five independent experiments. (C) Btk-deficient DT40 B cells were reconstituted with Btk-Wt, S180A, E41K or GFP vector control. FACS sorted cell populations with equivalent GFP expression were obtained and Ca2+ mobilization was monitored by spectrofluorimetry as in (A). GFP expression by FACS and Btk expression by western blot analysis are shown (upper panels). (D) MAPK activation was examined in BMMC derived from Btk–/– mice reconstituted with retroviruses expressing vector alone, Btk-Wt or Btk-S180A. BMMCs were sensitized overnight with 1 µg/ml anti-DNP monoclonal IgE antibody, then stimulated with DNP-HAS (100 ng/ml) for the indicated times. For JNK assay, in vitro kinase assay was performed with GST–c-Jun as substrate. p38 and ERK activity were evaluated using phosphospecific antibodies against phosphorylated p38 (anti-pp38) or ERK (anti-pERK). Protein expression was analyzed by immunoblotting with anti-JNK, anti-p38 or anti-ERK. Equal Btk protein expression was examined by immunoblotting with anti-Btk antibody (right).
We hypothesized that S180A would be ‘hyperactive’ due to the elimination of a negative regulatory site. Indeed, when compared with Btk-Wt, the phosphotyrosine content of Btk-S180A was ∼5-fold higher upon activation by Lyn (Figure 5A). Moreover, the basal tyrosine phosphorylation level of Btk-S180A was also significantly higher compared with the Btk-Wt (Figure 5A). This phenotype is very similar to that of Btk-E41K (Li et al., 1995).
The activated Btk allele, E41K, is characterized by increased tyrosine phosphorylation, enhanced membrane recruitment and increased downstream activation of PLCγ2. As a result, expression of E41K-Btk leads to a markedly enhanced BCR-induced Ca2+ signal (Li et al., 1995; Fluckiger et al., 1998). To determine whether Btk-S180A also enhances BCR-induced Ca2+ signaling, A20 B cells were infected with vaccinia virus expressing Btk-Wt, Btk-S180A or Btk-E41K (Figure 5B). Indeed, B cells expressing Btk-S180A exhibited an enhanced BCR-induced Ca2+ signal compared with Btk-Wt. This effect was less pronounced than for Btk-E41K expressing cells. This pattern was reproduced over multiple experiments (n = 5). Consistent with these results, nearly identical results were also obtained in A20 cell lines stably expressing either Btk-Wt, Btk-S180 or Btk-E41K (data not shown).
To evaluate the effect of mutation of S180 in the absence of endogenous Btk, we used a Btk-deficient DT40 chicken lymphoma cell line (DT40-Btk–/–) (Takata and Kurosaki, 1996). Wt and Btk mutant proteins were stably expressed in this line and the functional consequence of the S180A mutation was evaluated (Figure 5C). Cells reconstituted with the vector control produced no detectable Ca2+ signal. However, consistent with the data above, while Btk-Wt successfully reconstituted the BCR-induced Ca2+ mobilization, DT40-Btk–/– cells reconstituted with either Btk-E41K or Btk-S180A displayed a significantly augmented Ca2+ signal. Together, the overexpression data and these genetic reconstitution data demonstrate that the Btk-S180A mutation leads to enhanced BCR-dependent Ca2+ signaling.
To further assess the functional significance of Btk-S180A, we generated bone marrow derived mast cells (BMMC) from Btk-deficient (Btk–/–) mice. While mast cells from these animals develop normally, FcεRI-mediated mast cell activation is significantly reduced as demonstrated by impaired degranulation and cytokine production (Hata et al., 1998). The defect in Btk–/– mast cell activation is associated with failure to fully activate the JNK signaling pathway (Kawakami et al., 1997). We determined whether genetic reconstitution of Btk–/– BMMC with various Btk isoforms led to a differential activation of JNK. Btk–/– BMMC populations stably transfected with vector alone, Btk-Wt or Btk-S180A were sensitized with anti-IgE, then stimulated with antigen. Kinetic analysis of mitogen-activated protein kinase (MAPK) activity was performed (Figure 5D). While no significant differences in p38 or extracellular signal regulated kinase (ERK) activity were observed, there was a marked enhancement of JNK activity in Btk-S180A compared with Btk-Wt reconstituted cells. Thus, in this alternative system, Btk-S180A also functions as a hyperactive allele. Collectively, our data strongly support the conclusion that Btk-S180 is a bona fide target of negative regulation by PKCβ.
Elimination of PKCβ phosphorylation of S180 leads to enhanced Btk membrane localization
As noted above, pharmacological inhibition of PKC results in enhanced membrane targeting of Btk. Conversely, PKCβ can potently down-modulate the gain-of-function phenotype of Btk-E41K. The proximity of the Tec-linker to the Btk PH domain suggests that phosphorylation of S180 residue might lead to conformational changes in the PH domain (Hyvonen and Saraste, 1997). Together, these observations suggested that these events might modulate Btk PH function, thereby altering the membrane targeting of Btk.
To test this possibility directly, NIH 3T3 cells expressing Btk-Wt or Btk-180A were subjected to subcellular fractionation and examined by immunoblotting with an anti-Btk antibody. Strikingly, Btk-S180A exhibited a 7- to 10-fold higher level of membrane-associated protein compared with the wild-type Btk protein (Figure 6A). Consistent with previous data, Btk-E41K also exhibited a marked increase in membrane localization (2- to 3-fold higher than Btk-S180A; data not shown). These results strongly correlate with the previous observation that Btk-S180A expressing B cells exhibited augmented Ca2+ signaling compared with Btk-Wt, but lower than that observed in Btk-E41K-expressing cells (Figure 5B and C). Together, these findings provide direct evidence that PKCβ phosphorylation of the Tec-linker results in an altered membrane targeting capacity of Btk and that this change is likely responsible for the enhanced signaling capacity of Btk-S180A.
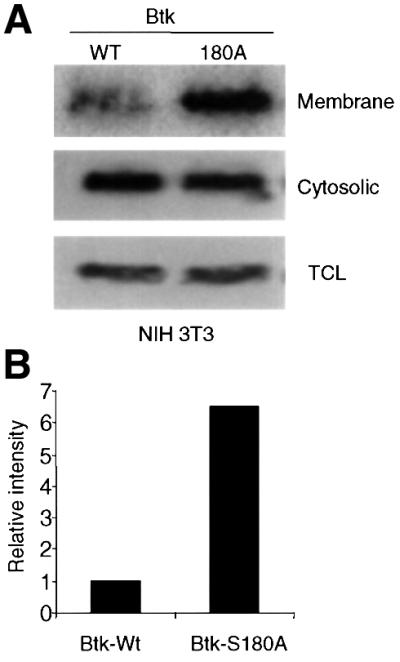
Fig. 6. Increased membrane targeting of Btk-S180A. (A) NIH 3T3 cells expressing Btk-Wt or Btk-S180A were fractionated by hypotonic lysis and Dounce homogenization (Li et al., 1995). Membrane and cytosolic fractions, and total cell lysates were analyzed by immunoblotting with anti-Btk antibody. (B) Graphical representation of Btk membrane localization. Relative intensity of Btk band was analyzed by densitometry. The data shown are representative of two independent experiments.
Discussion
B-cell antigen receptor engagement simultaneously triggers both positive and negative signals (Buhl and Cambier, 1997; Cyster and Goodnow, 1997). The consequence of antigen receptor engagement depends on the graded response resulting from the convergence of these signals. While negative feedback signaling has generally been attributed to inhibitory co-receptors, direct inhibitory feedback mechanisms may also exist and are likely to be essential for setting and maintaining the BCR activating threshold (Sidorenko et al., 1996). Consistent with this prediction, our work provides direct evidence that the classical PKC isoform PKCβ functions as a negative regulator of BCR signaling. These data demonstrate that PKCβ uniquely modulates Btk membrane recruitment and that PKCβ-dependent serine phosphorylation is a key physiological mechanism regulating the strength and duration of Btk/Tec family-dependent signals.
Btk/Tec activation is regulated by PKCβ phosphorylation of a single conserved serine in the Tec-linker region
Pharmacological inhibition of PKCβ led to a marked enhancement of the sustained phase of the BCR-dependent calcium signal (Figure 1A). This phenotype was remarkably similar to the enhanced calcium signal present in B cells expressing the activated Btk allele, Btk-E41K, where the altered calcium signal correlates with enhanced Btk membrane recruitment and phosphorylation and increased PLCγ2 activation (Li et al., 1995; Fluckiger et al., 1998; Scharenberg et al., 1998). Inhibition of PKCβ led to nearly identical biochemical changes (Figure 1B). These observations were also consistent with previous data demonstrating enhanced Btk tyrosine phosphorylation in PKCβ-deficient splenic B cells (Leitges et al., 1996). Notably, PKC inhibitor treatment led to no detectable change in the activity of two other non-receptor tyrosine kinases required for BCR function (Syk or Lyn; Figure 1C). Together, these findings suggest that PKCβ specifically modulates Btk function.
The present work defines the biochemical basis for the alterations in Btk activation observed in the presence of PKC activity. Using a fibroblast expression system, we demonstrate that increasing dosage of PKCβ results in a marked diminution of Lyn-dependent Btk activation (Figure 2). Using a combination of phosphopeptide mapping, radioactive sequencing and mutagenesis, we identified the PKCβ target phosphorylation site on Btk. These results revealed a single major site of PKCβ phosphorylation at S180 within the Btk TH domain (Figures 3 and 4). Notably, the S180 site is highly conserved within nearly all Tec family kinases and is located within a short linker, the Tec-linker, connecting the ‘Btk motif’ (BM) and the Btk proline-rich region (PRR; Figure 4B). This observation, and the analogous down-modulation of Tec activation by PKCβ (Figure 4C), indicates that the inhibitory effects of PKC are likely to be conserved among Tec family proteins.
Btk S180 phosphorylation was dependent on the kinase activity of PKCβ (Figure 2D). Similar effects were not observed with the serine/threonine kinase, Akt, or with PKCµ, a PKC-related protein previously implicated in the regulation of BCR-dependent Syk activity (Figure 2B) (Sidorenko et al., 1996). Thus, the inhibitory effects of PKCβ are relatively specific. It remains possible that other PKC isoforms expressed in hematopoietic cells may also modify Btk/Tec function and this possibility is currently being evaluated.
Elimination of PKC-dependent phosphorylation alters Btk membrane association and leads to enhanced Btk signaling
To evaluate the consequences of S180 phosphorylation we generated the mutant Btk-S180A. This mutation maintains Btk in its PKCβ-unmodified state. Strikingly, Btk-S180A was hyper-phosphorylated and resistant to PKC-mediated inhibition (Figure 5A). The increase in Btk-S180A phosphorylation was analogous to that observed with Btk-E41K. Indeed, B cells expressing Btk S180A exhibited enhanced BCR-dependent calcium signaling in two independent B-cell models including murine B cells and Btk–/– chicken B cells reconstituted with Btk-WT versus Btk-S180A (Figure 5). The calcium signal in cells expressing Btk-S180A was similar to that observed following BCR engagement in the presence of moderate doses of PKC inhibitors (Figures 1 and 5). This intermediate phenotype suggests that other Tec kinases present in B cells (including Tec) are also likely to be regulated by PKC (Ellmeier et al., 2000). Btk-S180A also exhibited an activated phenotype in non-B-lineage cells. Expression of Btk-S180A in mast cells led to a significant increase in FcεRI-dependent JNK activation, an independent assay of Btk-dependent signaling (Figure 5). Thus, PKCβ-dependent phosphorylation down-regulates both Btk-dependent calcium signaling and JNK activation, and is operative in at least two independent cell lineages and immunoreceptor signaling pathways.
It is remarkable that the modification of a single residue in the TH domain can so significantly alter Btk function. The TH domain is conserved in all the Tec kinases (Yang et al., 2000). Our observations demonstrate an additional level of complexity in the functional significance of the TH domain and highlight the importance of the ∼20 residue Tec-linker. Co-expression with PKCβ led to down-modulation of both trans- and autophosphorylation of Btk (residues Y551 and Y223, respectively; Figure 2C). This observation suggested that PKCβ might limit Btk activation by altering its accessibility for Src kinase-dependent activation. Since Btk transphosphorylation can be enhanced through PH domain-dependent membrane recruitment, we tested whether mutating the S180 phosphorylation site altered these events (Figure 6). Strikingly, the proportion of Btk-S180A within the membrane fraction was significantly increased relative to Btk-Wt. Consistent with this result, Btk-S180A also exhibited increased phosphorylation of both Y551 and Y223 (data not shown).
The Btk motif contains a novel fold stabilized by a zinc ion bound to two conserved cysteines and a histidine (Hyvonen and Saraste, 1997). In previous structural studies, the capacity to generate crystals of the Btk PH domain required the expression of both the PH and BM domains. S180 in the Tec-linker is located in close proximity to the BM zinc binding cleft. Introduction of a bulky, charged phospho group at this site is likely to significantly alter the interaction of the Tec-linker with the BM/PH domains. Taken together, these observations support a model whereby S180 phosphorylation leads to conformational changes in the BM/PH domain. These events are predicted to interfere with BM/PH function, possibly by altering the affinity for membrane phosphatidylinositol lipids. Ultimately, these structural changes result in reduced Btk recruitment, Btk transphosphorylation and subsequent activation.
There are several other mechanisms by which S180 phosphorylation might also alter Btk function. First, the Btk PRR can form an intramolecular loop with the SH3 domain. This is predicted to maintain Tec kinases in a closed, inactive conformation and disruption of this interaction may promote enzyme activation (Andreotti et al., 1997). While it is possible that S180 phosphorylation might further stabilize Btk in its inactive conformation, this seems improbable because introduction of a charged group would more likely disrupt the PRR/SH3 interaction. Secondly, S180 phosphorylation might promote dephosphorylation of the regulatory tyrosines on Btk or increase the rate of Btk degradation. While this remains a possibility, we have been unable to detect any significant differences in the relative half-lives of Btk-S180A, Btk-S180D/E or Btk-Wt. Thirdly, PKCβ may directly induce an allosteric change in the kinase domain of Btk. Studies directly comparing the relative Km and Vmax of wild-type and various S180 mutant proteins in vitro are currently being conducted to address this possibility. Structural analysis including the crystal structure of the combined PH-TH domains or full-length Btk will be required to elucidate the precise conformational change(s) induced by phosphorylation of the conserved Tec-linker serine residue.
The PKC/Btk inhibitory signal is likely to be a key determinant of the BCR signaling threshold
Our findings significantly expand on previous work suggesting that PTK activity can be modulated by serine/threonine phosphorylation. Receptor tyrosine kinases, including epidermal growth factor recoptor (EGFR) and the insulin receptor, are targets of PKC-mediated negative regulation (Bagowski et al., 1999). Similarly, the PKC-related protein, PKCµ, has been suggested to negatively regulate BCR signaling by inhibition of Syk activation (Sidorenko et al., 1996). The biochemical and cell signaling events characterized in the current study highlight an emerging paradigm of auto-inhibitory, negative feedback regulation applicable to diverse receptor signaling systems.
The dosage of Btk is critical for the generation, activa tion and maintenance of the mature pool of B cells (Satterthwaite et al., 1997). The threshold and specificity of Btk-dependent signaling events is mediated by precise spatial and temporal regulation of co-localizing, activating and inhibitory signals. PH domain-dependent recruitment of Btk is a key element in these events. This is clearly demonstrated by non-functional and gain-of-function Btk alleles (R28C and E41K) and their respective affinities for PIP3 (Baraldi et al., 1999). The PtdIns kinase, PI3K, and the SH2-containing inositol polyphosphate phosphatase, SHIP, exert counter-regulatory effects on wild-type Btk function via modulation of the levels of membrane PIP3 (Bolland et al., 1998; Fluckiger et al., 1998; Scharenberg et al., 1998).
The PKC/Btk inhibitory feedback loop provides a mechanism for modulating Btk membrane localization that is independent of the events that regulate PIP3 content (see model shown in Figure 7A). This mechanism for the regulation of Btk localization and activation probably permits appropriate fine-tuning of receptor-mediated signaling and the threshold for lymphocyte selection and function. Moreover, the conservation of PKC phosphorylation site in other Tec kinases suggests that regulation of signaling threshold by PKC may be a common mechanism shared by other cell lineages.

Fig. 7. Modulation of antigen receptor signaling by PKCβ. (A) Model for the PKCβ-mediated inhibition of Btk membrane localization. (B) Schematic representation of the Btk activity requirement for B-cell survival and function. Btk-XLAi (inactivated) represents loss-of-function Btk mutations. Btk-XLAa (activated) represents gain-of-function Btk mutations.
The altered signals leading to the ‘xid-like’ phenotype of the PKCβ-deficient mice have not been defined (Tarakhovsky, 1997). Interestingly, transgenic expression of an activated Btk allele, Btk-E41K, results in an overlapping but more severe phenotype than xid (Dingjan et al., 1998). The developmental anomaly in the latter case, however, arises because of exaggerated BCR signaling and enhanced negative selection of immature B cells. It is possible that PKCβ-deficient cells might manifest a common developmental defect through a similar mechanism. Consistent with this hypothesis, we have recently identified an activating mutation in XLA (Btk-XLAa; Figure 7B). When reintroduced into Btk-deficient DT40 B cells, this mutant exhibits an elevated tyrosine phosphorylation content and augmented Ca2+ signaling (M.I.Wahl, S.W.Kang, O.N.Witte and D.J.Rawlings, unpublished data). These observations suggest that an exaggerated Btk signal may be equally as detrimental as deficient Btk signaling at specific points in B-lineage development (see model shown in Figure 7B). The data presented here demonstrate that PKC-mediated inhibitory autoregulation of BCR signaling may function as a cellular ‘rheostat’ to maintain the optimal Btk activity.
Materials and methods
Cells and reagents
The murine B-cell line A20 and the human B-cell line Ramos were cultured in RPMI-1640 media containing 10% fetal calf serum (FCS). HEK 293T and NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% calf serum. Antibody reagents included: polyclonal goat anti-µ heavy chain (Southern Biotechnology); anti-phosphotyrosine mAb 4G10 (Upstate Biotechnology); anti-PLCγ2, anti-PKCβI, anti-PKCµ, anti-JNK1, anti-p38 and anti-Syk polyclonal antibodies (Santa Cruz Biotechnology, CA); anti-ERK and anti-transferrin receptor (Zymed Laboratories); anti-pp38 and anti-pERK (Cell Signaling Technology); anti-Akt and anti-PS473-Akt (New England Biolabs); rabbit polyclonal anti-Lyn was provided by Dr Andrew Scharenberg (University of Washington, Seattle, WA); anti-Tec antibody was provided by Dr Evan Parganas (St Jude Children’s Hospital, Memphis, TN); rabbit anti-murine Btk antibody (Wahl et al., 1997); anti-Btk phosphotyrosine site-specific mAbs directed against phospho-Y551 and phospho-Y223 (Wahl et al., 1997; Nisitani et al., 1999); and HRP-conjugated anti-mouse and anti-rabbit antibodies (Bio-Rad Laboratories). Ro318425 was purchased from Calbiochem. LY379196 was kindly provided by Dr Toshiaki Kawakami. PKD construct was kindly provided by Dr Enrique Rozengurt (UCLA). BMMC were generated from Btk–/– mice on a mixed C57BL/6 × 129/Sv genetic background. Genotyping was done by PCR analysis of mouse tail-derived DNAs. Mast cells were cultured as described previously (Hata et al., 1998).
Cell stimulation, lysis, immunoprecipitation and immunoblotting
Prior to stimulation, cells were washed once in phosphate-buffered saline (PBS) and incubated in serum-free RPMI-1640 medium for 1 h at 37°C. A20 and Ramos cells were stimulated with mouse or human specific goat anti-µ heavy chain F(ab)2 antibodies (10 µg/ml), and lysates were prepared and analyzed as described previously (Rawlings et al., 1996). FcεRI stimulation experiments were performed as described previously (Hata et al., 1998). Immunoprecipitation and immunoblotting were performed as described previously (Guo et al., 2000).
Retrovirus and vaccinia virus gene transfer
Recombinant vaccinia virus expressing murine Wt and mutant Btk, wild-type porcine Syk, murine Lyn, murine PKCµ, human PKCβI and PKCβII, and murine Akt were generated as described previously (Rawlings et al., 1996). Vaccinia infections were carried out in 5 ml of DMEM containing 10% calf serum. For cis-green fluorescent protein (GFP) Btk cell lines, full-length Btk-Wt, Btk-S180A and Btk-E41K were subcloned into MSCV-IRES-GFP, then transfected with helper constructs into 293T cells to generate recombinant retrovirus. Cells were infected with the 10A1 pseudotyped retroviruses (Imgenex). Cells were sorted by fluorescence-activated cell sorter (FACS) based on GFP fluorescence intensity. Equivalent Btk expression was confirmed by western blot analysis. BMMC stably expressing Btk-Wt and Btk-S180A were generated as described previously (Kawakami et al., 1997). Mass populations of puromycin-resistant cells were grown and then cultured in the absence of selection drug for 48 h before FcεRI stimulation.
Site-directed mutagenesis
Bluescript II plasmid containing full-length Btk was used as a template to perform mutagenesis using a QuikChange site-directed mutagenesis kit (Stratagene). Oligomer containing serine to alanine substitution at S180 (or other mutants as noted) was used to introduce single substitution mutations. DNA sequencing was performed to verify the presence of the correct mutations.
In vivo 32PO4 labeling, two-dimensional tryptic phosphopeptide mapping, phosphoamino acid analysis and peptide sequencing
Vaccinia viruses encoding Btk were infected into 1 × 107 NIH 3T3 cells 12 h before in vivo 32P labeling (Rawlings et al., 1996). Phosphopeptide mapping and phospho-amino acid analysis of tryptic peptides extracted from thin-layer chromatography (TLC) plates were performed as described previously (Park et al., 1996; Rawlings et al., 1996). 32P-labeled tryptic peptides were scraped from the TLC plates and extracted with pH 1.9 buffer at 55°C for 30 min. Extracted material was subjected to 11 cycles of automated Edman degradation sequence analysis.
IgA protease cleavage
Btk was expressed by vaccinia virus and labeled with [32P]orthophosphate as described above. Btk was partially purified and subjected to IgA protease analysis as described previously (Park et al., 1996).
In vitro kinase assays
Lyn was immunoprecipitated from the equivalent of 5 × 107 NIH 3T3 cells using 5 µl of polyclonal rabbit anti-human Lyn antibody. Kinase assay was performed as described previously (Guo et al., 2000). JNK kinase assays were performed as described previously (Kawakami et al., 1997).
Ca2+ mobilization assays
Cells were loaded with 1 mM indo-1 acetoxymethylester (indo-1 AM; Molecules Probes) and analyzed as described previously (Guo et al., 2000). To test PKC inhibitor effect on BCR-mediated Ca2+ mobilization, cells were pretreated with either Ro318425 or LY379196 for 5 min before stimulation with goat F(ab)2 anti-µ heavy chain (10 µg/ml). Intracellular calcium measurements were monitored as a ratio of 400:488 nm fluorescence emission following 350 nm excitation using a bulk spectrofluorimeter (Photon Technology International, Monmouth Junction, NJ).
Subcellular fractionation
5 × 107 B cells were serum starved for 1 h and then stimulated for 2 min at 37°C with 10 µg/ml polyclonal goat anti-µ antibody. 2 × 107 NIH 3T3 cells were infected with recombinant vaccinia virus encoding Btk-Wt or Btk-S180A for 8 h; subcellular fractionation was performed at 4°C as described previously (Li et al., 1995). Equal cell equivalents were subjected to 8% SDS–PAGE and transferred to a nitrocellulose membrane. Membranes were subjected to western blot analysis using anti-Btk antibody. Signals were quantified by densitometric analysis (Scion Corp.).
Acknowledgments
Acknowledgements
We thank John Colicelli and Phyllis Yu for critical reading of the manuscript and members of the Rawlings laboratory for technical assistance and thoughtful discussion. We thank Fiona Willis for FACS sorting. M.I.W is a senior associate and O.N.W. is an investigator of the Howard Hughes Medical Institite. D.J.R. is the recipient of a McDonnell Scholar Award, a Leukemia and Lymphoma Society Scholar Award and a Joan J. Drake Grant for Excellence in Cancer Research. This work was supported by NIH grant HD37091 and the facilities of the UCLA Jonsson Comprehensive Cancer Center.
References
- Andreotti A.H., Bunnell,S.C., Feng,S., Berg,L.J. and Schreiber,S.L. (1997) Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature, 385, 93–97. [DOI] [PubMed] [Google Scholar]
- Bagowski C.P., Stein-Gerlach,M., Choidas,A. and Ullrich,A. (1999) Cell-type specific phosphorylation of threonines T654 and T669 by PKD defines the signal capacity of the EGF receptor. EMBO J., 18, 5567–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi E., Carugo,K.D., Hyvonen,M., Surdo,P.L., Riley,A.M., Potter,B.V., O’Brien,R., Ladbury,J.E. and Saraste,M. (1999) Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure, 7, 449–460. [DOI] [PubMed] [Google Scholar]
- Bolland S., Pearse,R.N., Kurosaki,T. and Ravetch,J.V. (1998) SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity, 8, 509–516. [DOI] [PubMed] [Google Scholar]
- Buhl A.M. and Cambier,J.C. (1997) Co-receptor and accessory regulation of B-cell antigen receptor signal transduction. Immunol. Rev., 160, 127–138. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. and Goodnow,C.C. (1997) Tuning antigen receptor signaling by CD22: integrating cues from antigens and the microenvironment. Immunity, 6, 509–517. [DOI] [PubMed] [Google Scholar]
- Dingjan G.M., Maas,A., Nawijn,M.C., Smit,L., Voerman,J.S., Grosveld,F. and Hendriks,R.W. (1998) Severe B-cell deficiency and disrupted splenic architecture in transgenic mice expressing the E41K mutated form of Bruton’s tyrosine kinase. EMBO J., 17, 5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R.E., Lewis,R.S., Goodnow,C.C. and Healy,J.I. (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration [published erratum appears in Nature (1997) 388, 308].Nature, 386, 855–858. [DOI] [PubMed] [Google Scholar]
- Ellmeier W., Jung,S., Sunshine,M.J., Hatam,F., Xu,Y., Baltimore,D., Mano,H. and Littman,D.R. (2000) Severe B-cell deficiency in mice lacking the Tec kinase family members Tec and Btk. J. Exp. Med., 192, 1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluckiger A.C., Li,Z., Kato,R.M., Wahl,M.I., Ochs,H.D., Longnecker,R., Kinet,J.P., Witte,O.N., Scharenberg,A.M. and Rawlings,D.J. (1998) Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J., 17, 1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Satterthwaite,A.B. and Witte,O.N. (2000) xid-like phenotypes: a B-cell signalosome takes shape. Immunity, 13, 1–3. [DOI] [PubMed] [Google Scholar]
- Guo B., Kato,R.M., Garcia-Lloret,M., Wahl,M.I. and Rawlings,D.J. (2000) Engagement of the human pre-B-cell receptor generates a lipid raft-dependent calcium signaling complex. Immunity, 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Haire R.N., Strong,S.J. and Litman,G.W. (1997) Identification and characterization of a homologue of Bruton’s tyrosine kinase, a Tec kinase involved in B-cell development, in a modern representative of a phylogenetically ancient vertebrate. Immunogenetics, 46, 349–351. [DOI] [PubMed] [Google Scholar]
- Hata D. et al. (1998) Involvement of Bruton’s tyrosine kinase in FcεRI-dependent mast cell degranulation and cytokine production. J. Exp. Med., 187, 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy J.I., Dolmetsch,R.E., Timmerman,L.A., Cyster,J.G., Thomas,M.L., Crabtree,G.R., Lewis,R.S. and Goodnow,C.C. (1997) Different nuclear signals are activated by the B-cell receptor during positive versus negative signaling. Immunity, 6, 419–428. [DOI] [PubMed] [Google Scholar]
- Hyvonen M. and Saraste,M. (1997) Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J., 16, 3396–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y. et al. (1997) Bruton’s tyrosine kinase regulates apoptosis and JNK/SAPK kinase activity. Proc. Natl Acad. Sci. USA, 94, 3938–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T. (1999) Genetic analysis of B-cell antigen receptor signaling. Annu. Rev. Immunol., 17, 555–592. [DOI] [PubMed] [Google Scholar]
- Leitges M., Schmedt,C., Guinamard,R., Davoust,J., Schaal,S., Stabel,S. and Tarakhovsky,A. (1996) Immunodeficiency in protein kinase C-β deficient mice. Science, 273, 788–791. [DOI] [PubMed] [Google Scholar]
- Li T., Tsukada,S., Satterthwaite,A., Havlik,M.H., Park,H., Takatsu,K. and Witte,O.N. (1995) Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity, 2, 451–460. [DOI] [PubMed] [Google Scholar]
- Nisitani S., Kato,R.M., Rawlings,D.J., Witte,O.N. and Wahl,M.I. (1999) In situ detection of activated Bruton’s tyrosine kinase in the Ig signaling complex by phosphopeptide-specific monoclonal antibodies. Proc. Natl Acad. Sci. USA, 96, 2221–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Wahl,M.I., Afar,D.E., Turck,C.W., Rawlings,D.J., Tam,C., Scharenberg,A.M., Kinet,J.P. and Witte,O.N. (1996) Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity, 4, 515–525. [DOI] [PubMed] [Google Scholar]
- Rawlings D.J. (1999) Bruton’s tyrosine kinase controls a sustained calcium signal essential for B lineage development and function. Clin. Immunol., 91, 243–253. [DOI] [PubMed] [Google Scholar]
- Rawlings D.J. et al. (1993) Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science, 261, 358–361. [DOI] [PubMed] [Google Scholar]
- Rawlings D.J., Scharenberg,A.M., Park,H., Wahl,M.I., Lin,S., Kato,R.M., Fluckiger,A.C., Witte,O.N. and Kinet,J.P. (1996) Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science, 271, 822–825. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A.B., Cheroutre,H., Khan,W.N., Sideras,P. and Witte,O.N. (1997) Btk dosage determines sensitivity to B-cell antigen receptor cross-linking. Proc. Natl Acad. Sci. USA, 94, 13152–13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharenberg A.M. and Kinet,J.P. (1998) PtdIns-3,4,5-P3: a regulatory nexus between tyrosine kinases and sustained calcium signals. Cell, 94, 5–8. [DOI] [PubMed] [Google Scholar]
- Scharenberg A.M. et al. (1998) Phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3)/Tec kinase-dependent calcium signaling pathway: a target for SHIP-mediated inhibitory signals. EMBO J., 17, 1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorenko S.P., Law,C.L., Klaus,S.J., Chandran,K.A., Takata,M., Kurosaki,T. and Clark,E.A. (1996) Protein kinase C-µ (PKCµ) associates with the B-cell antigen receptor complex and regulates lymphocyte signaling. Immunity, 5, 353–363. [DOI] [PubMed] [Google Scholar]
- Takata M. and Kurosaki,T. (1996) A role for Bruton’s tyrosine kinase in B-cell antigen receptor-mediated activation of phospholipase C-γ 2. J. Exp. Med., 184, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakhovsky A. (1997) xid and xid-like immunodeficiencies from a signaling point of view. Curr. Opin. Immunol., 9, 319–323. [DOI] [PubMed] [Google Scholar]
- Vihinen M., Nilsson,L. and Smith,C.I. (1994) Tec homology (TH) adjacent to the PH domain. FEBS Lett., 350, 263–265. [DOI] [PubMed] [Google Scholar]
- Wahl M.I., Fluckiger,A.C., Kato,R.M., Park,H., Witte,O.N. and Rawlings,D.J. (1997) Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl Acad. Sci. USA, 94, 11526–11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.C., Collette,Y., Nunes,J.A. and Olive,D. (2000) Tec kinases: a family with multiple roles in immunity. Immunity, 12, 373–382. [DOI] [PubMed] [Google Scholar]
- Yao L., Kawakami,Y. and Kawakami,T. (1994) The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc. Natl Acad. Sci. USA, 91, 9175–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]