

Abstract
NK1 is a splice variant of the polypeptide growth factor HGF/SF, which consists of the N-terminal (N) and first kringle (K) domain and requires heparan sulfate or soluble heparin for activity. We describe two X-ray crystal structures of NK1–heparin complexes that define a heparin-binding site in the N domain, in which a major role is played by R73, with further contributions from main chain atoms of T61, K63 and G79 and the side chains of K60, T61, R76, K62 and K58. Mutagenesis experiments demonstrate that heparin binding to this site is essential for dimerization in solution and biological activity of NK1. Heparin also comes into contact with a patch of positively charged residues (K132, R134, K170 and R181) in the K domain. Mutation of these residues yields NK1 variants with increased biological activity. Thus, we uncover a complex role for heparan sulfate in which binding to the primary site in the N domain is essential for biological activity whereas binding to the K domain reduces activity. We exploit the interaction between heparin and the K domain site in order to engineer NK1 as a potent receptor agonist and suggest that dual (positive and negative) control may be a general mechanism of heparan sulfate-dependent regulation of growth factor activity.
Keywords: heparan sulfate/hepatocyte growth factor/scatter factor/MET/NK1/protein engineering
Introduction
The polypeptide growth factor hepatocyte growth factor/scatter factor (HGF/SF) (Stoker et al., 1987; Gherardi et al., 1989; Miyazawa et al., 1989; Nakamura et al., 1989) and its receptor MET, the product of the c-MET proto-oncogene (Bottaro et al., 1991), play essential roles in the development of epithelial organs such as the placenta and liver (Schmidt et al., 1995; Uehara et al., 1995) and in the migration of myogenic precursor cells (Bladt et al., 1995) and motor neurons (Ebens et al., 1996; Caton et al., 2000).
HGF/SF and MET are also involved in the spreading of a variety of epithelial tumours as a result of MET chromosomal rearrangements (Yu et al., 2000), somatic and/or germline mutations in the MET kinase (Schmidt et al., 1997) or, more often, overexpression in tumour cells of an unrearranged and unmutated MET gene (reviewed in Jeffers et al., 1996).
HGF/SF has a unique domain structure that resembles that of the blood proteinase precursor plasminogen and consists of six domains: an N-terminal (N) domain (homologous to plasminogen activation peptide) four copies of the kringle (K) domain and a catalytically inactive serine proteinase domain (Donate et al., 1994). Two products of alternative splicing of the primary HGF/SF transcript encode NK1, a fragment containing the N and the first K domain (Cioce et al., 1996), and NK2, a fragment containing the N, K1 and K2 domains (Chan et al., 1991; Miyazawa et al., 1991; Hartmann et al., 1992). Both NK1 (Lokker and Godowski, 1993) and NK2 (Chan et al., 1991) were initially characterized as MET antagonists, although experiments in transgenic mice have subsequently indicated that NK1 behaves in vivo as a bona fide receptor agonist (Jakubczak et al., 1998).
There is an important difference in the mechanism of receptor binding and activation by HGF/SF and NK1. HGF/SF is fully active in cells lacking heparan sulfate, while NK1 is only active in cells that display heparan sulfate or in the presence of soluble heparin (Schwall et al., 1996). Thus, NK1, but not HGF/SF, resembles FGF (Rapraeger et al., 1991; Yayon et al., 1991) in terms of a requirement for heparan sulfate for receptor binding and/or activation.
Early domain deletion experiments indicated that the N domain is important for heparin binding (Mizuno et al., 1994), and site-directed mutagenesis identified residues in this domain essential for binding (Hartmann et al., 1998). Thus, reverse-charge mutation of R73 and R76 decreased the affinity of HGF/SF for heparin by >50-fold (Hartmann et al., 1998). A role for several other positively charged residues, such as K58, K60 and K62, was suggested from the solution structure of the N domain, as these residues are clustered in close proximity to R73 and R76 (Zhou et al., 1998), and recent NMR experiments have provided experimental support for an involvement of K60, K62, R73, R76, R78 and several other residues in heparin binding to the N domain (Zhou et al., 1999).
Despite this progress, the mechanism through which heparin and heparan sulfate confer agonistic activity to NK1 remains incompletely understood. NK1 crystallizes as a dimer in the absence of heparin (Ultsch et al., 1998; Chirgadze et al., 1999), and the features of this dimer suggested that it could represent the biologically active form of NK1 (Chirgadze et al., 1999). No experimental evidence, however, supports this interpretation as yet.
We present here two X-ray crystal structures of NK1–heparin complexes that define the heparin-binding site of NK1 and demonstrate by mutagenesis that this site is essential for NK1 dimerization and biological activity. We also show that heparin binding to a patch of positively charged residues in the K domain inhibits activity. Thus, we provide a structural basis for the biological activity of NK1 and use this knowledge to engineer novel and potent agonists of the MET receptor.
Results
Structure determination
The NK1 fragment of HGF/SF (amino acids 28–210) was expressed in the methylotrophic yeast Pichia pastoris as described (Chirgadze et al., 1999) and crystallized in complex with a tetradecameric (14mer) heparin fragment. The heparin fragment was prepared by digestion and purification from polydisperse heparin of bovine lung origin. The NK1–heparin complex crystallized in two different crystal forms referred to as A and B. Type A crystals were dodecahedral-shaped and belonged to the tetragonal P43212 space group (cell parameters: a = b = 86.6, c = 117.8 Å). Type B crystals were needle-shaped, with orthorhombic P21212 space group and cell parameters a = 59.9, b = 174.2, c = 179.7 Å. Diffraction data to a resolution of 2.3 Å (for crystal type A) and 3.0 Å (for crystal type B) were collected using a synchrotron radiation source (PX 14.1/14.2, SRS, Daresbury, UK). The structures of both crystal types were solved by molecular replacement using the X-ray structures of NK1 (1nk1 and 1bht) (Ultsch et al., 1998; Chirgadze et al., 1999) as search probes. The refined structures of crystal types A and B have crystallographic R-factors of 25.5 (Rfree of 29.5%) and 26.2 (Rfree of 27.9%), respectively (Table I).
Table I. Crystallographic statistics.
| Data set statistics | Crystal type A | Crystal type B |
|---|---|---|
| Space group | P43212 | P21212 |
| Resolution limits (Å) | 21–2.3 | 20–2.95 |
| Unit cell parameters | a = b = 86.6 Å, c = 117.8 Å, α = β = γ = 90° | a = 179.7 Å, b = 174.1 Å, c = 59.9 Å, α = β = γ = 90° |
| Unique reflections | 20 349 | 37 651 |
| Rsym (%) (last shell) | 7.7 (37.5) | 6.3 |
| Completeness (%) (last shell) | 99.5 (99.0) | 95.2 (91.4) |
| Reflections with I/σ >3 (%) (last shell) | 79.3 (48.3) | 81.3 (53.1) |
| I/σ | 11 | 12 |
| Redundancy | 13 | 6 |
| Mosaicity (°) |
0.37 |
0.27 |
| Molecular replacement statistics | ||
| Resolution range (Å) | 8–3.5 | 8–3.5 |
| Rcryst/Corr. Coeff. for solution 1 (search probe) | 51.9/25.9 (Kringle A) | 52.5/21.9 (dimer 1) |
| Rcryst/Corr. Coeff. for solution 2 (search probe) | 48.4/37.3 (+ N domain A) | 49.1/33.4 (+ dimer 2) |
| Rcryst/Corr. Coeff. for solution 3 (search probe) | 45.4/45.4 (+ Kringle B) | 45.7/43.5 (+ dimer 3) |
| Rcryst/Corr. Coeff. for solution 4 (search probe) | 43.7/48.4 (+ dimer 4) | |
| Overall Rcryst/Corr. Coeff. after rigid-body refinement by AMoRe (8–3.5 Å) |
40.2/59.0 |
41.7/53.9 |
| Refinement statistics | ||
| Resolution limits | 21–2.3 | 20–3.0 |
| Number of reflections (test set) | 19 273 (1040) | 36 626 (1787) |
| Total protein atoms in structure | 2170 | 10 477 |
| Total heparin molecules (sugar units) | 86 (5) | 735 (41) |
| Total HEPES (molecules) | 30 (2) | 120 (8) |
| Water | 69 | 184 |
| Rcryst (%) | 26.2 | 25.5 |
| Rfree (%) | 27.9 | 29.5 |
| Average B-factor | 56.7 | 58.6 |
Structure description
The asymmetric unit of crystal type A contains two NK1 protomers assembled into a head-to-tail dimer (Figure 1A), as in the crystal structures of NK1 described previously (Ultsch et al., 1998; Chirgadze et al., 1999). A HEPES molecule is bound to each of the K domains in the putative lysine-binding pocket, as in the 1bht structure (Ultsch et al., 1998). A heparin molecule (H) is clearly seen bound to the N domain of protomer A, but the N domain of protomer B is partially disordered and poorly defined at the periphery (residues 28–41, 52–83, 88–97 and 105–117 shown in lighter colour in Figure 1A). Thus, it cannot be seen whether a heparin molecule is bound. The heparin molecule bound to protomer A also makes contacts with the K domain of protomer A′ from the neighbouring asymmetric unit. The final refined structure contains five heparin monosaccharide units: two glycosamine (GlcN) and three iduronic acid (IdoA) units (Figure 1A).

Fig. 1. Contents of the asymmetric unit (A and B) and electron density for heparin (C). (A) Content of the asymmetric unit of crystal type A. The NK1 dimer is shown in ribbon representation, the view is along the non-crystallographic 2-fold axis. Protomer A is shown in green, protomer B in blue, and their N- and C-termini are labelled in the corresponding colours. Weakly defined regions in the N-domain are transparent. Heparin (H) is shown in stick representation bound to the N-domain of protomer A. (B) The asymmetric unit of crystal type B contains: four NK1 dimers coloured in blue (protomers A and B), green (protomers C and D), red (protomers E and F) and yellow (protomers G and H); and six heparin molecules (labelled M, N, O, Q, X and Y). Heparin molecule Q is bound to the N domain of protomer A, heparin Y to the N domain of protomer B, heparin X to the N domain of protomer C, heparin N to the N domain of protomer F, heparin O to the N domain of protomer G and heparin M to the N domain of protomer H. (C) 2Fo – Fc electron density contoured at 1σ for the longest heparin molecule observed (heparin X), consisting of nine sugar units. (A) and (B) were produced with MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merritt and Murphy, 1994). (C) was produced with BOBSCRIPT (Esnouf, 1997) and RASTER3D.
In contrast, the asymmetric unit of crystal type B contains an assembly of four NK1 dimers with six bound heparin molecules. The dimers in the asymmetric unit are positioned in a circle with a pseudo-2-fold axis running through the centre (Figure 1B). The NK1 dimer arrangement is identical to that observed in crystal type A and described earlier (Ultsch et al., 1998; Chirgadze et al., 1999). Unlike the structure of crystal type A, all residues between 38 and 208 are well ordered and show clear electron density in all protomers. All N domains interact with heparin molecules. The N domains of protomers A and E share one heparin molecule as do N domains of protomers D and G. The longest heparin fragment that could be built into electron density maps is nine monosaccharide units in length (heparin molecule X in Figure 1B); it is bound to the N domain of protomer C and the K domain of protomer F (Figure 1C). Other heparin molecules are less ordered, with the shortest fragment having only five monosaccharide units well defined (heparin molecule N). Each K domain has a HEPES molecule bound in the same binding pocket as in the structure of crystal type A. The dimers within the asymmetric unit show a good agreement with root mean square deviation (r.m.s.d.) values of Cα atoms between 0.50 Å (protomers A and B versus G and H) and 1.32 Å (protomers A and B versus C and D). The NK1 dimers in crystal type B are very similar to the dimer in crystal type A, with the largest r.m.s.d. of Cα atoms amounting to 1.19 Å for the dimer consisting of protomers A and B.
The heparin-binding site of NK1
Analysis of the 10 independent N domain structures provided by crystal types A and B allowed identification of the heparin-binding site of NK1. In nine N domains the binding site is occupied; in the remaining (the N domain of protomer B in Figure 1A) it could not be seen whether a heparin molecule is bound because the domain is partially disordered. Six of the N domains in crystal type B (protomers B, C and E–H) and one in crystal type A interact with heparin in a nearly identical manner (Figure 2A). The N domains of protomers A and D in crystal type B interact with opposite sides of heparin molecules Q and O, which also interact in the consensus mode with the N domains of protomers E′ and G′ from neighbouring asymmetric units. Thus, all seven heparin molecules defined in the two crystal structures interact in a very conserved manner with one N domain; in addition, heparin molecules Q and O interact with a second N domain in opposite directionality (A and D). The region of the N domain of protomers A and D involved in heparin contacts is the same as shown in Figure 2A; however, the protein–heparin interactions differ and do not involve the crucial contact with R73 (see below). Thus, this second mode of binding, which is only observed in one of the two crystal structures (crystal type B), is probably the result of crystal packing.

Fig. 2. The heparin-binding site of NK1 and the interaction between heparin and the K domain. (A) Superposition of the N domain of protomer A from crystal type A and protomers B, C, F, G and H from crystal type B. The resulting position of the heparin molecules bound to these N domains shows a highly conserved binding motif for GlcN-5, IdoA-6, GlcN-7 and IdoA-8. The heparin molecule of the high-resolution structure of crystal type A is coloured in green. (B) Superposition of the N domain of protomer A in crystal type A (green), and protomer C in crystal type B (red). The resulting position of the corresponding K domains (the K domain of a symmetry-related protomer A′ in crystal type A and the K domain of protomer F in crystal type B) shows different positions relative to heparin, although the same patch of residues is involved in the contacts. (C) Surface electrostatic potential of NK1 at the site of interaction with heparin molecule X. Surface areas corresponding to positive potential are shown in blue, areas with negative potential are shown in red. The heparin is positioned in a positively charged groove between the N domain of protomer C and the K domain of protomer F. Electrostatic potential was calculated with GRASP (Nicholls et al., 1991).
The consensus heparin-binding site defined by the seven conserved N domain–heparin complexes consists of a cleft, the edges of which are created by the β2 strand of the central β-sheet, the helix and the loop connecting the helix with the β3 strand (Figure 2A). The buried surface area at the N domain–heparin interface averages 680 Å2, and a stretch of four monosaccharide units (GlcN-5, IdoA-6, GlcN-7 and IdoA-8) provides the key contact (Figure 2A). All glycosidic linkages within this tetrasaccharide adopt geometries similar to those found in other heparin–protein complex structures and in the solution conformation of heparin (Mulloy and Forster, 2000). The tetrasaccharide length of ∼17 Å is close to the value of 16.5 Å found by fibre diffraction experiments (Atkins and Nieduszynski, 1975). The pyranose ring of IdoA-6 is always in an approximate 1C4 chair, and IdoA-8 an approximate 2S0 skew boat conformation.
Within the interacting heparin tetramer, GlcN-5 represents the focal point of the heparin–N domain interface by making a specific two-point interaction with residues T61 and K63 of the β2 strand (Figure 3A and B). This involves hydrogen bonds between the 6-O-sulfate of GlcN-5 and the main chain N atom of K63 (2.66 ± 0.10 Å), as well as between the N-sulfate and the main chain N atom (2.85 ± 0.22 Å) and side chain OG1 atom of T61 (3.13 ± 0.31 Å) (Figure 3A and B). The 2-O-sulfate of IdoA-6 does not make any contacts with the residues of the N domain, whereas the 2-O-sulfate of IdoA-8 makes a hydrogen bond with the NH1 side chain atom of R73 (2.99 ± 0.2 Å) (Figure 3A and B). GlcN-7, although well defined in the electron density maps, is not involved in hydrogen bonds to the N domain but it is stabilized through a hydrophobic interaction between the C6 atom in GlcN-7 and the CZ atom of R73 (Figure 3B), as well as through ionic interactions between the NH2 side chain atom of R73 and the 6-O-sulfate of GlcN-7 (3.64 ± 0.5 Å) and a polar contact between the main chain N atom of G79 and the 6-O-sulfate of GlcN-7 (3.50 ± 0.26 Å). R73, the key residue of the heparin-binding site, also makes two hydrogen bonds with the carboxylate group of IdoA-6 (NH1–O62, 3.40 ± 0.50 Å; NH2–O61, 3.06 ± 0.25 Å). The above interactions are most specific and occur in all seven structures examined (T61, K63 and G79) or in six out of seven (R73). Additional interactions occur in some but not other heparin-binding sites. Side chain atom NZ of K60 forms hydrogen bonds in three binding sites (protomer A in crystal type A, protomers B and C in crystal type B) with the carboxylate group of IdoA-6 (3.43 ± 0.68 Å) and the NH1 atom of R76 forms salt bridges with the N-sulfate of GlcN-9 (3.84 ± 0.57 Å) in three structures (protomers B, C and E of crystal type B). K58 and K62 are only seen in the high-resolution structure of crystal type A to form hydrogen bonds to the N-sulfate of GlcN-5 (K58) and the ring oxygen of IdoA-6 (K62) (Figure 3B). The side chain of K78 is disordered in all seven heparin-binding sites.

Fig. 3. Interaction of NK1 with heparin. (A) Stereoview of interactions between heparin monosaccharide units GlcN-5, IdoA-6, GlcN-7 and IdoA-8 with the N domain of NK1. The side chains of R73, K60 and T61 are shown along with conserved hydrogen bonds (in green). (B) Hydrogen bonding network and hydrophobic contacts between the N domain and heparin in crystal type A (left) and type B (right). Most hydrogen bonds occur in both crystal forms; crystal type A shows additional bonds from the side chains of Lys58 and Lys62. (C) The interface between the K domain of protomer F and heparin chain X. Side chains contributing to ionic interactions are shown. The interaction between K170 and heparin is not apparent in this structure, but is seen in several others.
Heparin–K domain interactions are seen in both crystal structures (Figures 1B and C, 2B and C and 3C) and involve a cluster of positively charged residues (K132, R134, K170, R181). These residues form a patch of positive electrostatic potential lining against the negatively charged heparin chain (Figures 2C and 3C). The K domain–heparin interface buries an average surface of 460 Å2. Unlike the N domain, however, the orientation adopted by the K domain relative to heparin differs significantly in different structures (see Figure 2B for an example). Thus, the functional significance of the heparin–K domain interactions was probed by mutagenesis.
Novel NK1 mutants
Two reverse-charge N domain mutants (HP11 and HP12) and two K domain mutants (1K1 and 1K2) (Figure 4A) were generated and characterized for heparin binding and biological activity.
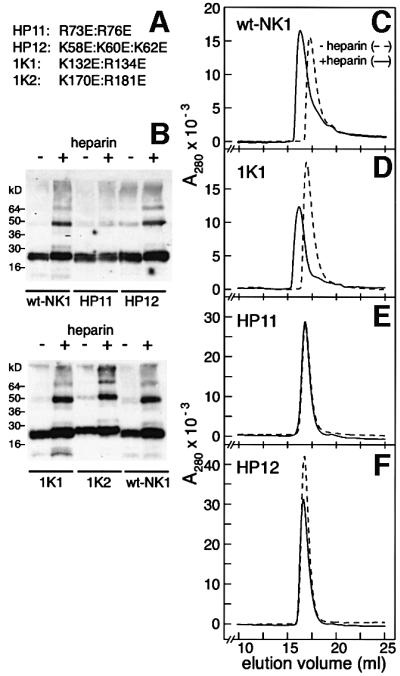
Fig. 4. Heparin-induced oligomerization of wt- and mutant-NK1. (A) The mutations introduced in the N (HP11, HP12) and K domain (1K1, 1K2) of NK1. (B) Cross-linking of wt- and mutant-NK1 in the absence or presence of equimolar concentrations of 14mer heparin. Cross-linked proteins were analysed by western blotting and detected with an anti-HGF/SF polyclonal antibody (1W53). The position of molecular weight markers is also shown. (C–F) Gel filtration of wt- and mutant-NK1 in the absence or presence of equimolar concentrations of 14mer heparin. Chromatography was carried out on an HR30 Superdex-200 column equilibrated in PBS adjusted to 300 mM NaCl. The elution profile of the 1K1 mutant was identical to that of 1K2 and is not shown. wt-NK1 without heparin shows slight retardation in elution volume due to residual interaction with the column. Heparin induced a shift in the elution volume of wt-NK1 and of the K domain mutants but not of the N domain mutants HP11 and HP12.
Cross-linking (Schwall et al., 1996) and gel filtration (Chirgadze et al., 1999) experiments were employed first in order to characterize heparin-mediated dimerization of wild-type (wt)-NK1 in solution. Heparin failed to induce both cross-linking (Figure 4B) and oligomerization in solution (Figure 4E) of the HP11 mutant. HP12, the second N domain mutant, was cross-linked by heparin (Figure 4B) but, like the HP11 mutant, failed to oligomerize in solution in the presence of heparin (Figure 4F). Thus, in these experiments, the behaviour of this mutant was intermediate between wt-NK1 and HP11. The two K domain mutants (1K1 and 1K2) behaved like wt-NK1 (Figure 4B and D). In conclusion, the amino acids that make crystallographic contact with heparin in the N domain (Figure 3A and B) are required for heparin-dependent dimerization of NK1 in solution (Figure 4E and F), suggesting that these amino acids may be responsible for heparan sulfate-dependent dimerization of NK1 on the cell surface.
Biological activity of NK1 mutants
Experiments with heparan sulfate-deficient cells have established an essential requirement for heparan sulfate or soluble heparin for the biological activity of NK1 (Schwall et al., 1996). Normal cells display membrane-bound heparan sulfate and thus, if they express the MET receptor, respond to NK1. They may fail, however, to respond to heparan sulfate-deficient NK1 mutants such as HP11 and HP12.
Figure 5 shows colony dispersion (scatter) assays with MDCK cells (Stoker et al., 1987; Gherardi et al., 1989) in the presence of full-length HGF/SF or NK1 proteins. The colonies in control cultures exhibited strong cell–cell adhesion and a typical epithelial, ‘cobblestone’ appearance (Figure 5A). HGF/SF (10–10 M), wt-NK1 or the K domain mutant 1K1 (10–8 M) (Figure 5B, C and F) induced full dissociation of MDCK colonies. In contrast, both the HP11 and HP12 mutants (10–8 M) were inactive (Figure 5D and E). Addition of soluble heparin (10–6 M) did not affect control cultures or cultures containing HGF/SF or NK1 proteins (data not shown).

Fig. 5. Biological activity of NK1 mutants. (A–F) Colony scatter assays on MDCK cells; (G) DNA synthesis assays on MK cells. (A–F) For colony dispersion (scatter) assays, MDCK cells were plated at low density in 60 mm dishes and cultured for 3 days in standard medium after which the medium was replaced with fresh medium (A) or medium containing 10–10 M HGF/SF (B) or 10–8 M NK1 proteins (C–F). After overnight incubation several colonies from each dish were photographed using phase-contrast optics. The HP11 and HP12 mutants are inactive. (G) For DNA synthesis assays, MK cells were cultured to confluence in keratinocyte serum-free medium and transferred in basal medium for 24 h before incubation with [3H]thymidine and HGF/SF or NK1 proteins at the concentrations indicated in the figure. DNA synthesis was measured as TCA-insoluble radioactivity. The HP11 mutant is inactive and the HP12 shows much reduced activity compared with wt-NK1. In contrast the 1K1 mutant is more active than wt-NK1 and full-length HGF/SF. The activity of the 1K2 mutant (not shown) is similar to that of the 1K2 mutant.
The biological activity of the NK1 mutants was studied further on a different target, the MK mouse keratinocyte line, which exhibits a strong mitogenic response to HGF/SF (Moorby et al., 1995). wt-NK1, but not the N domain mutants HP11 and HP12, induced appreciable stimulation of DNA synthesis at concentrations of 10–10 M or higher (Figure 5G). Remarkably, the K domain mutant 1K1 exhibited activity much higher than wt-NK1 and comparable to that of full-length HGF/SF (Figure 5G). 1K2, the second K domain mutant, behaved like the 1K1 mutant (data not shown). Thus, the HP11 and HP12 mutations that failed to induce dimerization of NK1 in solution (Figure 4E and F) also caused loss of biological activity (Figure 5), presumably due to the inability of these mutants to bind cell-associated heparan sulfate on the surface of MDCK or MK cells. In contrast, the K domain mutations conferred increased biological activity to NK1 and converted it to a full receptor agonist (Figure 5G).
In order to establish whether the loss of activity of the HP11 and HP12 mutants was due to defective receptor binding or activation, competition experiments were carried out in which MDCK cells were cultured in the presence of HGF/SF alone (10–10 M) or in the presence of HGF/SF and excess concentrations (10–8 or 10–7 M) of wt-NK1 or the two N domain mutants. As expected, wt-NK1 behaved as a partial antagonist (compare Figure 6C and D with B) but HP11 and HP12 exhibited no (HP11) or very little (HP12) antagonistic activity (Figure 6E and F, and G and H), implying that the lack of activity of these mutants is due to reduced receptor binding rather than failure to induce receptor activation.

Fig. 6. Effect of wt- or mutant-NK1 on HGF/SF-induced scatter of MDCK colonies. MDCK cells were cultured for 3 days in standard medium before incubation with fresh medium (A), 10–10 M HGF/SF (B) or 10–10 M HGF/SF plus the concentrations of wt- or mutant-NK1 indicated in the figure (C–H). After overnight incubation, colonies were photographed using phase-contrast optics. wt-NK1 but not the HP11 or HP12 mutant partially inhibits the activity of full-length HGF/SF.
Mechanism of heparan sulfate-induced NK1 activity
The role of heparan sulfate in the binding of NK1 to the MET receptor could depend entirely on dimer formation, without a further involvement in the binding of the NK1 dimer to the MET receptor, or could involve both dimer formation and binding of the NK1 dimer to MET. In order to clarify this, binding experiments were carried out with NK1 or HP11 and a soluble, dimeric fragment of the MET receptor encompassing the whole ligand-binding region (E.Gherardi, unpublished data). wt-NK1–MET or HP11– MET complexes were formed at room temperature using a 2:2 MET:ligand molar ratio in the absence or presence of a 5-fold molar excess of heparin and analysed by gel filtration (Figure 7A and C).

Fig. 7. Effect of heparin on the binding of wt-NK1 or HP11 mutant to the MET receptor (A–D) and binding of NK1 and MET to immobilized heparin (E). (A and C) For binding of NK1 or HP11 to a soluble, dimeric fragment of the MET receptor, proteins were incubated for 3 h at room temperature, at a 2:2 molar ratio (2 × 10–5 M NK1 or HP11 and 1 × 10–5 M dimeric MET) in the absence (dashed line) or presence (solid line) of 1 × 10–4 M heparin (Calbiochem 375097) before gel filtration chromatography on an HR30 Superdex-200 column (Amersham Pharmacia Biotech). The column was equilibrated and eluted with PBS, adjusted to 300 mM NaCl at 0.5 ml/min, and 0.5 ml fractions were collected. (B and D) Fractions 21–39 were analysed by western blotting for MET and NK1. Heparin induces dimerization of wt-NK1 and partial association of wt-NK1 dimer with MET. No binding of the N domain mutant HP11 to MET could be detected with or without heparin. (E) Binding of NK1 and MET to immobilized heparin (Heparin-HiTrap; Amersham Pharmacia Biotech) equilibrated in PBS. Bound proteins were eluted at 0.25 ml/min with a linear gradient of NaCl (dashed line).
In the absence of heparin both MET and wt-NK1 eluted at positions corresponding to the free species (Figure 7A) and there was no evidence for complex formation, as determined by western blot analysis, of fractions eluted from the gel filtration column (Figure 7B). When wt-NK1 and MET were incubated in the presence of heparin, the majority of NK1 eluted as a dimer (Figure 7A and B), but some was clearly associated with the MET peak (Figure 7B). When the HP11 mutant was incubated with MET, the elution profile was unaffected by the absence or presence of heparin (Figure 7C), and western blot analysis of individual column fractions showed no significant association of HP11 with MET in either case (Figure 7D).
The ability of the MET receptor to bind heparin was next studied. The soluble MET receptor fragment bound to immobilized heparin and eluted at ∼0.5 M NaCl, a concentration slightly lower than that required for eluting NK1 from the same column (Figure 7E). Similar experiments with a second form of the MET protein that corresponds to the entire ectodomain showed that the latter bound heparin with the same apparent affinity as the MET fragment comprising the ligand-binding region alone (data not shown). These results indicate that: (i) MET, as well as NK1, can bind heparin directly; and (ii) the heparin-binding site of MET is located in the region of the receptor that binds NK1 or HGF/SF. They also demonstrate that the role of heparan sulfate in the NK1–MET system is dual: ligand dimerization, on the one hand, and promotion of an NK1–heparan sulfate–MET ternary complex on the other.
Discussion
The HGF/SF–MET signalling system is a complex one. The ligand is a high molecular weight protein with a multidomain structure that requires proteolytic processing from a single chain precursor (Naldini et al., 1992; Mars et al., 1993; Miyazawa et al., 1993; Shimomura et al., 1995) in order to yield the biologically active heterodimer. Furthermore, at least two products of alternative splicing occur in vivo, NK1 (Cioce et al., 1996) and NK2 (Chan et al., 1991; Miyazawa et al., 1991; Hartmann et al., 1992); both can interact with the MET receptor and modulate the cell response to HGF/SF through partial agonistic and antagonistic activities.
That HGF/SF does not require heparan sulfate for activity is clear from experiments on heparan-deficient cells (Schwall et al., 1996), from the biological activity of HGF/SF mutants deficient in heparin binding (Hartmann et al., 1998) and from the phenotype of mice lacking key enzymes involved in heparan sulfate biosynthesis (Bullock et al., 1998; Ringvall et al., 2000), which bear no resemblance to the phenotype of the HGF/SF and MET mutant mice (Bladt et al., 1995; Schmidt et al., 1995; Uehara et al., 1995). However, while heparan sulfate is not required for MET binding and activation by HGF/SF, the case is entirely different for the NK1 splice variant. NK1 contains the first of the two heparan sulfate-binding sites of HGF/SF (the second is located in Kringle 2) (Mizuno et al., 1994; Hartmann et al., 1998) and requires heparan sulfate or soluble heparin for activity (Schwall et al., 1996).
This study: (i) defines the structure of the heparan sulfate-binding site in NK1 (Figures 1–3); (ii) demonstrates through mutational studies an essential role of this site in NK1 dimerization (Figure 4) and biological activity (Figures 5 and 6); (iii) provides evidence for a further role of heparan sulfate in the binding of the NK1 dimer to the MET receptor (Figure 7); and (iv) has led to the generation of potent receptor agonists through mutagenesis of a cluster of positively charged residues on the K domain (Figure 5).
The heparan sulfate-binding site of NK1
The three-dimensional structures of seven independent copies of the NK1 protomer complexed with heparin, each having different packing relationships in the crystal asymmetric units, have allowed unambiguous definition of the heparin-binding site of NK1 (Figures 2A, 3A and B). Main contributors to this binding site are the side chains of residues R73, K60 and T61 and main chain atoms of residues T61 and K63. All these residues make specific hydrogen bonds to negatively charged moieties of heparin, with R73 carrying the pivotal role. This residue makes two hydrogen bonds with NH1 and NH2 to the two oxygens in the carboxylate group of IdoA-6, another hydrogen bond with NH1 to the 2-O-sulfate of IdoA-8, a salt bridge with NH2 to the 6-O-sulfate of GlcN-7 and a van der Waals contact to carbon-6 of GlcN-7. Additional contributions to the heparin-binding site are made by the main chain N atom of G79 and side chains of R76 and to a lesser extent K58 and K62.
The main involvement of R73 in interactions with heparin is in agreement with early mutagenesis work (Hartmann et al., 1998) and NMR studies with the N domain that showed largest chemical shifts for this residue (Zhou et al., 1999). The latter studies also assigned K60 and K62 to the primary heparin-binding site, with additional contributions from R76 and K78. A role for K60 and R76 is confirmed here; a role for K62 and K58 is apparent in the high-resolution structure of crystal type A, whereas the side chain of K78 is disordered in all seven structures. It remains possible, though, that the side chain of K78 contributes to the positively charged patch on the surface of the N domain that attracts heparin in the proximity of the binding site.
It has been suggested that the heparan sulfate sequence that binds HGF/SF differs from the one that binds FGF. The crystal structure of FGF2–heparin complexes (Faham et al., 1996), as well as previous studies of FGF2–heparan sulfate complexes (Habuchi et al., 1992; Turnbull et al., 1992), indicated a major contribution for 2-O-sulfates in IdoA, N-sulfates in GlcN and the carboxylate group in IdoA units, whereas 6-O-sulfates in GlcN units seem to be of lesser importance. In contrast, analysis of heparan sulfate–HGF/SF interactions indicated that 6-O-sulfates in GlcN units are the most important determinant of binding, with 2-O-sulfates in IdoA and N-sulfates in GlcN playing minor roles (Lyon et al., 1994). From the data presented here, it is conceivable that the loss of the sole hydrogen bond involving an IdoA-2 SO4 (Figure 3A and B) would not significantly alter the binding affinity of heparin for NK1 or HGF/SF. The N-sulfate in GlcN, however, is involved in the central two-point attachment of GlcN-5, thus suggesting an important contribution (Figure 3A and B). This is in agreement with DNA synthesis studies on heparan sulfate-deficient cells (BaF3) with NK1 and de-N-sulfated heparin, which showed no stimulation compared with NK1 alone (Schwall et al., 1996).
The heparin-induced NK1 dimer and role of heparan sulfate in the NK1–MET system
A striking conclusion of this study is that the NK1 dimer complexed with heparin (Figure 1A and B) is essentially the same yielded by NK1 alone under crystallization conditions (Ultsch et al., 1998; Chirgadze et al., 1999). Comparison of NK1 dimers complexed with heparin (Figure 1A and B) with unbound NK1 (Ultsch et al., 1998; Chirgadze et al., 1999) shows good superimposition of the Cα atoms with r.m.s.d. values of not more than 1.1 Å. Even in comparison with the heparin-binding site, the backbone shows no large deviations. Some significant deviations in the loop composed of residues 51–57 in the structure of crystal type A (r.m.s.d. of Cα atoms up to 4 Å) are clearly the result of crystal packing, with the proline ring of P55 packing against the identical residue in a neighbouring asymmetric cell.
These results establish conclusively that heparin binding does not induce a conformational change in NK1 and confirm the earlier suggestion (Chirgadze et al., 1999) that heparin promotes the formation of this protein dimer. This may involve initial binding to an NK1 monomer and a local change in the electrostatic potential that results in dimer formation. This interpretation is supported by the finding that several other polyanions, such as dextran sulfate, can induce dimerization and biological activity of NK1 (Schwall et al., 1996). However, other sulfated polymers, such as chondroitin sulfate, are inactive, suggesting a requirement for a specific pattern or spacing of negatively charged groups.
Heparin binding to NK1, however, does not involve, as predicted, a second cluster of positively charged amino acids in the N domain (K91, R93 and K94) (Chirgadze et al., 1999) or the polar side chains of N72, T75 and N77 (Zhou et al., 1999), nor does heparin link two N domains by bridging their heparin-binding sites (Zhou et al., 1999). Thus, the crystal structures of NK1–heparin complexes considerably extend the conclusions of NMR solution experiments (Zhou et al., 1999) by unequivocally defining the binding site in the N domain (Figure 2A) and unveiling additional contacts with the K domain (Figures 1C, 2C and 3C).
We note that the NK1 dimer has features found in other dimers (Perisic et al., 1994; Schiering et al., 2000; Zhang et al., 2000) that result from domain swapping (Bennett et al., 1995). There is conclusive evidence from other systems (for example, single chain Fv fragments of antibodies; Holliger et al., 1993) that dimerization through domain swapping depends critically on the length of the inter-domain linker. It would be interesting to determine the consequences of shorter or longer N–K domain linkers in terms of their propensity to form dimers, the arrangements of the N and K domains and ultimately the biological activity of such dimers.
The role of heparan sulfate in growth factor signalling is best understood in another system, namely the FGF one. There are important similarities and differences between the FGF–FGF receptor and NK1–MET systems. In both instances heparin promotes ligand dimerization in solution. However, the heparin-induced FGF dimer is bridged by heparin alone and lacks a protein–protein interface (DiGabriele et al., 1998). In the case of NK1, heparin induces the formation of a dimer that exhibits a protein interface in excess of 2000 Å (Figure 1A) (Ultsch et al., 1998; Chirgadze et al., 1999). Two crystal structures of FGF–heparin–FGFR ternary complexes (Pellegrini et al., 2000; Schlessinger et al., 2000) demonstrate that heparin contacts both ligand and receptor although the stoichiometry and overall architecture of the two structures differ considerably. The data presented here demonstrate that NK1, heparin and MET may also form a ternary complex (Figure 7), although under the conditions used only a small amount of NK1 associated with the receptor and the precise stoichiometry of such a complex remains to be defined.
There is evidence that, in cells expressing MET, the receptor is localized in discrete patches on the cell surface where it is present in the form of large complexes (>500 kDa) (Faletto et al., 1992). Such clusters exist independently of ligand (Faletto et al., 1992) and conceivably result from MET–heparan sulfate interactions. They may also form the basis for the ability of target cells to respond to NK1. Thus, while MET alone and its downstream pathways are sufficient to dictate the ability of a cell to respond to full-length HGF/SF, tissue-specific heparan sulfate variants (Park et al., 2000), and their affinity for MET and NK1, may control the ability of different cell types to respond to NK1.
Novel MET agonists
A major effort is underway to develop MET antagonists and agonists for therapy. MET antagonists are expected to find applications in a variety of epithelial tumours overexpressing MET, while receptor agonists may be valuable in liver regeneration, the repair of skin wounds and therapeutic angiogenesis. The structural and mutagenesis data presented here have enabled the generation of potent MET agonists.
NK1 was first characterized as a receptor antagonist (Lokker and Godowski, 1993) but subsequent studies highlighted significant agonistic activity on cells in culture (Cioce et al., 1996) and in vivo (Jakubczak et al., 1998). Such agonistic activity of NK1 has not received adequate attention although the phenotype of NK1-overexpressing transgenic mice (Jakubczak et al., 1998) clearly provided a strong reminder of it. In the studies presented here, wt-NK1 behaved as a partial agonist, as expected. It produced dispersion of MDCK colonies (Figure 5C) and stimulation of DNA synthesis in MK cells (Figure 5G). Interestingly, maximal stimulation of DNA synthesis by wt-NK1 occurred at concentrations as low as 10–10 M (Figure 5G), a concentration much lower than those required in other studies (see for example, Schwall et al., 1996). The higher potency of NK1 observed in our studies may reflect the source (yeast versus bacterial), and hence the activity, of the protein used.
While wt-NK1 remains less active than full-length HGF/SF (Figure 5G), remarkably, the two K domain mutants exhibited biological activity much greater than wt-NK1 and equal to or greater than full-length HGF/SF (Figure 5F and G). Although we do not offer formal proof for the mechanism responsible for the increased activity of these mutants, preliminary data suggest that the K domain mutations result in increased net affinity of NK1 for heparin. Thus, it is possible that reverse-charge mutations of K132, R134, K170 and R181 may lead to a local re-orientation of the heparin chain across the N and K domains (Figure 2C), increasing binding to the main site in the N domain (Figure 2A).
Regardless of the mechanism, we have demonstrated that substitution of only two amino acids in the K domain (K132E:R134E or K170E:R181E) is sufficient for converting NK1 into a full receptor agonist. NK1, but not HGF/SF, can be produced in yeast and is expected to exhibit favourable in vivo kinetics and tissue distribution compared with full-length HGF/SF. These engineered fragments of HGF/SF thus strengthen the concept that dissecting receptor- and co-receptor-binding sites by protein engineering offers a potent route for the development of growth factors for therapy (Hartmann et al., 1998).
Materials and methods
Cloning, mutagenesis, expression and purification
Cloning, expression and purification of wt-NK1 were carried out as described in Chirgadze et al. (1999), except that final purification of NK1 by cation exchange chromatography was carried out on a Source15S column (Amersham Pharmacia Biotech). For expression of NK1 mutants, an EcoRI–NotI fragment from the wt-NK1 expression construct in pPIC9K was cloned into the pBluescript KS– vector (Stratagene) and DNA amplification reactions were carried out using complementary pairs of mutagenic oligonucleotides. The N domain mutants were produced by DNA amplification of the relevant fragments from full-length human HGF/SF cDNA, which carried R73E:R76E mutations (mutant HP11) or the K58E:K60E:K62E mutations (mutant HP12). N and K domain mutants were finally cloned in the expression vector pPIC9K. Transformation and selection of P.pastoris was carried out as described previously (Chirgadze et al., 1999).
Purification of heparin fragments
Sodium heparin from bovine lung (material remaining from the 2nd International Standard Heparin) was digested with heparinase I (a kind gift of Leo Pharmaceuticals) for 14 min at 37°C in 10 mM phosphate buffer, 1.25 mM CaCl2 pH 7.0. The water was evaporated, the residue dissolved in 20 g/l ammonium bicarbonate and loaded on to a Biogel P-10 (Bio-Rad) column. Fractions containing heparin fragments of the same length (up to hexadecasaccharide) were combined and water and ammonium bicarbonate evaporated on a rotavapor (Buchi). The heparin fragments were then dissolved in 0.1 M ammonium acetate and an aliquot of each was analysed by gel permeation chromatography using TSK G3000 SW XL (30 cm × 7.8 mm) and G2000 SW XL (30 cm × 7.8 mm) columns in series in order to assess purity and concentration. The fragments were next lyophilized and redissolved in water (at least three cycles) in order to eliminate ammonium acetate.
Characterization of wt- and mutant-NK1–heparin complexes
This was carried out by gel filtration chromatography and cross-linking experiments. For gel filtration, wt- or mutant-NK1 (5 × 10–5 M) was incubated for 2 h in the presence or absence of an equimolar concentration of 14mer heparin in phosphate-buffered saline (PBS) adjusted to 300 mM NaCl. One hundred microlitres were then loaded on to an HR30 Superdex-200 column (Amersham Pharmacia Biotech) and eluted at 0.5 ml/min.
For cross-linking, 10 µl of wt- or mutant-NK1 (0.1 mg/ml) were incubated in the absence or presence of an equimolar concentration of 14mer heparin in PBS. After 2 h incubation at room temperature, 1 µl of crosslinker (BS3; Pierce) was added at 100-fold molar excess, the reaction was continued for 30 min and then quenched with 1 µl of 1 M Tris–HCl pH 7.4. Reaction products were loaded on to 15% SDS–polyacrylamide gels and blotted on to a nitrocellulose membrane (Schleicher & Schuell). The membrane was blocked in 2% skimmed milk, incubated for 1 h in the presence of sheep anti-HGF/SF polyclonal antibody (1W53, 1:1000), washed with PBS + 0.2% Tween-20 and next incubated for 1 h with HRP-conjugated anti-sheep immunogolobulin antibody (Dako). HRP activity was detected after three further washes in PBS + 0.2% Tween-20 using a chemiluminescent substrate (Pierce).
Formation of wt- and mutant-NK1–MET complexes
wt-NK1 or HP11 (2 × 10–5 M) was incubated with 1 × 10–5 M dimeric MET (a fusion protein containing the N-terminal region of the MET receptor fused to the hinge, CH2 and CH3 domains of human γ1 immunoglobulin) in the presence or absence of 1 × 10–4 M heparin (mean mol. wt = 5000 Da; Calbiochem) for 3 h in PBS (adjusted to 300 mM NaCl). One hundred microlitres were loaded on to an HR30 Superdex-200 column (Amersham Pharmacia Biotech), eluted at 0.5 ml/min and 0.5 ml fractions were collected. Twelve microlitres of alternate fractions were run on pre-cast Tris–glycine 4–20% polyacrylamide gels (Invitrogen) and then blotted on to nitrocellulose membranes. The membranes were blocked in 2% skimmed milk (Marvel) in PBS and then incubated for 1 h with sheep serum containing polyclonal 1W53 anti-HGF/SF antibody diluted 1:1000 in blocking agent. Membranes were washed in PBS containing 0.1% Tween-20, then incubated with a mixture of HRP-conjugated rabbit anti-sheep-immunogloblulin (Dako) diluted 1:1000 and HRP-conjugated rabbit anti-human-immunoglobulin (Dako) diluted 1:5000 in blocking agent. Membranes were washed as before and developed with ECL western blotting detection reagents (Amersham Pharmacia).
MDCK colony scatter assays
Scatter assays were carried out as described in Stoker et al. (1987) and Gherardi et al. (1989). Briefly, MDCK cells were plated at 1–2.5 × 103 cells/60 mm dish and cultured in 5% fetal calf serum in Dulbecco’s modified Eagle’s medium for 2–3 days before addition of HGF/SF, wt- or mutant-NK1 as detailed in the legends to Figures 5 and 6. After overnight incubation, plates were inspected and several colonies from each plate were photographed using a Leica DM IRB inverted microscope equipped with phase contrast optics and a Hamamatsu colour chilled 3CCD camera.
DNA synthesis in MK cells
The mouse keratinocyte line MK was cultured to confluence in keratinocyte SFM medium (Gibco) supplemented with 5 ng/ml EGF-53 and 50 µg/ml bovine pituitary extract (BPE) in 24-well tissue culture plates (Costar). At confluence, complete medium was replaced with basal medium (no EGF and BPE) for 24 h before addition of 1 µCi/well [3H]thymidine in basal medium containing 1 mg/ml bovine serum albumin and HGF/SF or NK1 proteins at the concentrations specified in the legend to Figure 5. After 16 h the cells were transferred on ice, washed with PBS and incubated in ice-cold, 5% trichloroacetic acid (TCA) for 30 min. TCA-insoluble radioactivity was measured by scintillation after two washes with water and lysis in 0.2 M NaOH for 30 min at 37°C.
Crystallization
Purified NK1 was dialysed against 0.05 M Tris–HCl, 0.1 M NaCl pH 8.5 and concentrated to ∼20–25 mg/ml (Ultrafree centrifugal concentrators, Millipore). For complex formation, NK1 was added slowly to a tetradecamer heparin solution in 0.05 M Tris–HCl, 0.1 M NaCl pH 8.5 to a final protein concentration of 15 mg/ml and a molar NK1:heparin ratio of 1:2. Typically some precipitate appeared at this stage that redissolved after incubation at room temperature for 16 h. The complex was then centrifuged for 10 min at 14 000 r.p.m. on a benchtop centrifuge and the supernatant transferred into a new tube. Crystallization was carried out in linbro plates (Molecular Dimensions Limited) using the vapour diffusion hanging drop technique and a complex:precipitant ratio of 1:1 in the hanging drop. Structure Screen 1 and Structure Screen 2 (Molecular Dimensions Limited) were used for initial trials. Precipitant solution of condition 19 from Structure Screen 2 (10% PEG 8000, 0.1 M Na–HEPES pH 7.5, 8% ethylene glycol) produced small crystals of low quality and thus precipitant formulations were optimized. Large dodecahedral-shaped crystals (crystal type A) with maximal dimensions of 0.7 × 0.3 × 0.3 mm were obtained using 10% PEG 8000 (Fluka), 8% ethylene glycol (Aldrich), 0.1 M Na–HEPES pH 7.5 (Fluka) and 0.55 M sodium dihydrogen phosphate (BDH) as precipitant solution. Needles of maximal 0.8 × 0.15 × 0.15 mm size (crystal type B) were obtained in 10% PEG 8000, 8% ethylene glycol, 0.1 M Na–HEPES pH 7.5 and 0.1 M sodium p-toluene sulfonate (Sigma).
Data collection and processing
Type A crystals were transferred into a new drop containing 15% PEG 8000, 8% ethylene glycol, 0.1 M HEPES pH 7.5 and 0.55 M BDH, and the drop was then replaced stepwise with a cryoprotectant solution that contained a higher ethylene glycol concentration (final concentrations: 15% PEG 8000, 0.1 M HEPES pH 7.5, 0.55 M sodium dihydrogen phosphate and 28% ethylene glycol). Crystals were then flash-frozen in liquid nitrogen. Type B crystals were transferred into a drop containing 15% PEG 8000, 0.1 M HEPES pH 7.5, 0.1 M sodium p-toluene sulfonate and 25% ethylene glycol as cryoprotectant, soaked for 30 s and then flash-frozen. Data were collected at the SRS (Daresbury Laboratories) beamline PX 14.1, using a Quantum 4R CCD detector (ADSC) with an active area diameter of 180 mm. During data collection, crystals were cooled to 100K using a Cryostream device (Oxford Instruments). From crystal type A, 180° of data were recorded from a single crystal in 180 frames of 1° oscillation with an exposure time of 10 s and a beam wavelength of 1.488 Å. The crystal diffracted to a maximum resolution of 2.3 Å. The space group was determined to be P43212 with unit cell parameters of a = b = 86.56, c = 117.75 and all angles 90°. Two NK1 and two heparin molecules per asymmetric unit yielded a Matthews’ coefficient of 2.21 Å3/Da and a water content of 44% (Matthews, 1968). Reflections to a resolution of 2.3 Å were indexed, integrated and scaled using DENZO and SCALEPACK (Otwinowski, 1996). Data from crystal type B were collected at a wavelength of 0.979 Å in 200 frames of 1.5° and 90 s exposure from a single crystal. The crystal had P21212 space group with cell parameters: a = 59.91, b = 174.15, c = 179.74 and all angles 90°. The water content was calculated to be 49% with a Matthew’s coefficient of 2.43 Å3/Da assuming that eight NK1 and six heparin molecules are present in the asymmetric unit. The diffraction data were processed including reflections to a maximal resolution of 3.0 Å.
Molecular replacement
All molecular replacement calculations were performed with AMoRe (Navaza, 1994) from the CCP4 suite (Collaborative Computational Project, 1994). For both crystal types, the diffraction data between 8 and 3.5 Å were used in calculations of the rotation and translation functions. In the case of crystal type A, the molecular replacement search was carried out using two search probes: the N domain (residues 38–120) and the K domain (residues 121–209), the structures of which were taken from NK1 crystal structures reported by Ultsch et al. (1998) and Chirgadze et al. (1999). The rotation function calculation using the K domain probe showed two peaks at the top of the ‘possible solutions’ list with signal-to-noise ratios of 0.62 and 0.59σ, while the rotation function search using the N domain yielded two peaks at the top of the list with signal-to-noise ratios of 0.72 and 0.45σ. (Signal-to-noise ratio was calculated in the following way: from the peak height the highest background peak was subtracted and the result divided by sigma.) For each of the top 20 peaks of the K domain rotation function the translation function was calculated. The first rotation function peak produced the highest translation function peak with a correlation coefficient of 25.9 and Rcryst of 51.9%. This solution was identified as the correct one and was included in the translation function calculation for the rest of the rotation function peaks. The rotation function peak that yielded best Rcryst and correlation coefficient values was then identified as the correct one and was included in the subsequent translation function calculation (Table I). As a result, the positions of two K domains and one N domain were identified, yielding the overall correlation coefficient of 45.4 and Rcryst of 45.4% (resolution range 8–3.5 Å). The K domain solutions corresponded to peaks 1 and 7, the solution for the N domain to peak 3 in the list of the rotation function peaks. No solution for a second N domain could be found at this stage. The molecular replacement search probe for the crystal type B included the whole NK1 dimer from the crystal structures 1bht and 1nk1. The search for the rotation and translation function solutions was performed in a similar way to that described for crystal type A. In total, four solutions were identified. The NK1 dimers were placed in the crystal cell according to the rotational and translational parameters obtained, and yielded overall values for correlation coefficient and Rcryst of 48.4 and 43.7%, respectively.
Refinement and rebuilding
Initial rigid-body refinement was carried out in AMoRe (Navaza, 1994), further refinement and electron density map calculations were performed with CNS (Brunger et al., 1998). After the rigid-body refinement, a set of reflections (test dataset, ∼5% of the total number of reflections in the resolution range used for refinement) from the X-ray diffraction data was selected for free R-factor calculation for both crystal types. The initial model for the structure from crystal type A was subjected to 10 rounds of refinement and manual rebuilding (including data for a resolution range of 21–2.3 Å), whereas the model for the structure from crystal type B had nine rounds (including data for a resolution range of 20–3.0 Å). Each refinement procedure of a round included the conventional positional refinement using the conjugate gradient minimization of maximum-likelihood target. Typically, 60 cycles of positional refinement were performed in each round. This was normally followed by the restrained isotropic B-factor refinement (30 cycles), except that in the initial round of refinement average B-factors for each domain were refined first followed by rigid-body refinement of the domains. Manual rebuilding was performed in program O (Jones et al., 1991) using 2Fo – Fc and Fo – Fc maps as guidelines. After each refinement procedure, the structures were subjected to various validation checks using the programs WHAT_CHECK PROCHECK (Laskowski et al., 1993) and OOPS (Kleywegt and Jones, 1996). The whole polypeptide chain was visually inspected and manually adjusted to fit the corresponding electron density maps. The stretches of residues that were violating various validation criteria (stereochemical, environmental, high temperature factors, poor electron density fit) were checked with particular attention. The second N domain of crystal type A structure was built into the structure after the second round of refinement by applying the non-crystallographic symmetry operation on to the existing N domain. The operator was calculated from the two K domains in the structure. Heparin monosaccharide units and HEPES were added in later refinement– rebuilding rounds, with water molecules added at the last two to three rounds. Addition of water molecules was stopped when the decrease of Rfree factor stopped. Each of the NK1 molecules in both structures has one amino acid in cis conformation, P157. A number of lysine and arginine residues that are located at the surface of molecules and had very weak or no electron density for side chain atoms were modelled as alanines. In addition, the following regions of the protein in the crystal type A structure had weak or no density and are not present in the final structure: chain A, residues 28–37 and 208–210; chain B, residues 28–41, 52–83, 88–97, 105–117 and 210. All residues included in the final models have good stereochemistry with no residues in disallowed regions of the Ramachandran plot.
Coordinates
The coordinates of crystal structures A and B have been deposited in the Protein Data Bank under ID codes 1gmn and 1gmo, respectively.
Acknowledgments
Acknowledgements
D.L. is grateful to L.Sheridan for advice on early data analysis. D.L. and E.G. also thank M.Bocci for mutant HGF/SF plasmids and other members of the MRC Growth Factors Group for discussions. Work in E.G’s laboratory is supported by the UK Medical Research Council (MRC) and a grant from the EU. Work in T.L.B’s laboratory is supported by the Wellcome Trust and the BBSRC. E.G. also has a part-time teaching appointment at the University of Pavia (Italy).
References
- Atkins E.D.T. and Nieduszynski,I.A. (1975) Crystalline structure of heparin. Adv. Exp. Med. Biol., 52, 19–37. [DOI] [PubMed] [Google Scholar]
- Bennett M.J., Schlunegger,M.P. and Eisenberg,D. (1995) 3D domain swapping: a mechanism for oligomer assembly. Protein Sci., 4, 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladt F., Riethmacher,D., Isenmann,S., Aguzzi,A. and Birchmeier,C. (1995) Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature, 376, 768–771. [DOI] [PubMed] [Google Scholar]
- Bottaro D.P., Rubin,J.S., Faletto,D.L., Chan,A.M., Kmiecik,T.E., Vande Woude,G.F. and Aaronson,S.A. (1991) Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science, 251, 802–804. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. et al. (1998) Crystallography and NMR system (CNS): a new determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bullock S.L., Fletcher,J.M., Beddington,R.S. and Wilson,V.A. (1998) Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev., 12, 1894–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton A., Hacker,A., Naeem,A., Livet,J., Maina,F., Bladt,F., Klein,R., Birchmeier,C. and Guthrie,S. (2000) The branchial arches and HGF are growth-promoting and chemoattractant for cranial motor axons. Development, 127, 1751–1766. [DOI] [PubMed] [Google Scholar]
- Chan A.M., Rubin,J.S., Bottaro,D.P., Hirschfield,D.W., Chedid,M. and Aaronson,S.A. (1991) Identification of a competitive HGF antagonist encoded by an alternative transcript. Science, 254, 1382–1385. [DOI] [PubMed] [Google Scholar]
- Chirgadze D.Y., Hepple,J.P., Zhou,H., Byrd,R.A., Blundell,T.L. and Gherardi,E. (1999) Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nature Struct. Biol., 6, 72–79. [DOI] [PubMed] [Google Scholar]
- Cioce V., Csaky,K.G., Chan,A.M.L., Bottaro,D.P., Taylor,W.G., Jensen,R., Aaronson,S.A. and Rubin,J.S. (1996) Hepatocyte growth factor (HGF)/NK1 is a naturally occurring HGF/scatter factor variant with partial agonist/antagonist activity. J. Biol. Chem., 271, 13110–13115. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- DiGabriele A.D., Lax,I., Chen,D.I., Svahn,C.M., Jaye,M., Schlessinger,J. and Hendrickson,W.A. (1998) Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature, 393, 812–817. [DOI] [PubMed] [Google Scholar]
- Donate L.E., Gherardi,E., Srinivasan,N., Sowdhamini,R., Aparicio,S. and Blundell,T.L. (1994) Molecular evolution and domain structure of plasminogen-related growth factors (HGF/SF and HGF1/MSP). Protein Sci., 3, 2378–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebens A., Brose,K., Leonardo,E.D., Hanson,M.G.,Jr, Bladt,F., Birchmeier,C., Barres,B.A. and Tessier-Lavigne,M. (1996) Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron, 17, 1157–1172. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 132–134, 112–113. [DOI] [PubMed] [Google Scholar]
- Faham S., Hileman,R.E., Fromm,J.R., Linhardt,R.J. and Rees,D.C. (1996) Heparin structure and interactions with basic fibroblast growth factor. Science, 271, 1116–1120. [DOI] [PubMed] [Google Scholar]
- Faletto D.L., Tsarfaty,I., Kmiecik,T.E., Gonzatti,M., Suzuki,T. and Vande Woude,G.F. (1992) Evidence for non-covalent clusters of the c-met proto-oncogene product. Oncogene, 7, 1149–1157. [PubMed] [Google Scholar]
- Gherardi E., Gray,J., Stoker,M., Perryman,M. and Furlong,R. (1989) Purification of scatter factor, a fibroblast-derived basic protein that modulates epithelial interactions and movement. Proc. Natl Acad. Sci. USA, 86, 5844–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi H., Suzuki,S., Saito,T., Tamura,T., Harada,T., Yoshida,K. and Kimata,K. (1992) Structure of a heparan sulphate oligosaccharide that binds to basic fibroblast growth factor. Biochem. J., 285, 805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann G., Naldini,L., Weidner,K.M., Sachs,M., Vigna,E., Comoglio,P.M. and Birchmeier,W. (1992) A functional domain in the heavy chain of scatter factor/hepatocyte growth factor binds the c-Met receptor and induces cell dissociation but not mitogenesis. Proc. Natl Acad. Sci. USA, 89, 11574–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann G. et al. (1998) Engineered mutants of HGF/SF with reduced binding to heparan sulphate proteoglycans, decreased clearance and enhanced activity in vivo [published erratum appears in Curr. Biol., 1998, 8, R739]. Curr. Biol., 8, 125–134. [DOI] [PubMed] [Google Scholar]
- Holliger P., Prospero,T. and Winter,G. (1993) ‘Diabodies’: small bivalent and bispecific antibody fragments. Proc. Natl Acad. Sci. USA, 90, 6444–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubczak J.L., LaRochelle,W.J. and Merlino,G. (1998) NK1, a natural splice variant of hepatocyte growth factor/scatter factor, is a partial agonist in vivo. Mol. Cell. Biol., 18, 1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers M., Rong,S. and Woude,G.F. (1996) Hepatocyte growth factor/scatter factor–Met signaling in tumorigenicity and invasion/metastasis. J. Mol. Med., 74, 505–513. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kleywegt G. and Jones,T. (1996) Efficient rebuilding of protein structures. Acta Crystallogr. D, 52, 829–832. [DOI] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R., MacArthur,M., Moss,D. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lokker N.A. and Godowski,P.J. (1993) Generation and characterization of a competitive antagonist of human hepatocyte growth factor, HGF/NK1. J. Biol. Chem., 268, 17145–17150. [PubMed] [Google Scholar]
- Lyon M., Deakin,J.A., Mizuno,K., Nakamura,T. and Gallagher,J.T. (1994) Interaction of hepatocyte growth factor with heparan sulfate. Elucidation of the major heparan sulfate structural determinants. J. Biol. Chem., 269, 11216–11223. [PubMed] [Google Scholar]
- Mars W.M., Zarnegar,R. and Michalopoulos,G.K. (1993) Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am. J. Pathol., 143, 949–958. [PMC free article] [PubMed] [Google Scholar]
- Matthews B. (1968) Solvent content of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Merritt E. and Murphy,M. (1994) RASTER 3D version 2.0, a program for photorealistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Miyazawa K. et al. (1989) Molecular cloning and sequence analysis of cDNA for human hepatocyte growth factor. Biochem. Biophys. Res. Commun., 163, 967–973. [DOI] [PubMed] [Google Scholar]
- Miyazawa K., Kitamura,A., Naka,D. and Kitamura,N. (1991) An alternatively processed mRNA generated from human hepatocyte growth factor gene. Eur. J. Biochem., 197, 15–22. [DOI] [PubMed] [Google Scholar]
- Miyazawa K., Shimomura,T., Kitamura,A., Kondo,J., Morimoto,Y. and Kitamura,N. (1993) Molecular cloning and sequence analysis of the cDNA for a human serine protease reponsible for activation of hepatocyte growth factor. Structural similarity of the protease precursor to blood coagulation factor XII. J. Biol. Chem., 268, 10024–10028. [PubMed] [Google Scholar]
- Mizuno K., Inoue,H., Hagiya,M., Shimizu,S., Nose,T., Shimohigashi,Y. and Nakamura,T. (1994) Hairpin loop and second kringle domain are essential sites for heparin binding and biological activity of hepatocyte growth factor. J. Biol. Chem., 269, 1131–1136. [PubMed] [Google Scholar]
- Moorby C.D., Stoker,M. and Gherardi,E. (1995) HGF/SF inhibits junctional communication. Exp. Cell Res., 219, 657–663. [DOI] [PubMed] [Google Scholar]
- Mulloy B. and Forster,M.J. (2000) Conformation and dynamics of heparin and heparan sulfate. Glycobiology, 10, 1147–1156. [DOI] [PubMed] [Google Scholar]
- Nakamura T., Nishizawa,T., Hagiya,M., Seki,T., Shimonishi,M., Sugimura,A., Tashiro,K. and Shimizu,S. (1989) Molecular cloning and expression of human hepatocyte growth factor. Nature, 342, 440–443. [DOI] [PubMed] [Google Scholar]
- Naldini L. et al. (1992) Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J., 11, 4825–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaza J. (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr. A, 50, 157–163. [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. (1996) Processing of X-ray diffraction data collected in the oscillation mode. Methods Enzymol., 276, 301–315. [DOI] [PubMed] [Google Scholar]
- Park P.W., Reizes,O. and Bernfield,M. (2000) Cell surface heparan sulfate proteoglycans: selective regulators of ligand–receptor encounters. J. Biol. Chem., 275, 29923–29926. [DOI] [PubMed] [Google Scholar]
- Pellegrini L., Burke,D.F., von Delft,F., Mulloy,B. and Blundell,T.L. (2000) Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature, 407, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Perisic O., Webb,P.A., Holliger,P., Winter,G. and Williams,R.L. (1994) Crystal structure of a diabody, a bivalent antibody fragment. Structure, 2, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Rapraeger A.C., Krufka,A. and Olwin,B.B. (1991) Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science, 252, 1705–1708. [DOI] [PubMed] [Google Scholar]
- Ringvall M., Ledin,J., Holmborn,K., van Kuppevelt,T., Ellin,F., Eriksson,I., Olofsson,A.M., Kjellen,L. and Forsberg,E. (2000) Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J. Biol. Chem., 275, 25926–25930. [DOI] [PubMed] [Google Scholar]
- Schiering N., Casale,E., Caccia,P., Giordano,P. and Battistini,C. (2000) Dimer formation through domain swapping in the crystal structure of the Grb2-SH2-Ac-pYVNV complex. Biochemistry, 39, 13376–13382. [DOI] [PubMed] [Google Scholar]
- Schlessinger J., Plotnikov,A.N., Ibrahimi,O.A., Eliseenkova,A.V., Yeh,B.K., Yayon,A., Linhardt,R.J. and Mohammadi,M. (2000) Crystal structure of a ternary FGF–FGFR–heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell, 6, 743–750. [DOI] [PubMed] [Google Scholar]
- Schmidt C., Bladt,F., Goedecke,S., Brinkmann,V., Zschiesche,W., Sharpe,M., Gherardi,E. and Birchmeier,C. (1995) Scatter factor/hepatocyte growth factor is essential for liver development. Nature, 373, 699–702. [DOI] [PubMed] [Google Scholar]
- Schmidt L. et al. (1997) Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genet., 16, 68–73. [DOI] [PubMed] [Google Scholar]
- Schwall R.H., Chang,L.Y., Godowski,P.J., Kahn,D.W., Hillan,K.J., Bauer,K.D. and Zioncheck,T.F. (1996) Heparin induces dimerization and confers proliferative activity onto the hepatocyte growth factor antagonists NK1 and NK2. J. Cell Biol., 133, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T., Miyazawa,K., Komiyama,Y., Hiraoka,H., Naka,D., Morimoto,Y. and Kitamura,N. (1995) Activation of hepatocyte growth factor by two homologous proteases, blood-coagulation factor XIIa and hepatocyte growth factor activator. Eur. J. Biochem., 229, 257–261. [DOI] [PubMed] [Google Scholar]
- Stoker M., Gherardi,E., Perryman,M. and Gray,J. (1987) Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature, 327, 239–242. [DOI] [PubMed] [Google Scholar]
- Turnbull J.E., Fernig,D.G., Ke,Y., Wilkinson,M.C. and Gallagher,J.T. (1992) Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J. Biol. Chem., 267, 10337–10341. [PubMed] [Google Scholar]
- Uehara Y., Minowa,O., Mori,C., Shiota,K., Kuno,J., Noda,T. and Kitamura,N. (1995) Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature, 373, 702–705. [DOI] [PubMed] [Google Scholar]
- Ultsch M., Lokker,N.A., Godowski,P.J. and de Vos,A.M. (1998) Crystal structure of the NK1 fragment of human hepatocyte growth factor at 2.0 Å resolution. Structure, 6, 1383–1393. [DOI] [PubMed] [Google Scholar]
- Yayon A., Klagsbrun,M., Esko,J.D., Leder,P. and Ornitz,D.M. (1991) Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell, 64, 841–848. [DOI] [PubMed] [Google Scholar]
- Yu J. et al. (2000) Frequency of TPR-MET rearrangement in patients with gastric carcinoma and in first-degree relatives. Cancer, 88, 1801–1806. [PubMed] [Google Scholar]
- Zhang Z., Zhang,R., Joachimiak,A., Schlessinger,J. and Kong,X.P. (2000) Crystal structure of human stem cell factor: implication for stem cell factor receptor dimerization and activation. Proc. Natl Acad. Sci. USA, 97, 7732–7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Mazzulla,M.J., Kaufman,J.D., Stahl,S.J., Wingfield,P.T., Rubin,J.S., Bottaro,D.P. and Byrd,R.A. (1998) The solution structure of the N-terminal domain of hepatocyte growth factor reveals a potential heparin-binding site. Structure, 6, 109–116. [DOI] [PubMed] [Google Scholar]
- Zhou H., Casas-Finet,J.R., Heath Coats,R., Kaufman,J.D., Stahl,S.J., Wingfield,P.T., Rubin,J.S., Bottaro,D.P. and Byrd,R.A. (1999) Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry, 38, 14793–14802. [DOI] [PubMed] [Google Scholar]