

Abstract
Cyclin A-mediated activation of cyclin-dependent kinases (CDKs) is essential for cell cycle transversal. Cyclin A activity is regulated on several levels and cyclin A elevation in a number of cancers suggests a role in tumorigenesis. In the present study, we used a modified DNA binding site selection and PCR amplification procedure to identify DNA binding proteins that are potential substrates of cyclin A–CDK. One of the sequences identified is the Sp1 transcription factor binding site. Co-immunoprecipitation experiments show that cyclin A and Sp1 can interact physically. In vitro and in vivo phosphorylation studies indicate that cyclin A–CDK complexes can phosphorylate Sp1. The phosphorylation site is located in the N-terminal region of the protein. Cells overexpressing cyclin A have elevated levels of Sp1 DNA binding activity, suggesting that cyclin A–CDK-mediated phosphorylation augments Sp1 DNA binding properties. In co-transfection studies, cyclin A expression stimulated transcription from an Sp1-regulated promoter. Mutation of the phosphorylation site abrogated cyclin A–CDK-dependent phosphorylation, augmentation of Sp1 transactivation function and DNA binding activity.
Keywords: cyclin A/cyclin-dependent kinase/Sp1/transcription
Introduction
In eukaryotic cells, cell cycle progression is regulated by the sequential activation of cyclin-dependent kinases (CDKs). Cyclin A acts in two distinct stages. In late G1 and early S phase, cyclin A complexes with and activates CDK2; later in the cell cycle, cyclin A activates the related kinase cdc2 (CDK1) (Pagano et al., 1992). A crucial role for cyclin A in late G1/S is supported by reports that antibodies to cyclin A block entry into S phase (Girard et al., 1991) and that ectopic expression of cyclin A advances S phase entry (Resnitzky et al., 1995). Cyclin A expression is tightly regulated at multiple levels. During G1, cyclin A gene transcription is repressed, but in late G1, gene expression is induced. This induction is mediated by an E2F site located in the cyclin A promoter (Schulze et al., 1995; Soucek et al., 1997). cAMP can also stimulate cyclin A transcription via a cAMP response element (CRE) (Desdouets et al., 1995), and TAFII250, a subunit of the transcription factor TFIID, stimulates transcription through the TSRE enhancer element (Wang et al., 1997). MDM2, a protein that inhibits the p53 tumor suppressor, stimulates transcription of cyclin A through an interaction with TAFII250, thus linking a transforming protein implicated in many human tumors, with this regulator of the cell cycle (Leveillard and Wasylyk, 1997). Transcriptional repression of cyclin A is mediated by the retinoblastoma gene product (RB)/E2F complexes (Spitkovsky et al., 1997). In addition transforming growth factor β represses cyclin A transcription through an ATF site (Yoshizumi et al., 1997). Cyclin A is also regulated post-transcriptionally by an alteration of mRNA stability during the cell cycle (Maity et al., 1997). The activity of cyclin A–CDK complexes is modulated by interactions with the CDK kinase inhibitors p21 and p27 (Harper et al., 1993; Xiong et al., 1993; Dulic et al., 1994; Polyak et al., 1994; Toyoshima and Hunter, 1994). The protein levels of cyclin A are also regulated by ubiquitin-mediated degradation (Murray, 1995), which occurs during mitosis. The multiple levels of regulation argue that it is essential to ensure that cyclin A is expressed in specific cell cycle states but repressed in others.
Deregulation of cyclin A in cultured cells does not result in transformation, but is associated with adhesion-independent growth (Hauser, 1998). Overexpression of cyclin A in mammary glands of transgenic mice results in mammary hyperplasia, nuclear abnormalities such as multinucleation and karyomegaly, and increased apoptosis, which are suggestive of preneoplastic alterations (Bortner and Rosenberg, 1995). Furthermore, cyclin A is overexpressed in some tumors including liver, colon, ovarian and esophageal (Barboule et al., 1998; Chao et al., 1998; Chetty and Simelane, 1999; Handa et al., 1999). Overexpression correlates with poor prognosis and earlier relapse (Chao et al., 1998), suggesting that cyclin A deregulation contributes to the tumorigenic process.
There are several known cyclin A–CDK substrates that function to regulate gene transcription, including RB, p107, E2F-1, Estrogen receptor α, the basic helix–loop– helix proteins Id2 and Id3, and B-Myb (Hinds et al., 1992; Hall et al., 1993; Peeper et al., 1993; Krek et al., 1994; Deed et al., 1997; Hara et al., 1997; Sala et al., 1997; Rogatsky et al., 1999; Ma et al., 2000). Phosphorylation of B-Myb stimulates its transactivation activity, thus promoting B-Myb-mediated transcription (Saville and Watson, 1998). Several of the cyclin A–CDK substrates are members of the RB or E2F family—a network of transcriptional regulators that act to both repress and activate transcription of genes whose products are needed for proliferation. The hyperphosphorylation of RB results in the release of E2F, converting E2F to a transcriptional activator (Nevins, 1992; Hatakeyama and Weinberg, 1995; Johnson and Schnieder-Broussard, 1998). RB phosphorylation is dependent on cyclins D and E as well as cyclin A (Hall et al., 1993; Hatakeyama and Weinberg, 1995; Sherr, 1996). E2F-1 can also be phosphorylated by cyclin A–CDK (Krek et al., 1995; Kitagawa et al., 1996). The phosphorylated form of E2F-1 cannot bind DNA, hence cyclin A prevents E2F-1-mediated gene regulation. Studies have shown that overexpression of E2F-1 in cells growth-arrested by serum starvation results in entry into S phase followed by apoptosis, but that cyclin A can rescue the cells from apoptosis (Qin et al., 1994; Krek et al., 1995). Overexpression of an E2F-1 cyclin A binding-defective mutant was effective in inducing apoptosis in a p53-dependent and -independent manner, arguing that cyclin A can function as a survival factor (Pruschy et al., 1999). In addition, cyclin A, along with E2F-4, p107 and CDK2, is a component of the S phase E2F transcription factor complex (Mudryj et al., 1991; Shirodkar et al., 1992). Cyclin A is also a component of the CDP–cut transcription complex that binds to the histone H4 promoter (van Wijnen et al., 1994). Recent studies have found that the cyclin A-containing CDP–cut complex represses transcription of osteocalcin, an osteoblast- specific differentiation protein (van Gurp et al., 1999).
Since several known substrates of cyclin A include proteins that directly or indirectly bind DNA and modulate transcription, we used a glutathione S-transferase (GST)–cyclin A chimera protein and a modified DNA binding site selection/PCR-mediated enrichment procedure to identify DNA sequences that interact with cyclin A. One of the identified sequences was the binding site of the Sp1 transcription factor. In vitro, in vivo and site-directed mutagenesis studies demonstrated that the cyclin A–CDK2 complex phosphorylates Sp1. Sp1 DNA binding activity was elevated in NIH 3T3 cyclin A overexpressing cells, suggesting that cyclin A-dependent phosphorylation augments Sp1 DNA binding activity. In vitro phosphorylation of the GST–Sp1 fusion protein further enhanced DNA binding activity. Incubation with identical extracts did not alter the binding activity of the GST–Sp1Mut protein. Transfection studies using Sp1-dependent promoter-CAT constructs indicated that cyclin A stimulated transcription 3- to 4-fold. In addition, cells that overexpress cyclin A have an elevation of endogenous dihydrofolate reductase (DHFR), thymidylate synthetase and adenosine deaminase mRNA. Finally, transfection of Sp1 into the Sp1-negative SL2 cells transactivated transcription from the DHFR promoter. Cyclin A further stimulated the Sp1-dependent transactivation, but mutation of the identified phosphorylation site abrogated responsiveness to cyclin A.
Results
Isolation of sequences that interact with cyclin A
To identify DNA binding proteins that interact with cyclin A, we use a modified DNA binding site selection and PCR amplification strategy (Kinzler et al., 1989; Chittenden et al., 1991). This method is based on the premise that a protein which has DNA recognition properties may be able to select a defined sequence from a totally random mixture of oligonucleotides. Since cyclin A does not bind DNA directly, the sequences that we identify are likely to interact with DNA binding proteins that in turn bind cyclin A. We employed a chimeric protein that consisted of cyclin A sequences fused to GST. This GST–cyclin A fusion protein functioned as an affinity reagent to bind cellular proteins, which interact in a sequence-specific manner with the synthetic oligonucleotides. A 56-base oligonucleotide with a central random 16-base core was synthesized. The 5′- and 3′-regions flanking the random core had defined sequences. Oligonucleotides that were 20 bases in length were synthesized to serve as primers for PCR amplification of the 56-mer. A double-stranded 56-mer oligonucleotide was generated by annealing the 56-mer to the reverse primer followed by extension with Taq polymerase. The 56-bp oligonucleotide was incubated with purified GST–cyclin A protein bound to glutathione– Sepharose beads and extracts prepared from WI-38 normal human fibroblasts. The reaction buffer was a gel shift reaction buffer and contained sheared salmon sperm DNA to minimize non-specific protein–DNA interactions. The beads were washed and the remaining DNA eluted and amplified by PCR. After six serial selection/amplification cycles, the amplified PCR products were cloned into a dT-tailed plasmid vector and introduced into bacterial cells. A total of 81 individual clones were isolated and sequenced.
Cyclin A interacts with several classes of sequences
Since cyclin A is a component of a stable DNA binding transcription factor complex that consists of E2F-4, p107 protein, cyclin A and CDK2 (Kitagawa et al., 1996), we predicted that our procedure would result in the isolation of E2F binding sequences and function as a positive control. Three of the isolates harbored the E2F recognition sequence within the central core of the cloned 56-mer oligonucleotide. The remaining sequences were analyzed using the GCG program (Genetics Computer Group, Madison, WI). Inspection of the consensus sites made it apparent that one of the classes was the binding site of the Sp1 transcription factor (Table I). Sp1 binds to GC-rich sequences (Azizkhan et al., 1993) as well as GT-rich sequences (Vindevoghel et al., 1997; Gory et al., 1998); both were represented several times.
Table I. Sp1 sequences identified by the DNA binding site selection and PCR amplification procedure.
| GC-rich sequences | GT-rich sequences |
|---|---|
| TCGGGGGCCGACAACC | CGGTTGGGTGGTACTC |
| CGGGCGGGGCTGTTGG | CGGGATGATGGGTGAG |
| CATCGGGGGCGGGCAG | AATTTGTGGGTGGTGA |
Cyclin A interacts with Sp1
To determine whether cyclin A associated with Sp1 in vivo, cell lysates were prepared from cells transfected with the cyclin A expression plasmid, labeled with [32P] orthophosphate and subjected to immunoprecipitation with either anti-cyclin A or anti-Sp1 antibodies. The immunoprecipitated proteins were re-immunoprecipitated with the reciprocal antibody. The cyclin A immunoprecipitates contained the Sp1 protein (Figure 1A, lane 3) and the Sp1 immunoprecipitates contained the cyclin A protein (Figure 1A, lane 4). Immunoprecipitation with an SV40 T-antigen-specific antibody served as a negative control (Figure 1A, lanes 1 and 2). Therefore, we concluded that cyclin A and Sp1 interact physically.

Fig. 1. Cyclin A interacts with and phosphorylates Sp1. (A) Interaction of Sp1 and cyclin A in vitro. Lane 3: in vivo-labeled proteins were immunoprecipitated by cyclin A, and following dissociation of the immune complexes, re-immunoprecipitated by Sp1-specific antibodies. Lane 4: labeled proteins were immunoprecipitated by Sp1; the immune complexes were dissociated and re-immunoprecipitated by cyclin A. Lane 5: cyclin A immune complexes were re-immunoprecipitated with cyclin A to provide a marker. Lane 6: Sp1 immunoprecipitates were re-immunoprecipitated with Sp1 antibodies to generate an Sp1 marker. Lanes 1 and 2: proteins immunoprecipitated with SV40 T-antigen- specific antibodies and re-immunoprecipitated with either cyclin A (lane 1) or Sp1 (lane 2) served as negative controls. (B) Sp1 is phosphorylated by cyclin A in vitro. NIH 3T3 cells were transfected with CDK2 in the absence (lanes 1, 2 and 5) or presence (lanes 3 and 4) of p21. Either 5 or 10 µg of p21 expression plasmid were used. Immunoprecipitation with SV40 T-antigen antibodies served as a negative control (lane 1). The cellular extracts were immuno precipitated with cyclin A and Sp1 antibodies. The immune complexes were resuspended in kinase buffer and [32P]ATP. After termination of the kinase reaction, Sp1 was re-immunoprecipitated with specific antibodies and analyzed by 10% SDS–PAGE. The bands were visualized by autoradiography. (C) In vivo phosphorylation of Sp1. NIH 3T3 cells were transfected with CDK2 as well as cyclin A expression plasmid (lanes 1, 2 and 4) and p21 (lane 4) expression plasmid. Following transfection, the cells were serum starved to reduce endogenous levels of cyclin A. Cellular extracts prepared from the transfected cells were immunoprecipitated with Sp1 antibodies, the proteins were separated by 10% SDS–PAGE and visualized by autoradiography.
Cyclin A–CDK2 phosphorylates Sp1 in kinase assays in vitro
Next, we examined whether cyclin A–CDK2 could phosphorylate Sp1 in vitro. NIH 3T3 cells were transfected with the CDK2 expression plasmid or with the CDK2 and the CDK inhibitor p21(WAF1, cip1, sdi-1) expression plasmids. As a control, cells were transfected with the pRC-CMV plasmid. Extracts were immunoprecipitated with cyclin A and Sp1 antibodies, and the immunoprecipitates were resuspended in kinase buffer containing radiolabeled [γ-32P]ATP. Sp1 is phosphorylated by cyclin A–CDK2 complexes (Figure 1B, lane 5). The extent of phosphorylation is enhanced when CDK2 levels are increased (Figure 1B, lane 2). The presence of p21 inhibited Sp1 phosphorylation (Figure 1B, lanes 3 and 4); hence the phosphorylation is cyclin dependent. Immuno precipitation with an SV40 T-antigen antibody served as a negative control (Figure 1B, lane 1).
In vivo phosphorylation of Sp1 by cyclin A–CDK is inhibited by p21
To determine whether Sp1 is phosphorylated by cyclin A–CDK2 in vivo, we transfected NIH 3T3 cells with cyclin A and CDK2 expression plasmids, cyclin A, CDK2 and p21 expression plasmids or with control vector pRC-CMV. To reduce the endogenous levels of cyclin A, the cells were serum starved after transfection. After labeling with [32P]orthophosphate, the Sp1 protein was immunoprecipitated and analyzed by SDS–PAGE. Cells transfected with both cyclin A and CDK2 have high levels of phosphorylated Sp1 (Figure 1C, lane 2). The levels of phosphorylation were reduced when p21 was included in the transfection (Figure 1C, lane 4). Cells transfected with CDK2 alone (Figure 1C, lane 3) also had lower levels of Sp1 phosphorylation, indicating that the phosphorylation was dependent on cyclin A. Cells that were transfected with a control plasmid had low levels of phosphorylated Sp1. In addition, the migration of the phosphorylated Sp1 in non-transfected cells was distinct from that observed in the other lanes (Figure 1C, lane 5). This study confirmed our in vitro results that cyclin A–CDK2 can phosphorylate Sp1.
Cyclin A phosphorylates the N-terminus of the Sp1 protein
CDKs can phosphorylate both serine and threonine residues. To identify the Sp1 amino acid(s) that are phosphorylated by cyclin A–CDK2, in vitro phosphorylated Sp1 was immunoprecipitated, degraded to amino acids and subjected to fast-performance liquid chromatography (FPLC) analysis. The radioactive peak coincided with the phosphoserine peak and we therefore concluded that a serine residue was phosphorylated (Figure 2A).

Fig. 2. Cyclin A phosphorylates a serine residue located in the N-terminal region of the Sp1 protein. (A) FPLC analysis of the Sp1 protein, which was in vitro phosphorylated by cyclin A–CDK2 complexes and acid hydrolyzed. The radioactivity co-eluted with the phosphoserine peak. (B) A schematic representation of the predicted mouse Sp1 CNBr cleavage-generated peptides. S and T designate the potential CDK phosphorylation sites. Sizes are given in kDa. (C) CNBr cleavage analysis of in vitro phosphorylated Sp1 protein. The phosphorylated protein was re-immunoprecipitated by Sp1-specific antibodies, CNBr cleaved and fractionated on a 12% SDS–PAGE. [14C]leucine-labeled, CNBr-cleaved Sp1 protein served as a marker (lane 1). The positions of commercially available peptide markers are noted on the left. Lanes 2 and 3 are duplicate samples; however, twice as much starting material was used in lane 2 than in lane 3. The arrow on the right hand side indicates the phosphorylated peptide. The arrows on the left indicate the locations of the 15.8/15.6 kDa and 13.7 kDa [14C]leucine-labeled Sp1 peptides. (D) Trypsin digest of in vitro phosphorylated Sp1 protein. The phosphorylated protein was re-immunoprecipitated by Sp1-specific antibodies, digested with trypsin and fractionated on a 16.5% Tris-tricine SDS–PAGE. The positions of the peptide markers are noted on the left. The arrow on the right hand side indicates the phosphorylated peptide.
Cyanogen bromide (CNBr) cleavage analysis was used to further delineate the site of phosphorylation. The predicted CNBr fragments are shown in Figure 2B. The immunoprecipitated Sp1 protein was phosphorylated in vitro by cyclin A–CDK2, re-immunoprecipitated with Sp1 antibodies, cleaved by CNBr and analyzed on 15% SDS–PAGE. In vivo [14C]leucine-labeled Sp1 protein was immunoprecipitated and cleaved with CNBr (Figure 2C, lane 1) to serve as a marker. In addition, commercially available peptide markers (Bio-Rad) were also used. Only one peptide, ∼16 kDa in size, was labeled (Figure 2C). As expected, the size of the phosphorylated peptide was greater than that of the unphosphorylated leucine marker. There are two CNBr-digested peptides with a predicted molecular weight of ∼16 kDa (15.8 and 15.6). However, only one contains the serine-proline (SP) sequences that are the minimum consensus required for CDK phosphorylation. The two SP sequences are located in the N-terminal region of the protein and are contained in a 15.8 kDa CNBr cleavage fragment that spans amino acids 8–160.
The two potential serine phosphorylation sites are separated by 13 amino acids, including an arginine residue. Therefore, trypsin digestion would cleave between the two potential phosphorylation sites. The predicted sizes of the tryptic peptides that harbor the potential phosphorylation site are 2.8 and 5.1 kDa. In vitro phosphorylated Sp1 was digested with trypsin and the peptides were separated on a 16.5% Tricine gel (Figure 2D). As shown in lane 2, an ∼3 kDa peptide is labeled. We conclude that Ser61 of the mouse Sp1 (which corresponds to Ser59 of human Sp1) is the cyclin A–CDK2 phosphorylation site.
Increased Sp1 binding activity is observed in extracts from cyclin A overexpressing NIH 3T3 cells
Cyclin A may modify Sp1-mediated transcription by altering Sp1 DNA binding activity or by altering the levels of Sp1 protein. To address this question, we transfected NIH 3T3 cells with a cyclin A expression plasmid and generated NIH 3T3 cell lines that had elevated levels of cyclin A (Figure 3A). Extracts prepared from cyclin A overexpressing cells and from parental NIH 3T3 cells (G0, G1 and S phase) were used in electrophoretic mobility shift analysis (EMSA) (Figure 3B). The DHFR promoter fragment isolated from pDHF-210ΔE2F (Figure 3A) served as a probe. This probe harbors only four Sp1 sites. The gel shift patterns obtain using G0, G1, S phase and cyclin A-NIH 3T3 cell extracts were identical. The intensity of the bands in the cyclin A overexpressing cellular extracts was 2- to 3-fold greater than that of the G1 or S phase extracts. All the bands were competed with an excess of a double-stranded oligonucleotide that contained the Sp1 binding site. Further analysis using Sp1-specific antibodies indicated that only one of the shifted bands contained Sp1. The remaining bands were eliminated by addition of Sp3-specific antibodies. Therefore, each band contained either Sp1 or Sp3. The bands were not shifted or diminished by cyclin A-specific antibodies, suggesting that cyclin A is not a component of the DNA–Sp1 complexes (data not shown).

Fig. 3. The level of Sp1 binding is increased in cells overexpressing cyclin A. (A) Western blot analysis of levels of cyclin A protein present in parental NIH 3T3 cells and a cell line overexpressing cyclin A. (B) Gel shift assays using extracts prepared from G0, G1 and S phase NIH 3T3 cells as well as from cells that overexpress cyclin A. The probe contains promoter sequences present in pDHF-210ΔE2F. Three shifted bands are present and all are competed by an Sp1-specific oligonucleotide. The middle band is supershifted by an Sp1 antibody, while the two other bands are supershifted by Sp3 antibodies. (C and D) Western blot analysis using Sp1- (C) and Sp3-specific (D) antibodies.
Next, we asked if elevation of DNA binding activity in the cyclin A overexpressing cells was due to an increase in Sp1 and Sp3 protein levels or a modification of the proteins. Western blot analysis using Sp1- or Sp3-specific antibodies indicated that in cyclin A overexpressing cells Sp3 levels were elevated while Sp1 levels were unchanged (Figure 3C and D). This suggested that the increase in Sp1 DNA binding activity was not due to an increase in protein levels.
Cyclin A stimulates transcription from an Sp1-dependent promoter
Since cyclin A–CDK2 phosphorylates Sp1, and cyclin A overexpressing cells have elevated levels of Sp1 DNA binding activity, we asked if cyclin A augments Sp1-mediated transcription. We utilized the hamster DHFR promoter, which has four Sp1 binding sites that have been shown to be crucial for transcription, as well as two overlapping E2F sites that contribute to the regulation of this TATA-less promoter (Blake et al., 1990). In our studies, we used both a wild-type promoter construct pDHF-210, pDHF-210ΔE2F (a construct where the E2F site is mutated) and pDHF-103 (a construct containing only the E2F site) (Figure 4A). The promoter-CAT plasmids were transfected into NIH 3T3 fibroblasts along with a cyclin A expression plasmid. The results of multiple transfections are summarized in Figure 4B. Cyclin A transactivated transcription ∼3-fold from promoters containing Sp1 sites. pDHF-210ΔE2F was transactivated, as was the wild-type promoter; this indicated that transactivation is not dependent on the E2F sites. Cyclin A also transactivated pDHF-103 but at a lower magnitude, arguing that cyclin A can transactivate transcription through E2F sites.

Fig. 4. Cyclin A transactivated the DHFR promoter. (A) Schematic presentation of the promoter–recorder construct used in the study. pDHF-210, a wild-type promoter that contains four Sp1 sites and two overlapping E2F sites; pDHF-210ΔE2F, a promoter that retains the four Sp1 sites but no E2F sites; pDHF-103, a promoter that contains the intact E2F sites. (B) Fold transactivation of the promoter–CAT plasmids by cyclin A. Cyclin A expression was under the control of the CMV major immediate early promoter.
Cyclin A overexpressing NIH 3T3 cells have higher levels of DHFR mRNA
Our transfection data demonstrated that, in transient transfections, cyclin A transactivated transcription from the Sp1-regulated DHFR promoter. However, it was unclear if cyclin A transactivated transcription from endogenous promoters. We utilized our cyclin A overexpressing NIH 3T3 cell line to determine if transcription of genes that were regulated in part by Sp1 was altered by cyclin A. The mRNA levels of DHFR, thymidylate synthetase, adenosine deaminase and collagen type VII were analyzed by northern blot analysis. As shown in Figure 5, cells overexpressing cyclin A have elevated levels of DHFR, thymidylate synthetase and adenosine deaminase. In contrast, the level of collagen VII mRNA was unchanged, as was the level of the negative control GAPDH. These results indicate that overexpression of cyclin A alters transcription of some, but not all, Sp1 responsive genes.
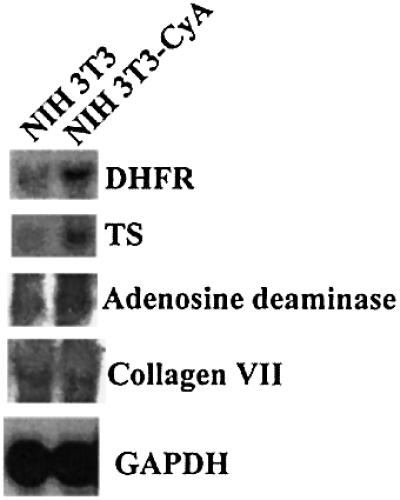
Fig. 5. Northern blot analysis of DHFR mRNA in cyclin A overexpressing 3T3 cells. RNA was prepared from NIH 3T3-CycA cells as well as from parental NIH 3T3 cells. Mouse DHFR, thymidylate synthetase, adenosine deaminase, collagen VII and GAPDH probes were used to detect transcripts after fractionation on an agarose gel and transfer to membrane.
Ser59 is required for phosphorylation by cyclin A–CDK complexes
To further demonstrate that Ser59 (Ser61 in mouse Sp1) is the phosphorylation site, we analyzed a human full-length GST–Sp1 fusion protein. Ser59 was mutated to alanine by site-directed mutagenesis. The wild-type and mutated GST fusion proteins were purified and subjected to an in vitro kinase assay using cyclin A–CDK2 complexes immunoprecipitated by cyclin A-specific antibodies from cells transfected with CDK2 or CDK2 and p21 expression plasmids. GST–Sp1 fusion protein as well as a GST– Sp1Mut protein were purified and used as a substrate in an in vitro phosphorylation assay. Wild-type Sp1 became phosphorylated; this phosphorylation was decreased when the extracts were prepared from cells transfected with p21 (Figure 6A). The mutant GST fusion protein was not phosphorylated (Figure 6A, lanes 4 and 5). SV40 T-antigen immunoprecipitates isolated from CDK2-transfected cells served as a negative control (Figure 6A, lane 1). This result further argues that Ser59 is the cyclin A–CDK phosphorylation site.
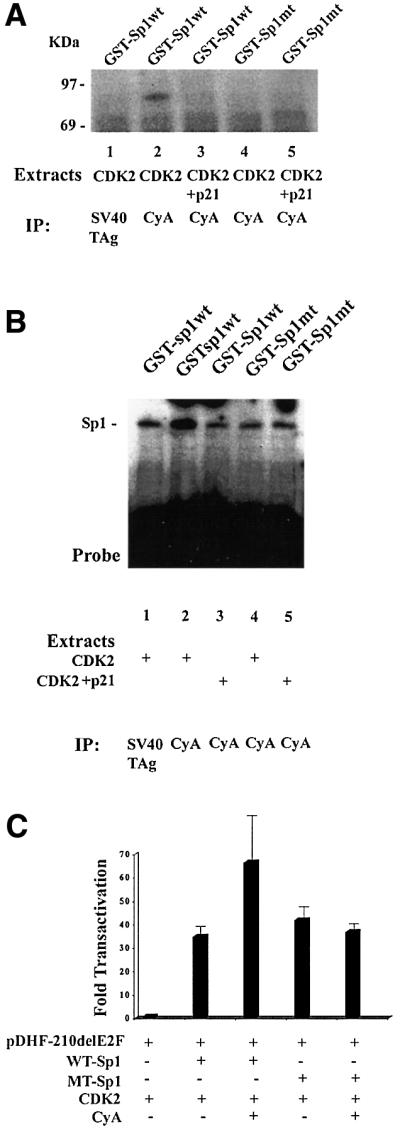
Fig. 6. Cyclin A–CDK phosphorylates the Ser59 of the human Sp1 protein. (A) Wild-type GST–Sp1 fusion protein (lanes 1, 2 and 3) and a GST–Sp1 mutant protein where Ser59 was mutated to alanine (lanes 4 and 5) were subjected to an in vitro kinase assay utilizing cyclin A–CDK immunoprecipitated from cells transfected with the CDK2 expression vector (lanes 1, 2 and 4) or with the CDK2 and p21 expression plasmids (lanes 3 and 5). SV40 T-antigen immunoprecipitates served as a negative control (lane 1). (B) Wild-type and mutant GST–Sp1 protein were subjected to in vitro phosphorylation followed by EMSA. The probe contains promoter sequences present in pDHF-210ΔE2F. Both mutant and wild-type protein have DNA binding activity; phosphorylation enhances DNA binding of the wild-type (lane 2), but not the mutant protein (lane 4). SV40 T-antigen immunoprecipiates served as negative controls. (C) Transfection of Drosophila SL2 cells with pDHF210ΔE2F and the wild-type or mutant Sp1 under the control of the Drosophila actin promoter with and without Drosophila actin promoter–cyclin A expression plasmid. The CMV-CDK2 expression plasmid was included in all the transfections.
This phosphorylation assay was repeated but with the radiolabeled ATP omitted to generate a phosphorylated GST–Sp1 protein. GST–Sp1 or GST–Sp1Mut protein was incubated with extracts prepared from cells transfected with CDK2 or the p21 expression plasmid. When the reaction was complete, DNA binding activity was analyzed by EMSA. As shown in Figure 6B, both wild-type and mutant protein were able to bind DNA. Phos phorylation of the wild-type protein further enhanced binding 2- to 3-fold, while phosphorylation of the mutant did not. These experiments are in agreement with our initial EMSA study, where cyclin A expression enhanced Sp1 DNA binding activity. This result further argues that phosphorylation of Ser59 is important for augmentation of Sp1 DNA binding activity by cyclin A–CDK.
Mutation of Sp1 Ser59 abrogates the cyclin A–CDK augmentation of Sp1-dependent transcriptional transactivation
To further demonstrate that cyclin A-dependent phosphorylation of Sp1 modified Sp1 activity, we conducted transient transfection studies in Sp1-negative Drosophila SL2 cells. Using an Sp1-negative line ensured that all the activity was due to transcription from the transfected construct. To facilitate this study, full-length Sp1 and cyclin A cDNAs were cloned under the control of the Drosophila actin promoter (PDros.Act.-). SL2 cells were co-transfected with the pDHF-210ΔE2F-CAT plasmid, a CMV-CDK2 expression plasmid, pPDros.Act.-Sp1 or pPDros.Act.-Sp1Mut (where Ser59 was mutated to an alanine residue) with and without the pPDros.Act.-cyclin A plasmid. The pDHF-210ΔE2F-CAT construct has very little activity in Drosophila SL2 cells, but was transactivated 25- to 40-fold by wild-type Sp1 (Figure 6C). Cyclin A expression further enhanced expression from the DHFR promoter ∼2-fold. pPDros.Act.-Sp1Mut transactivated transcription from the DHFR promoter as well as the wild-type protein, but this activity was not enhanced further by cyclin A–CDK, demonstrating that cyclin A augmentation of Sp1 activity is dependent on phosphorylation of this N-terminal serine residue.
Discussion
Cyclin A overexpression is a feature of many tumors, but it is unclear how cyclin A overexpression contributes to the transformed phenotype. We used a modified DNA site selection and PCR-mediated amplification protocol to identify DNA binding proteins that are potential cyclin A–CDK substrates. Since cyclin A interacts with the E2F transcription factor, identification of the E2F binding site served as a positive control. We also identified the binding site of the Sp transcription factor family. Members of the Sp family are ubiquitously expressed proteins that recognize GC-rich sequences (Azizkhan et al., 1993) and GT-rich sequences (Vindevoghel et al., 1997; Gory et al., 1998) present in a wide variety of housekeeping genes and genes involved in growth regulation. Sp1, Sp2 and Sp3 are ubiquitously expressed, but the expression of Sp4 is limited to the brain (Supp et al., 1996). Thus, Sp4 was not considered as a potential cyclin A–CDK substrate. Sp2 recognizes a GT sequence rather than the GC sequence recognized by Sp1 (Kingsley and Winoto, 1992). Sp1 and Sp3 both recognize GC-rich sequence (Kingsley and Winoto, 1992). It has also been reported that Sp1 recognizes GT sequences (Vindevoghel et al., 1997; Gory et al., 1998). While Sp1 appears to be an activating transcription factor, Sp3 also contains a repression domain and can act as an activator or repressor, depending on the cell type or promoter (Birnbaum et al., 1995; De Luca et al., 1996; Majello et al., 1997). Increased binding of both Sp1 and Sp3 in NIH 3T3 cells expressing cyclin A was observed during EMSA. Sp1 and Sp3 levels were identical in extracts prepared from G1 and S phase extracts. Sp1 protein levels in 3T3-CycA cells were also unaltered, but Sp3 levels were elevated. Increased Sp3 binding activity correlates with an increase in Sp3 present in the cell. It is unclear if the elevation of Sp3 depends on transcriptional or post-transcriptional mechanisms such as an alteration in stability. In contrast, Sp1 levels are not altered; therefore, the increase in DNA binding is dependent on a modification of the Sp1 protein. Cyclin A overexpression alters the DNA binding activity of two members of the Sp family, but the mechanisms employed are distinct.
A number of protein kinases phosphorylate Sp1. Sp1 can be phosphorylated by DNA-dependent protein kinase (Jackson et al., 1990), casein kinase II (Armstrong et al., 1997), protein kinase A (Rohlff et al., 1997) and by cell cycle-regulated, Sp1-associated kinase activity (Black et al., 1999). DNA-dependent protein kinase has been reported to increase Sp1 activity, while phosphorylation of the C-terminus of Sp1 by casein kinase II decreases its DNA binding property. Protein kinase A phosphorylation activates Sp1 site-dependent transcription. But in vivo, this activation appears to be independent of alterations in DNA binding activity or protein levels. Black et al. (1999) described an Sp1-associated kinase activity that is regulated during the cell cycle and increases in late G1/early S phase. The kinase activity elevation correlates with the induction of the DHFR gene, which is regulated in part by Sp1. Haidweger et al. (2001) reported that cyclin A–CDK interacts with and phosphorylates Sp1, and enhances Sp1 activity. While Sp1 is a ubiquitous transcription factor, multiple mechanisms that augment its activity allow for regulation by different transduction pathways to promote appropriate expression of specific genes.
In the current study, co-immunoprecipitation and in vitro and in vivo kinase experiments established that cyclin A physically interacts with and phosphorylates Sp1. In the in vitro phosphorylation studies, both cyclin A and Sp1 were immunoprecipitated from cells that were transfected with CDK2 expression plasmid. Elevating the level of the catalytic component of the cyclin A–CKD2 complex ensured a high level of kinase activity. Inclusion of the p21 CDK kinase inhibitor decreased the level of Sp1 phosphorylation, indicating that phosphorylation was dependent on a cyclin. The in vivo studies recapitulate our in vitro results. In order to demonstrate that cyclin A is involved in the phosphorylation of Sp1, cells were serum starved to reduce endogenous levels of cyclin A. Low levels of phosphorylation were detected only when the CDK2 expression plasmid was introduced into the cells. This phosphorylation is probably dependent on the residual levels of cyclins present in serum-starved cells. Sp1 phosphorylation was greatly increased when cyclin A was present. But this increase in phosphorylation was inhibited when p21 was introduced into the cells together with cyclin A. It is interesting to note that in the absence of both CDK2 and cyclin A, the sizes of the phosphorylated Sp1 protein are slightly different, suggesting that the phosphorylation is distinct from the cyclin A–CDK-dependent phosphorylation. We also noted that these Sp1 species were not detected if Sp1 was phosphorylated by cyclin A–CDK2. Perhaps cyclin A–CDK2 phosphorylation precludes phosphorylation by other protein kinases. In summary, these studies demonstrate that Sp1 is a substrate for cyclin A–CDK2.
The minimal CDK kinase consensus sequence has been defined as serine or threonine followed by a proline. FPLC analysis determined that a serine residue was phosphorylated. There are only two serine–proline sites in the mouse Sp1; both are in the N-terminal region of the protein. The two sites are contained within a 16 kDa CNBr cleavage peptide. Our CNBr cleavage analysis confirmed that a 16 kDa peptide is phosphorylated. Analysis of the trypsin digest localized the site to Ser61 of the mouse Sp1 (which corresponds to Ser59 in human Sp1). Therefore, the phosphorylation site was within the N-terminal domain of the molecule. The location of the phosphorylation site was confirmed by site-directed mutagenesis where Ser59 of the human GST–Sp1 fusion protein was mutated to alanine. The mutated protein was not phosphorylated by cyclin A–CDK2 in an in vitro kinase assay. The mutated GST–Sp1 protein bound DNA, but subjecting the mutant protein to a cyclin A–CDK2-dependent kinase assay did not enhance DNA binding further. In contrast, wild-type Sp1 binding was increased by cyclin A–CDK2 phosphorylation. This is consistent with our findings that cells overexpressing cyclin A have an increase in Sp1 DNA binding activity.
The increase in binding activity is also consistent with our functional studies, which established that cyclin A enhances Sp1-mediated transcription. Three independent experiments support this conclusion. The first used a DHFR–CAT promoter construct that contained only Sp1 sites. In transient transfection studies, cyclin A stimulated transcription from this promoter. The second piece of evidence that supports our conclusion is that expression of several endogenous genes is stimulated in cells overexpressing cyclin A. Finally, mutation of the identified phosphorylation site abrogated cyclin A-dependent modification of Sp1 transactivation in Drosophila SL2 cells. Transfection of wild-type and mutant Sp1 stimulated transcription from the Sp1-dependent promoter, and inclusion of the cyclin A expression vector further increased transcription from wild-type but not the mutant Sp1. This study argues that while cyclin A–CDK-mediated phosphorylation enhances Sp1 DNA binding and transactivation, it is not required for either. Our study of cyclin A overexpressing NIH 3T3 cells indicated that expression from some Sp1 regulated promoters is stimulated, that the magnitude of transactivation from the endogenous promoter is promoter specific and that some promoters that harbor Sp1 sites are not affected at all. While the thymidylate synthetase promoter is transactivated very well, the collagen type VII promoter, which has been shown to be regulated by Sp1, is not transactivated at all. We speculate that while Sp1 (and/or Sp3) is required for transcription, there is an interplay between Sp1 and other transcription factors to coordinate transcription. It is interesting to note that cyclin A can interact with the two transcription factors that regulate the DHFR gene, Sp1 and E2F. The interaction of cyclin A with each of these two factors is different. While we demonstrated that cyclin A and Sp1 interact physically, we did not detect the cyclin A protein in the Sp1–DNA complexes. This suggests that the interaction of Sp1 and cyclin A—unlike that of cyclin A and p107/E2F-4 complex—is transient. In contrast, cyclin A is a component of a stable E2F-4/p107/cyclin A–CDK2 complex that can bind DNA. The role of the E2F site in the DHFR promoter may be 2-fold. When an E2F-1/RB complex is bound to the E2F site, transcription of the gene is repressed. However, when cyclin A is present, E2F-1 is phosphorylated and replaced by the E2F-4/p107/cyclin A–CDK2 complex. In this way, cyclin A is brought into close proximity to Sp1.
What are the consequences of cyclin A deregulation? Stimulation of Sp1 by cyclin A is within the 3–4 fold range. A 3- to 4-fold increase in activity could significantly alter the pattern of gene transcription. The cyclin A overexpressing cell line mimics the cyclin A overexpression characteristic of certain tumors. It is unclear whether Sp1 is a substrate of cyclin A–CDK in cells that do not have de-regulated cyclin A expression. However, when cyclin A levels are elevated, or possibly present at inappropriate times of the cell cycle, cyclin A–CDK apparently mediates phosphorylation of proteins like Sp1. The consequences of this phosphorylation can be significant. When the substrate is a transcription factor that regulates the expression of a wide variety of genes (including genes that promote growth) the outcome of cyclin A deregulation is the transcriptional activation of growth-promoting genes. Our results are consistent with previous research (Hauser et al., 1998), which demonstrated that inappropriate expression of cyclin A correlates with anchorage-independent growth. In these studies, the inappropriate expression of cyclin A overrides the signals that mediate transcriptional repression. In conclusion, our study suggests that an alteration of the ubiquitous Sp1/Sp3 transcription factors is one mechanism involved in this growth-promoting process.
Materials and methods
Purification of GST fusion proteins
The fusion proteins were isolated as described in the manufacturer’s instructions (Promega). The GST–Sp1 and GST–SpMut protein were affinity-purified on a calf thymus DNA-cellulose column before use in EMSA studies.
Cell culture and cellular extracts
Early passage WI-38 normal human fibroblasts (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with penicillin/streptomycin (P/S) and 10% fetal bovine serum (FBS) (Invitrogen Life Technologies) at 37°C and 5% CO2. NIH 3T3 cells (ATCC) were cultured in DMEM supplemented with P/S, and 10% calf serum (CS) (Invitrogen Life Technologies). NIH 3T3 cells were synchronized by incubation in DMEM supplemented with P/S, and 0.5% CS for 48 h. Cells were released from G0 by incubation in media containing 10% CS and harvested at 0, 4 and 16 h. Cells harboring a pRC-CMV-cyclin A expression plasmid were maintained in media containing 400 µg/ml geneticin. Drosophila SL2 cells were cultured in Schnieder’s Drosophila media (Invitrogen Life Technologies) supplemented with P/S and 10% FBS.
Transfections and CAT assays
The GST–cyclin A plasmid was generously provided by Drs T.Hunter and J.Pines. The human cyclin A cDNA was cloned into the multilinker region of pGEX 2T (fusing the cyclin A sequence initiating at codon 1 to the GST domain). The plasmids pDHF-103, pDHF-210, pDHF-210ΔE2F, pCMV-cyclin A and pCMV-CDK2 were as described previously (Swick et al., 1989; Blake et al., 1990; Latham et al., 1996). The p21 expression vector was a gift from Dr David Beach. The pRC-CMV plasmid was obtained from Invitrogen. Cells were transfected using the Superfect reagent (Qiagen). CAT assays were performed as described previously (Afshari et al., 1997). Cellular extracts for CAT assays were prepared as described previously (Sambrook et al., 1989).
Oligonucleotides
The following oligonucleotides were utilized: forward primer: 5′-GCGTCGACAAGCTTTCTAGA-3′; reverse primer: 5′-CGCTCGAGGGATCCGAATTC –3′; 56-mer: 5′-GCGTCGACAAGCTTTCTAGANNNNNNNNNNNNNNNNGAATTCGGATCCCTCGAGCG. Sp1 binding site: 5′-ATTCGATCGGGGCGGGGCGAGC-3′, 5′-GCTCGCCCCGCCCCGATCGAAT-3′.
DNA site selection and PCR amplification
To generate a double-stranded 56 bp oligonucleotide, the 56-mer and the reverse primer were placed in a buffer containing 20 mM Tris–HCl pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 200 µM dNTPs and Taq polymerase, and subjected to the following cycle: 95°C for 1 min, 50°C for 1 min and 72°C for 20 min. The double-stranded oligonucleotide was purified on a 3% low melting agarose gel and using a Wizard kit (Promega). The double-stranded oligonucleotide was resuspended in 50 µl of NET-N buffer [20 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.05% (w/v) non-fat dried milk]. GST–cyclin A was bound to glutathione–Sepharose beads according to the manufacturer’s instructions (Oncogene Science). A mixture of 50 µg of cellular extracts, 50 µl of GST–cyclin A coupled to glutathione–Sepharose beads, 50 µl of the double-stranded oligonucleotide and 4 µg of sonicated salmon sperm DNA were incubated at room temperature for 1 h. The beads were washed four times with NET-N buffer at 4°C. The remaining material was resuspended in 400 µl of proteinase K buffer [500 mM Tris–HCl pH 8.8, 20 mM EDTA, 10 mM NaCl, 1% SDS, 200 µg/ml proteinase K (Sigma)] and digested at 52°C for 3 h. Following incubation, the mixture was extracted with phenol/chloroform (1:1), precipitated with ethanol and resuspended in 20 µl of water. Ten microliters of the resuspension were PCR-amplified using the following conditions: 94°C for 1 min, 50°C for 1 min and 72°C for 1 min for 35 cycles. The PCR products were separated on a 3% agarose gel and the oligonucleotide was purified as described above. The purified oligonucleotide was used in the subsequent DNA binding site selection. This process was repeated five more times.
Cloning
The pBluescript SKII vector (Stratagene) was digested with SmaI and HindIII and the ends were dT-tailed by incubation with 1 mM dTTP, 1× PCR buffer and Taq polymerase for 2 h at 70°C. The 56 bp oligonucleotide was ligated into the plasmid vector, transfected into XL-1 blue competent cells and plated. Colonies were isolated and plasmid DNA was isolated from the bacterial cultures and analyzed by sequencing. The bacterial plasmid harboring the GST–Sp1 fusion (pGEX1SP1FLU) was a gift form Dr Jon Horowitz (North Carolina State University, NC). The Sp1 cDNA was excised from the plasmid by digestion with EcoRI and ligated into the EcoRI site of the Drosophila PromoterAct. expression vector (a gift from Dr Ken Burgess, University of California, Davis, CA). The cyclin A cDNA was excised from the pCMV-cyclin A plasmid by digestion with HindIII and XbaI, the end was filled in using Klenow polymerase and cloned into the EcoRI site (filled in by the Klenow polymerase) of the Drosophila PromoterAct. expression vector. A Stratagene Quick Change kit was used for site-directed mutagenesis. The following primer was used: 5′-GGGGGCAATGGTAATGGTGG-3′. The mutations were verified by sequencing.
Sequencing
Clones were sequenced using T7 and T3 primers and the Sequenase Kit (US Biochemical) according to manufacturer’s instructions.
Co-immunoprecipitation of cyclin A and Sp1
NIH 3T3 cells were transfected with appropriate plasmids. Forty-eight hours after transfection, the cells were incubated in phosphate-free DMEM (Gibco-BRL) for 1 h, labeled with 1 mCi 32P/dish (Amersham-Pharmacia) for 4 h and harvested in cold NP40+ buffer with protease inhibitors [50 mM Tris–HCl pH 8.0, 250 mM NaCl, 5 mM EDTA, 0.2% NP-40, 5% glycerol, 1 mM dithiothreitol (DTT), 0.4 mM Pefablock, 5 mg/ml leupeptin, 5 mg/ml pepstatin, 5 mg/ml aprotinin, 10 mM NaF, 0.1 mM Na3VO4]. Protein extract (900 µg) was pre-cleared and incubated with Sp1 antibodies (Santa Cruz, sc-59-G), cyclin A antibodies (Santa Cruz, sc-239) or SV40 T-antigen antibodies (Oncogene Science, dp 01) for 4 h at 4°C. The protein–antibody complex was isolated on protein A/G (Oncogene Science) beads. After washing (4×) the beads were resuspended in 50 µl of 1× SDS–PAGE loading buffer without dye [0.06 M Tris–HCl pH 8.0, 1.71% (w/v) SDS, 6% (w/v) glycerol, 0.1 M DTT]. After adding 900 µl of 2% BSA, the mixture was incubated with the appropriate antibodies. The beads were washed and resuspended in SDS–PAGE loading buffer, and proteins were separated on a 10% SDS–PAGE gel. The gel was fixed and dried. Proteins were visualized using a phosphorimaging analysis.
In vitro kinase assays
NIH 3T3 cells were washed with cold PBS and lysed in 1.5 ml per 150 mm dish of NP40+ lysis buffer with protease inhibitors. Protein solution (800 µg) was pre-cleared and incubated with Sp1 antibody and cyclin A antibody. After addition of protein A/G beads, the precipitates were washed, resuspended in 50 µl kinase buffer (20 mM HEPES pH 7.0, 80 mM β-glycerolphosphate, 1 mM ATP, 20 mM EGTA, 50 mM MgCl2, 5 mM MnCl2, 0.1 mM BSA, 1 mM DTT, 10 µM cAMP protein kinase inhibitor) with 4 µCi/µl [γ-32P]ATP (Amersham-Pharmacia) and incubated for 30 min at 30°C. The reaction was terminated by the addition of 10 µl of 6 × SDS–PAGE loading buffer without dye [0.35 M Tris–HCl pH 8.0, 10.28% (w/v) SDS, 36% (w/v) glycerol, 0.6 M DTT]. For phosphorylation of the GST fusion proteins, equal amounts of protein (as assessed by western blot analysis) were used in the assay. Cyclin A–CDK complexes were isolated from 500 µg of cellular extracts from cells transfected with CDK2 expression plasmid or equal amounts of CDK2 and p21 expression plasmid. Immuno precipitation with SV40 T-antigen served as a negative control. Equal amounts of affinity purified GST–Sp1 or GST–Sp1Mut were subjected to the kinase assay. The kinase assay was terminated by the addition of loading buffer, boiled and loaded on a 10% SDS–PAGE. The gel was dried and autoradiographed.
In vivo phosphorylation analysis
NIH 3T3 cells were starved 20–24 h after transfection, incubated in phosphate-free DMEM (Gibco-BRL) for 1 h, labeled with 1 mCi 32P (Amersham-Pharmacia) per dish for 4 h and harvested in cold RIPA buffer with protease inhibitors (150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris–HCl pH 8.0, 1 mM DTT, 0.4 mM Pefablock, 5 mg/ml leupeptin, 5 mg/ml pepstatin, 5 mg/ml aprotinin, 10 mM NaF, 0.1 mM Na3VO4). Protein solution (700 µg) was pre-cleared and incubated with Sp1 or SV40 T-antigen antibody. The immunoprecipitates were isolated with protein A/G beads, the beads were washed four times with NP40+ buffer, resuspended in 50 µl of 1× SDS–PAGE loading buffer [0.06 M Tris–HCl pH 8.0, 1.71% (w/v) SDS, 6% (w/v) glycerol, 0.1 M DTT, 0.002% Bromophenol blue] and boiled. The released proteins were separated on a 10% SDS–PAGE gel. Pre-stained SDS–PAGE standards from Bio-Rad were used. The gel was fixed, dried and autoradiographed.
Immunoblot analysis
NIH 3T3 cells were lysed in RIPA buffer. After addition of loading buffer, the proteins were loaded on a 10% SDS–PAGE gel. The gel was transferred to BA-85 membrane (Schleicher and Schuell), blocked with 5% non-fat dry milk in PBS and 1% Tween. After addition of cyclin A antibody (Santa Cruz sc239), the membrane was incubated overnight at 4°C in PBS and 1% Tween. The membrane was washed with PBS and 1% Tween incubated with mouse monoclonal horseradish-peroxidase-linked NIF 825 antibodies (Amersham Pharmacia). Proteins were detected using the ECL (Amersham) following the manufacturer’s instructions.
Electophoretic gel shift analysis
Electrophoretic gel shift analyses were performed as previously described (Mudryj et al., 1991). The promoter sequence of pDHFΔE2F was excised by digestion with Asp718 and HindIII and end labeled. Five to ten micrograms of cellular extracts were used in each gel shift reaction. A 100-fold excess of Sp1-specific oligonucleotides was used in the competition assays. Gel shift analysis of GST–Sp1 and SGT-Sp1mut was performed in a similar manner. In vitro phosphorylation of the GST–Sp1 protein was performed as described above except that the [32P]ATP was omitted and 10–12 µl of the GST fusion proteins (equal amounts) were included in the assay. The beads were removed by centrifugation and 5 µl of the supernatant were used in the gel shift analysis. The protein–DNA complexes were analyzed on a 4% non-denaturing acrylamide gel in 0.25× TBE buffer. After drying, the gel was autoradiographed.
Preparation of RNA and northern blot analysis
RNA was prepared using Trizol reagent (Gibco-BRL) following the manufacturer’s instructions. RNA was size-fractionated on a 1% agarose denaturing gel, transferred to membranes and hybridized to probes using previously described protocols (Sambrook et al., 1989). The mouse probes were generated by RT–PCR amplification of mouse RNA using the following primers: dihydrofolate reductase; 5′-CAGATATTTCCAGAGAATGACCACAA-3′ and 5′-TTCTTATAAACAGAACTGCCACCAA-3′; thymidylate synthetase: 5′-TCCAGGCACACATGATGATT-3′ and 5′-TTTCTGGACAGCTTGGGATT-3′; adenosine deaminase: 5′-TAACCATGTCCCACCCTCTC-3′ and 5′-CAGAAGACCGTGGTGGTAT-3′; collagen type VII: 5′-TCCCAGAGCCAAGTGTATCC-3′ and 5′-ATGCTGCTGACATCGTGTTC-3′; GAPDH: 5′-TGGTATCGTGGAAGGACTCATGAC-3′ and 5′-ATGCCAGTGAGCTTCCCGTTCAGC-3′.
Fast performance liquid chromatography
An in vitro kinase assay, as previously described, was performed on 4 mg of total protein. The sample was hydrolyzed for 1 h in 6 N HCl at 100°C after re-immunoprecipitation of Sp1 and combined with phosphoserine and phosphothreonine standards. The mixture was applied to a 25 cm3 Dowex 50 cation exchange column using 0.05 N HCl. Each fraction was scanned for radioactivity using a liquid scintillation counter. Phospho-serine and phospho-threonine standard peaks were detected with a fluorescence monitor as described previously (Bradbury et al., 1973).
Cyanogen bromide digestion of 32P-labeled Sp1
Sp1 protein was in vitro-labeled and immunoprecipitated as described above. Immunoprecipitated 32P-labeled Sp1 was washed briefly in 70% formic acid. The beads were re-suspended in 100 µl of 5 mg/ml CNBr (Sigma) in 70% formic acid and incubated at room temperature overnight in darkness. The peptide fragments were separated on a 12% SDS–PAGE gel, fixed, dried and autoradiographed.
Trypsin digestion of 32P-labeled Sp1
Sp1 protein was kinased in vitro and immunoprecipitated. Immuno precipitated material was resuspended in 60 µl digestion buffer containing 50 mg trypsin (Roche). The reaction was incubated at 37°C for 24 h, centrifuged briefly and the peptide fragments separated on a 16.5% Tris–tricine polyacrylamide gel. The gel was fixed, dried and autoradiographed.
Acknowledgments
Acknowledgements
We are grateful to Dr H.Matthews for his expert advice and assistance with the FPLC. We thank Navdeep Dhillon for reading the manuscript. This work was supported by a VA Merit Award (to M.M.).
References
- Afshari C.A., Rhodes,N., Paules,R.S. and Mudryj,M. (1997) Deregulation of specific E2F complexes by the v-mos oncogene. Oncogene, 14, 3029–3038. [DOI] [PubMed] [Google Scholar]
- Armstrong S.A., Barry,D.A., Leggett,R.W. and Mueller,C.R. (1997) Casein kinase II-mediated phosphorylation of the C-terminus of Sp1 decreases its DNA binding activity. J. Biol. Chem., 272, 13489–13495. [DOI] [PubMed] [Google Scholar]
- Azizkhan J.C., Jensen,D.E., Pierce,A.J. and Wade,M. (1993) Transcription from TATA-less promoters: dihydrofolate reductase as a model. Crit. Rev. Eukaryot. Gene Expr., 3, 229–254. [PubMed] [Google Scholar]
- Barboule N., Baldin,V., Jozan,S., Vidal,S. and Valette,A. (1998) Increased level of p21 in human ovarian tumors is associated with increased expression of cdk2, cyclin A and PCNA. Int. J. Cancer, 76, 891–896. [DOI] [PubMed] [Google Scholar]
- Birnbaum M.J., van Wijnen,A.J., Odgren,P.R., Last,T.J., Suske,G., Stein,G.S. and Stein,J.L. (1995) Sp1 trans-activation of cell cycle regulated promoters is selectively repressed by Sp3. Biochemistry, 34, 16503–16508. [DOI] [PubMed] [Google Scholar]
- Black A.R., Jensen,D., Lin,S.Y. and Azizkhan,J.C. (1999) Growth/cell cycle regulation of Sp1 phosphorylation. J. Biol. Chem., 274, 1207–1215. [DOI] [PubMed] [Google Scholar]
- Blake M.C., Jambou,R.C., Swick,A.G., Kahn,J.W. and Azizkhan,J.C. (1990) Transcriptional initiation is controlled by upstream GC-box interactions in a TATAA-less promoter. Mol. Cell. Biol., 10, 6632–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortner D.M. and Rosenberg,M.P. (1995) Over-expression of cyclin A in the mammary glands of transgenic mice results in the induction of nuclear abnormalities and increased apoptosis. Cell Growth Differ., 6, 1579–1589. [PubMed] [Google Scholar]
- Bradbury E.M., Inglis,R.J., Matthews,H.R. and Sarner,N. (1973) Phosphorylation of very lysine-rich histone in Physarum polycephalum. Correlation with chromosome condensation. Eur. J. Biochem., 33, 131–139. [DOI] [PubMed] [Google Scholar]
- Chao Y., Shih,Y.L., Chiu,J.H., Chau,G.Y., Lui,W.Y., Yang,W.K., Lee,S.D. and Huang,T.S. (1998) Over-expression of cyclin A but not Skp 2 correlates with the tumor relapse of human hepatocellular carcinoma. Cancer Res., 58, 985–990. [PubMed] [Google Scholar]
- Chetty R. and Simelane,S. (1999) p53 and cyclin A protein expression in squamous carcinoma of the esophagus. Pathol. Oncol. Res., 5, 193–196. [DOI] [PubMed] [Google Scholar]
- Chittenden T., Livingston,D.M. and Kaelin,W.G. (1991) The T/E1A-binding domain of the retinoblastoma product can interact selectively with a sequence-specific DNA-binding protein. Cell, 65, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Deed R.W., Hara,E., Atherton,G.T., Peters,G. and Norton,J.D. (1997) Regulation of Id3 cell cycle function by Cdk-2-dependent phosphorylation. Mol. Cell. Biol., 17, 6815–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca P., Majello,B. and Lania,L. (1996) Sp3 represses transcription when tethered to promoter DNA or targeted to promoter proximal RNA. J. Biol. Chem., 271, 8533–8536. [DOI] [PubMed] [Google Scholar]
- Desdouets C., Matesic,G., Molina,C.A., Foulkes,N.S., Sassone-Corsi,P., Brechot,C. and Sobczak-Thepot,J. (1995) Cell cycle regulation of cyclin A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol. Cell. Biol., 15, 3301–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V., Kaufmann,W.K., Wilson,S.J., Tlsty,T.D., Lees,E., Harper,J.W., Elledge,S.J. and Reed,S.I. (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell, 76, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Girard F., Strausfeld,U., Fernandez,A. and Lamb,N.J. (1991) Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell, 67, 1169–1179. [DOI] [PubMed] [Google Scholar]
- Gory S., Dalmon,J., Prandini,M.H., Kortulewski,T., de Launoit,Y. and Huber,P. (1998) Requirement of a GT box (Sp1 site) and two Ets binding sites for vascular endothelial cadherin gene transcription. J. Biol. Chem., 273, 6750–6755. [DOI] [PubMed] [Google Scholar]
- Haidweger E., Novy,M. and Rotheneder,H. (2001) Modulation of Sp1 activity by a cyclin A/CDK complex. J. Mol. Biol., 306, 201–212. [DOI] [PubMed] [Google Scholar]
- Hall F.L., Williams,R.T., Wu,L., Wu,F., Carbonaro-Hall,D.A., Harper,J.W. and Warburton,D. (1993) Two potentially oncogenic cyclins, cyclin A and cyclin D1, share common properties of subunit configuration, tyrosine phosphorylation and physical association with the Rb protein. Oncogene, 8, 1377–1384. [PubMed] [Google Scholar]
- Handa K., Yamakawa,M., Takeda,H., Kimura,S. and Takahashi,T. (1999) Expression of cell cycle markers in colorectal carcinoma: superiority of cyclin A as an indicator of poor prognosis. Int. J. Cancer, 84, 225–233. [DOI] [PubMed] [Google Scholar]
- Hara E., Hall,M. and Peters,G. (1997) Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J., 16, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J.W., Adami,G.R., Wei,N., Keyomarsi,K. and Elledge,S.J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell, 75, 805–816. [DOI] [PubMed] [Google Scholar]
- Hatakeyama M. and Weinberg,R.A. (1995) The role of RB in cell cycle control. Prog. Cell Cycle Res., 1, 9–19. [DOI] [PubMed] [Google Scholar]
- Hauser P.J., Agrawal,D. and Pledger,W.J. (1998) Primary keratinocytes have an adhesion dependent S-phase checkpoint that is absent in immortalized cell lines. Oncogene, 17, 3083–3092. [DOI] [PubMed] [Google Scholar]
- Hinds P.W., Mittnacht,S., Dulic,V., Arnold,A., Reed,S.I. and Weinberg,R.A. (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell, 70, 993–1006. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., MacDonald,J.J., Lees-Miller,S. and Tjian R. (1990) GC box-binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell, 63, 155–165. [DOI] [PubMed] [Google Scholar]
- Johnson D.G. and Schneider-Broussard,R. (1998) Role of E2F in cell cycle control and cancer. Front. Biosci., 3, d447–d448. [DOI] [PubMed] [Google Scholar]
- Kingsley C. and Winoto,A. (1992) Cloning of GT box-binding proteins: a novel Sp1 multigene family regulating T-cell receptor gene expression. Mol. Cell. Biol., 12, 4251–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler K.,W. and Vogelstein,B. (1989) Whole genome PCR: application to the identification of sequences bound by gene regulatory proteins. Nucleic Acids Res., 17, 3645–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M. et al. (1996) The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J., 15, 7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Krek W., Ewen,M.E., Shirodkar,S., Arany,Z., Kaelin,W.G. and Livingston,D.M. (1994) Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell, 78, 161–172. [DOI] [PubMed] [Google Scholar]
- Krek W., Xu,G. and Livingston,D.M. (1995) Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S-phase checkpoint. Cell, 83, 1149–1158. [DOI] [PubMed] [Google Scholar]
- Latham K.M., Eastman,S.W., Wong,A. and Hinds,P.W. (1996) Inhibition of p53-mediated growth arrest by over-expression of cyclin-dependent kinases. Mol. Cell. Biol., 16, 4445–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveillard T. and Wasylyk,B. (1997) The MDM2 C-terminal region binds to TAFII250 and is required for MDM2 regulation of the cyclin A promoter. J. Biol. Chem., 272, 30651–30661. [DOI] [PubMed] [Google Scholar]
- Ma T. et al. (2000) Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in cajal bodies promotes histone gene transcription. Genes Dev., 14, 2298–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity A., McKenna.,W.G. and Muschel,R.J. (1997) Cyclin A message stability varies with the cell cycle. Cell Growth Differ., 8, 311–318. [PubMed] [Google Scholar]
- Majello B., De Luca,P. and Lania,L. (1997) Sp3 is a bifunctional transcription regulator with modular independent activation and repression domains. J. Biol. Chem., 272, 4021–4026. [DOI] [PubMed] [Google Scholar]
- Mudryj M., Devoto,S.H., Hiebert,S.W., Hunter,T., Pines,J. and Nevins,J.R. (1991) Cell cycle regulation of the E2F transcription factor involves an interaction with cyclin A. Cell, 65, 1243–1253. [DOI] [PubMed] [Google Scholar]
- Murray A. (1995) Cyclin ubiquitylation: the destructive end of mitosis. Cell, 81, 149–152. [DOI] [PubMed] [Google Scholar]
- Nevins J.R. (1992) E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science, 258, 424–429. [DOI] [PubMed] [Google Scholar]
- Pagano M., Pepperkok,R., Verde,F., Ansorge,W. and Draetta,G. (1992) Cyclin A is required at two points in the human cell cycle. EMBO J., 11, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeper D.S., Parker,L.L., Ewen,M.E., Toebes,M., Hall,F.L., Xu,M., Zantema,A., van der Eb,A.J. and Piwnica-Worms,H. (1993) A- and B-type cyclins differentially modulate substrate specificity of cyclin-cdk complexes. EMBO J., 12, 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K., Lee,M.H., Erdjument-Bromage,H., Koff,A., Roberts,J.M., Tempst,P. and Massague,J. (1994) Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell, 78, 59–66. [DOI] [PubMed] [Google Scholar]
- Pruschy M., Wirbelauer,C., Glanzmann,C., Bodis,S. and Krek,W. (1999) E2F-1 has properties of a radiosensitizer and its regulation by cyclin A kinase is required for cell survival of fibrosarcoma cells lacking p53. Cell Growth Differ., 10, 141–146. [PubMed] [Google Scholar]
- Qin X.Q., Livingston,D.M., Kaelin,W.G. and Adams,P.D. (1994) Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl Acad. Sci. USA, 91, 10918–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D., Hengst,H. and Reed,S.I. (1995) Cyclin A-associated kinase activity is rate-limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell. Biol., 15, 4347–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I., Trowbridge,J.M. and Garabedian,M.J. (1999) Potentiation of human estrogen receptor α-transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J. Biol. Chem., 274, 22296–22302. [DOI] [PubMed] [Google Scholar]
- Rohlff C., Ahmad,S., Borellini,F., Lei,J. and Glazer,R.I. (1997) Modulation of transcription factor Sp1 by cAMP-dependent protein kinase. J. Biol. Chem., 272, 21137–21141. [DOI] [PubMed] [Google Scholar]
- Sala A. et al. (1997) Activation of human B-MYB by cyclins. Proc. Natl Acad. Sci. USA, 94, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Saville M.K. and Watson,R.J. (1998) The cell-cycle regulated transcription factor B-Myb is phosphorylated by cyclin A/Cdk2 at sites that enhance its transactivation properties. Oncogene, 17, 2679–2689. [DOI] [PubMed] [Google Scholar]
- Schulze A., Zerfass,K., Spitkovsky,D., Middendorp,S., Berges,J., Helin,K., Jansen-Durr,P. and Henglein,B. (1995) Cell cycle regulation of the cyclin A gene promoter is mediated by a variant E2F site. Proc. Natl Acad. Sci. USA, 92, 11264–11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- Shirodkar S., Ewen,M., DeCaprio,J.A., Morgan,J., Livingston,D.M. and Chittenden,T. (1992) The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell, 68, 157–166. [DOI] [PubMed] [Google Scholar]
- Soucek T., Pusch,O., Hengstschlager-Ottnad,E., Adams,P.D. and Hengstschlager,M. (1997) Deregulated expression of E2F-1 induces cyclin A- and E-associated kinase activities independently from cell cycle position. Oncogene, 14, 2251–2257. [DOI] [PubMed] [Google Scholar]
- Spitkovsky D., Schulze,A., Boye,B. and Jansen-Durr,P. (1997) Down-regulation of cyclin A gene expression upon genotoxic stress correlates with reduced binding of free E2F to the promoter. Cell Growth Differ., 8, 699–710. [PubMed] [Google Scholar]
- Supp D.M., Witte,D.P., Branford,W.W., Smith,E,P. and Potter,S.S. (1996) Sp4, a member of the Sp1-family of zinc finger transcription factors, is required for normal murine growth, viability and male fertility. Dev. Biol., 176, 284–299. [DOI] [PubMed] [Google Scholar]
- Swick A.G., Blake,M.C., Kahn,J.W. and Azizkhan,J.C. (1989) Functional analysis of GC element binding and transcription in the hamster dihydrofolate reductase gene promoter. Nucleic Acids Res., 17, 9291–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima H. and Hunter,T. (1994) p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell, 78, 67–74. [DOI] [PubMed] [Google Scholar]
- van Gurp M.F. et al. (1999) The CCAAT displacement protein/cut homeodomain protein represses osteocalcin gene transcription and forms complexes with the retinoblastoma protein-related protein p107 and cyclin A. Cancer Res., 59, 5980–5988. [PubMed] [Google Scholar]
- van Wijnen A.J. et. al. (1994) Transcription of histone H4, H3 and H1 cell cycle genes: promoter factor HiNF-D contains CDC2, cyclin A and an RB-related protein. Proc. Natl Acad. Sci. USA, 91, 12882–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindevoghel L., Chung,K.Y., Davis,A., Kouba,D., Kivirikko,S., Alder,H., Uitto,J. and Mauviel,A. (1997) A GT-rich sequence binding the transcription factor Sp1 is crucial for high expression of the human type VII collagen gene (COL7A1) in fibroblasts and keratinocytes. J. Biol. Chem., 272, 10196–10204. [DOI] [PubMed] [Google Scholar]
- Wang E.H., Zou,S. and Tjian,R. (1997) TAFII250-dependent transcription of cyclin A is directed by ATF activator proteins. Genes Dev., 11, 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Hannon,G.J., Zhang,H., Casso,D., Kobayashi,R. and Beach,D. (1993) p21 is a universal inhibitor of cyclin kinases. Nature, 366, 701–704. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M., Wang,H., Hsieh,C.M., Sibinga,N.E., Perrella,M.A. and Lee,M.E. (1997) Down-regulation of the cyclin A promoter by transforming growth factor-β1 is associated with a reduction in phosphorylated activating transcription factor-1 and cyclic AMP-responsive element-binding protein. J. Biol. Chem., 272, 22259–22264. [DOI] [PubMed] [Google Scholar]