

Abstract
We show that, dependent on serine hyperphosphorylation, protein tyrosine phosphatase α (PTPα) is activated by two different mechanisms during mitosis: its specific activity increases and its inhibitory binding to Grb2 decreases. The latter effect probably abates Grb2 inhibition of the phosphotyrosine displacement process that is required specifically for Src dephosphorylation and causes a mitotic increase in transient PTPα-Src binding. Thus, part of the increased protein tyrosine phosphatase activity may be specific for Src family members. These effects cease along with Src activation when cells exit mitosis. Src is not activated in mitosis in PTPα-knockout cells, indicating a unique mitotic role for this phosphatase. The activation of PTPα, combined with the effects of mitotic Cdc2-mediated phosphorylations of Src, quantitatively accounts for the mitotic activation of Src, indicating that PTPα is the membrane-bound, serine phosphorylation-activated, protein tyrosine phosphatase that activates Src during mitosis.
Keywords: cell cycle control/mitosis/protein tyrosine phosphatase α/signal transduction/Src–Grb2 association
Introduction
Protein tyrosine phosphatase (PTP) α (PTPα) is an ∼130 kDa transmembrane phosphatase expressed in a variety of cell types (Kaplan et al., 1990; Krueger et al., 1990). Its cytoplasmic region contains two catalytic domains, D1 and D2, although only the former appears to be active on peptide substrates (Wang and Pallen, 1991; den Hertog et al., 1993; Wu et al., 1997; Harder et al., 1998). While analogous in primary structure to some receptor PTPs, no ligand for the extracellular domain has been found (for reviews see Neel and Tonks, 1997; Schaapveld et al., 1997). However, the extracellular domain associates in cis with contactin, an extracellular membrane-anchored neuronal receptor, suggesting that the two proteins together may function as a receptor complex (Zeng et al., 1999). PTPα activity can also be stimulated by protein kinase C (PKC)-mediated serine phosphorylation (den Hertog et al., 1995; Tracy et al., 1995), and may be negatively regulated by dimerization (Bilwes et al., 1996; Majeti et al., 1998; Jiang et al., 1999, 2000; Tertoolen et al., 2001) or intermolecular associations with other PTPs (Blanchetot and den Hertog, 2000).
PTPα is a physiological regulator of the Src proto-oncoprotein and other Src-family members (Zheng et al., 1992; den Hertog et al., 1993; Harder et al., 1998; Arnott et al., 1999; Jiang et al., 1999; Ponniah et al., 1999; Su et al., 1999; Zheng et al., 2000; Petrone and Sap, 2000). Src is a membrane-localized proto-oncoprotein whose protein tyrosine kinase activity is negatively regulated by intramolecular association between its SH2 domain and phosphoTyr (pTyr) 527, a residue near its C-terminus (for reviews see Brown and Cooper, 1996; Thomas and Brugge, 1997). (This residue is at position 527 in chicken Src and in 529 in mammalian Src; we refer to it generically as Tyr527.) This association protects pTyr527 from dephosphorylation and renders it resistant to many phosphatases. Yet PTPα, like only a few other PTPs, can dephosphorylate pTyr527 and thus activate Src. The fact that overexpression of PTPα results in Src activation, neoplastic transformation and a concomitant increase in total cell protein tyrosine phosphorylation implies that PTPα activity must be restricted in vivo to Src family members and a limited number of other proteins (Zheng et al., 1992; den Hertog et al., 1993).
PTPα itself can be phosphorylated at Tyr789, which is close to its C-terminus (den Hertog et al., 1994; Su et al., 1994). This phosphorylated residue participates in a phosphotyrosine displacement mechanism that is required for PTPα-mediated dephosphorylation of pTyr527 in Src: pTyr789 displaces pTyr527 from the Src SH2 domain, thus deprotecting it while transiently binding PTPα to Src to facilitate dephosphorylation (Zheng et al., 2000). Dephosphorylation of Tyr789 or Tyr789→Phe site- directed mutagenesis abrogates PTPα’s ability to selectively dephosphorylate pTyr527 and to transform cells without diminishing PTPα’s ability to dephosphorylate pTyr416 in Src (an unprotected autophosphorylation site) or phosphotyrosines in other proteins. That is, phosphorylation of Tyr789 regulates PTPα substrate specificity but not its intrinsic enzymatic activity (Zheng et al., 2000). Src can phosphorylate Tyr789 in cotransfected cells that overexpress both proteins (den Hertog et al., 1994; X-M.Zheng and D.Shalloway, unpublished results), suggesting that these two proteins may constitute a positive feedback loop in vivo.
About 20% of the PTPα in NIH 3T3 cells is phosphorylated at Tyr789, and most of this is bound by Grb2 (den Hertog et al., 1994). Grb2 binding involves the association of the Grb2 SH2 domain with pTyr789 and the carboxyl Grb2 SH3 domain with other regions of PTPα (den Hertog et al., 1994; den Hertog and Hunter, 1996; Su et al., 1994, 1996). Binding of the Grb2 SH2 domain to pTyr789 blocks the phosphotyrosine displacement mechanism and the transient binding of PTPα to Src, and thus prevents PTPα from dephosphorylating pTyr527. Therefore, Grb2 can act as a negative regulator of PTPα activation of Src (Zheng et al., 2000).
Src (and other Src-family members) are activated during, and possibly shortly before, mitosis by a dual mechanism that involves both their phosphorylation by Cdc2 and the mitotic activation of an unknown PTP (for reviews see Taylor and Shalloway, 1993, 1996). The Cdc2-mediated phosphorylations of serine and threonine residues in the N-proximal region of Src weaken the intramolecular Src SH2-pTyr527 association, thereby rendering pTyr527 more susceptible to dephosphorylation (Bagrodia et al., 1991, 1994; Kaech et al., 1991; Stover et al., 1994). However, a Src mutant that lacks the Cdc2 phosphorylation sites is still partially activated during mitosis (Shenoy et al., 1992). This suggests that there is either an increase in pTyr527-directed phosphatase activity and/or a decrease in Tyr527-directed kinase activity during mitosis. The former is likely to be involved since orthovanadate, a PTP inhibitor, blocks the mitotic activation (Bagrodia et al., 1993). Membrane-localization is required for mitosis-specific pTyr527 dephosphorylation and Src activation (Bagrodia et al., 1993; Kaech et al., 1993), suggesting that these phenomena result from the mitotic activation of a membrane-localized PTP.
In studying this hypothesis, we have discovered that the ability of PTPα to dephosphorylate Src is increased during mitosis and that this involves two separate mechanisms: increased specific activity and decreased inhibitory Grb2 binding with a concomitant increase in PTPα-Src binding. These changes correlate with and require hyperphosphorylation of PTPα at serine. Both effects enhance dephosphorylation of pTyr527, supporting the hypothesis that PTPα activation is a proximal cause of the mitotic activation of Src. Moreover, we found that Src kinase activity is not increased during mitosis in PTPα–/– cells, indicating that PTPα is required for Src mitotic activation.
Results
We have previously characterized genetically-modified NIH 3T3 cell lines that inducibly overexpress (under repressive control of doxycycline) wild-type (wt) human PTPα and PTPα having a Tyr789→Phe mutation, PTPα(Y789F), as well as the same proteins with a nine-residue hemagglutinin (HA) tag at their C-termini, PTPα-HA and PTPα(Y789F)-HA (Zheng et al., 2000). A cell line that had been transfected with an empty vector system and G418-coselected in the same manner as the overexpressor cell lines was used as a control (for analyzing endogenous PTPα). We found that the localization and catalytic activity of overexpressed PTPα was similar to that of endogenous PTPα in unsynchronized cells. The Tyr789 →Phe mutation eliminated both Grb2 binding and the ability of PTPα to dephosphorylate pTyr527 in Src, without reducing its ability to dephosphorylate other substrates.
When fully induced, the overexpressor cells express ∼15× the endogenous level of PTPα. At that level, the overexpressed wt PTPα is phosphorylated at Tyr789 only to ∼40% the extent of endogenous PTPα, presumably because the endogenous Tyr789 kinase is saturated (Zheng et al., 2000). For the experiments reported here, transgene PTPα was only induced to ∼5–9 × endogenous levels (Table I), so that equal levels of overexpression could be obtained in both unsynchronized and mitotic cells (see Materials and methods). At these levels, the relative extent of Tyr789 phosphorylation was similar in both endogenous and overexpressed PTPα (compare lanes 1, 3 and 5 of Figure 1B and C).
Table I. Src specific kinase activity in unsynchronized and mitotic PTPα expressor and knockout cell lines.
| PTPα | Cell line | PTPα expression levela | Relative Src kinase specific activityb |
|
|---|---|---|---|---|
| Unsynchronized | Mitotic | |||
| Endogenous | NIH(pTet-Splice/cos)1 | 1 | 1 | 2.3 ± 0.1 |
| Overexpressed PTPα | NIH(pNTPTPα/cos)1 | 5.8 ± 0.4 | 1.8 ± 0.1 | 3.3 ± 0.1 |
| Overexpressed PTPα-HA | NIH(pTPTPα/cos)1 | 8.6 ± 0.5 | 4.2 ± 0.2 | 7.5 ± 0.3 |
| Overexpressed PTPα(Y789F)-HA | NIH(pTPTPα/Y789F/cos)1 | 8.0 ± 0.5 | 1.2 ± 0.1 | 2.3 ± 0.1 |
| PTPα+/+ | 3αP9 | 1.05 ± 0.05 | 1.1 ± 0.1 | 1.9 ± 0.1 |
| PTPα–/– | 3BP2 | <0.1c | 0.6 ± 0.1 | 0.7 ± 0.1 |
aFor the overexpressor lines, the expression levels under the induced experimental conditions are listed. Values are averages (± SEM) of at least two experiments normalized to the level of endogenous PTPα.
bSrc specific activity for phosphorylation of enolase was measured in experiments like those shown in Figures 3 and 9. Values are averages (± SEM) of two experiments normalized to the specific activity of Src in control NIH 3T3 cells that had been transfected with an empty vector.
cNo expression was detectable.

Fig. 1. Phosphatase activity of PTPα from unsynchronized and mitotic cells. Endogenous mouse PTPα (Endog) and overexpressed human PTPα (WT), PTPα-HA (WT-HA), PTPα(Y789F) (Y789F) and PTPα(Y789F)-HA (Y789F-HA) were immunoprecipitated with anti-PTPα(D2) antibody from lysates from non-overexpressor or overexpressor NIH 3T3-derived cells that were either unsynchronized (U) or arrested in mitosis (M). (The lysates were adjusted to contain approximately equal amounts of PTPα.) Aliquots of immunoprecipitates were incubated with [32P]pTyr-containing MBP in phosphatase buffer for 15 min at 30°C or subjected to anti-pTyr or-PTPα immunoblots. (A) Amount of 32P released per molecule of PTPα after 15 min incubation, normalized to the amount released by overexpressed PTPα from unsynchronized cells. Error bars indicate the SEM. (B) Anti-pTyr immunoblot of the immunoprecipitated PTPα. (C) Anti-PTPα immunoblot of the immunoprecipitated PTPα. SDS–PAGE was in 10% gels. The positions of molecular weight standards are indicated in kDa.
Increased phosphatase activity of PTPα during mitosis
Endogenous mouse PTPα, overexpressed human PTPα and overexpressed human PTPα-HA were immunoprecipitated with anti-PTPα antibody from the NIH 3T3-derived cell lines, either unsynchronized or arrested in mitosis by 10 h treatment with nocodazole. Comparing anti-PTPα and anti-pTyr immunoblots of aliquots of the immunoprecipitates showed that there was either no change or a very small increase (10 ± 10% for three experiments) in the level of tyrosine phosphorylation of PTPα during mitosis (Figure 1B and C). No phosphotyrosine was detected in the Tyr789→Phe mutants in either unsynchronized or mitotic cells, indicating that there is no other significant tyrosine phosphorylation site in PTPα even during mitosis. A mitosis-specific reduction in the electrophoretic mobility of a fraction of all the PTPα variants was observed (Figure 1B and C), suggesting that PTPα may be hyperphosphorylated at serine and/or threonine during mitosis.
Aliquots of the immunoprecipitates were incubated with the dephosphorylation substrate myelin basic protein (MBP), which had previously been Tyr phosphorylated by v-Src using [γ-32P]ATP. Measuring the amounts of [32P]phosphate released per molecule of PTPα showed that the catalytic activity of PTPα approximately doubled during mitosis (data not shown). The same increase occurred with the Tyr789→Phe mutants, indicating that Tyr789 phosphorylation was not required for this increase in specific activity. There were no significant differences between the activities of endogenous, overexpressed PTPα and overexpressed PTPα(Y789F), and no differences due to the addition of the HA tag. To exclude the possibility that these changes resulted from changes in the association of another protein, the same experiments were performed with immunoprecipitates that were subjected to a high-salt wash that removed associated Grb2 and, presumably, any other non-covalently associated proteins. The same results were obtained (Figure 1A), indicating that the mitotic increase in specific activity did not result from a mitotic change in the binding of other proteins.
Phosphatase experiments were also performed using pTyr527 in wt chicken Src as substrate. Since the Src target was immobilized as an immunoprecipitate, these experiments required the use of immunopurified PTPα. PTPα-HA and PTP(Y789F)-HA were immunopurified with anti-HA antibody from unsynchronized or nocodazole-arrested mitotic overexpressor cells and equal amounts were incubated in phosphatase buffer with chicken Src that had been immunoprecipitated from unsynchronized NIH 3T3-derived Src overexpressor cells (Figure 2). [The only detectable tyrosine phosphorylation in Src in these cells is at Tyr527 (Kmiecik and Shalloway, 1987), so anti-pTyr immunoblots can be used to assay the extent of Tyr527 phosphorylation.] Mock-immunopurified protein from cells that lacked HA-tagged PTPα was used as a negative control (Figure 2, lanes 1 and 2). Immunoblotting confirmed that the immunopurification procedure stripped co-associated Grb2 from the PTPα (data not shown).
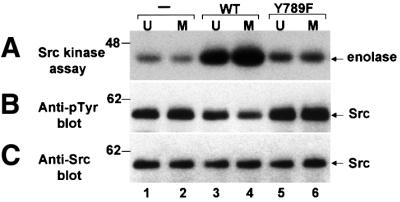
Fig. 2. Tyrosine-dephosphorylation and activation of Src in vitro by PTPα from unsynchronized and mitotic cells. Src that had been immunoprecipitated from chicken-Src overexpressing NIH 3T3-derived cells was incubated in phosphatase buffer with PTPα-HA (lanes 3 and 4) and PTPα(Y789F)-HA (lanes 5 and 6) that had been immuno purified from unsynchronized or mitotic PTPα-overexpressor cells using anti-HA antibody or with mock-immunopurified protein from control cells that did not express any HA-tagged protein (–, lanes 1 and 2). The partially dephosphorylated Src immunoprecipitates were washed to remove PTPα and then incubated with enolase and [γ-32P]ATP in kinase buffer. (A) Autoradiograph of [32P]enolase after the Src kinase assay. (B) Anti-pTyr immunoblot of the Src immuno precipitates after PTPα treatment. (C) Anti-Src immunoblot of the immunoprecipitates. SDS–PAGE was in 10% gels. The positions of molecular weight standards are indicated in kDa.
As previously shown (Zheng et al., 2000), incubation with wt PTPα-HA immunopurified from unsynchronized cells significantly increased Src activity (∼3.4-fold; Figure 2A, lanes 1 and 3). We now discovered that PTPα from mitotic cells activated Src to a greater extent than did PTPα from unsynchronized cells (Figure 2A, lanes 3 and 4). The activation of Src was correlated with dephosphorylation of pTyr527: incubation of Src with PTPα from unsynchronized cells reduced phosphorylation of Tyr527 by ∼27% while incubation with the same amount of PTPα from mitotic cells reduced phosphorylation by ∼44% (Figure 2B, lanes 1–4). Adjusting for nonlinearities resulting from the fact that much of the pTyr527 substrate is dephosphorylated in the course of these assays (see Materials and methods), these results imply that the intrinsic specific activity of PTPα for dephosphorylation of pTyr527 is increased by 2.0 ± 0.2-fold (in three experiments) during mitosis. This increase, observed in PTPα that had been stripped of associated Grb2, is essentially the same as the increase observed using MBP as substrate.
PTPα(Y789F)-HA from either unsynchronized or mitotic cells was unable to dephosphorylate and activate Src (Figure 2, lanes 5 and 6). This shows that the mechanism responsible for mitotic activation of PTPα does not abrogate the requirement that Tyr789 be phosphorylated if pTyr527 is to be dephosphorylated (Zheng et al., 2000).
Dephosphorylation and activation of Src in vivo by PTPα in unsynchronized and mitotic cells
We studied the effect of PTPα overexpression on Src activity in vivo during interphase and mitosis by measuring the effect of PTPα overexpression on the ability of immunoprecipitated Src to phosphorylate enolase (Figure 3; Table I). As expected from the in vitro results, overexpression of PTPα(Y789F) did not affect the tyrosine phosphorylation or kinase activity of Src in either unsynchronized or mitotic cells (Figure 2, compare lanes 1 and 2 with 5 and 6). As previously reported (Chackalaparampil and Shalloway, 1988; Shenoy et al., 1989), the electrophoretic mobility of about half the mitotic Src was retarded due to mitotic Cdc2-mediated serine/threonine phosphorylations (Figure 3C). Also in agreement with previous results (Zheng et al., 1992, 2000; den Hertog et al., 1993), overexpression of wt PTPα increased Src specific kinase activity 2- to 4-fold in unsynchronized cells, depending on expression level (Figure 3A, lanes 1 and 3; Table I). In agreement with other results (Chackalaparampil and Shalloway, 1988), the specific kinase activity of Src from control cells that did not overexpress PTPα was increased ∼2.3-fold during mitosis (Figure 3A, lanes 1 and 2). We found that these two enhancements combined to result in an even greater increase in Src activity and decrease in Src tyrosine phosphorylation in mitotic PTPα-overexpressor cells (Figure 3A and B, lane 4; Table I). pTyr527 is the only detectably phosphorylated tyrosine in mitotic Src; i.e. the autophosphorylation site Tyr416 is not phosphorylated even in this activated situation (Chackalaparampil and Shalloway, 1998; Bagrodia et al., 1991), possibly because of PTPase activity. Therefore, the observed total tyro sine dephosphorylation reflects dephosphorylation of this residue.
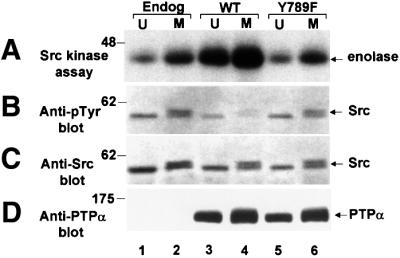
Fig. 3. Src kinase activity and Tyr527 phosphorylation in unsynchronized and mitotic normal and PTPα-overexpressor cells. Endogenous Src was immunoprecipitated from unsynchronized (U) or mitotic (M) non-overexpressor cells (lanes 1–2), or cells overexpressing wt PTPα-HA (lanes 3 and 4) or PTPα(Y789F)-HA (lanes 5 and 6). The immunoprecipitates were split into aliquots and used in the assays shown in A–D. (A) The in vitro kinase activity of the Src immunoprecipitate was measured by incubating it with enolase substrate and [γ-32P]ATP in phosphorylation buffer, separating the reaction products by electrophoresis and autoradiography. (B) Anti-phosphotyrosine immunoblot of the immunoprecipitate. (C) Anti-Src immunoblot of the immunoprecipitate. (D) Anti-PTPα immunoblot of cell lysates containing 10 µg total cell protein. SDS–PAGE was in 9% gels. The positions of molecular weight standards are indicated in kDa.
Reduced PTPα-Grb2 association during mitosis
We have previously shown that the ability of a population of PTPα molecules to dephosphorylate pTyr527 is governed by two factors: (i) its intrinsic specific activity, and (ii) the fraction of the population that contains pTyr789 that is not bound by Grb2. To see if the extent of PTPα-Grb2 binding changed during mitosis, we immunoblotted anti-Grb2 immunoprecipitates with anti-PTPα antibody to measure the amounts of associated protein. In contrast with the experiments described above, in which the immunoprecipitates from 1% Nonidet-40 (NP-40) lysates were washed with 500 mM NaCl to remove non-covalently associated proteins, no high-salt wash was used in these experiments (see Materials and methods). In agreement with den Hertog et al. (1994), about one-fifth of the endogenous PTPα co-immunoprecipitated with Grb2 (data not shown). This amount decreased ∼3.7-fold (two experiments) during mitosis (Figure 4, lanes 3 and 4), even though the total amount of Grb2 was similar in both unsynchronized and mitotic cell lysates (Figure 4, lanes 1 and 2) and in the anti-Grb2 immunoprecipitates (Figure 4, lanes 3 and 4). (The smaller reduction in PTPα electrophoretic mobility observed in these experiments results from the shorter electrophoresis times that were used to keep the small molecular weight Grb2 protein on the gels.)

Fig. 4. Decreased in vivo binding of Grb2 to PTPα during mitosis. The association of endogenous PTPα with Grb2 in unsynchronized and mitotic cells was measured by complementary co-immunoprecipitation experiments and by Grb2 SH2 domain affinity-precipitation experiments. Immunoprecipitates made with either anti-Grb2 (lanes 3 and 4) or -PTPα (lanes 5 and 6) antibody and lysates from unsynchronized (U) or mitotic (M) non-overexpressor cells were analyzed by 10% SDS–PAGE and immunoblotted with anti-PTPα or -Grb2 antibody as indicated. Lanes 7 and 8: PTPα was affinity-precipitated from lysates from unsynchronized or mitotic cells using GST–Grb2 SH2 domain fusion protein bound to Sepharose beads and immunoblotted with anti-PTPα antibody only. Direct immunoblots of the whole cell lysates used in lanes 5 and 6 (lanes 1 and 2, bottom panel) or lanes 7 and 8 (lanes 1 and 2, top panel) are also shown. To optimize detection, the different experiments used lysates containing differing amounts of total cell protein: lanes 1 and 2, top, 0.05 mg; lanes 1 and 2 bottom, 0.025 mg; lanes 3 and 4, 0.4 mg; lanes 5 and 6, 1.5 mg; lanes 7 and 8, 1 mg. The positions of molecular weight standards are indicated in kDa.
Reciprocal experiments, in which anti-PTPα immunoprecipitates were immunoblotted with anti-Grb2 antibody, showed that the amount of co-immunoprecipitated Grb2 decreased ∼2.9-fold in mitotic cells, with no change in the amount of immunoprecipitated PTPα (Figure 4, lanes 5 and 6). The mitotic decreases in PTPα-Grb2 association were not caused by decreases in the amount of PTPα or phosphorylation of Tyr789 since, as already indicated in Figure 1B and C and verified in this set of experiments, there was no significant mitotic change in either of these levels during mitosis. [Because of modest anti-PTPα antibody affinity, not all the PTPα was immunoprecipitated; so the fraction of total cellular Grb2 that was co-immunoprecipitated in these experiments (∼2%) underestimates the fraction that is co-associated in the cell.]
The mitotic reduction in PTPα-Grb2 binding in vivo could result from an intrinsic change in PTPα or Grb2, or from other causes such as sequestration of the proteins in different locations or interference by binding of a third protein. To test the possibilities that changes in cellular Grb2 or sequestration were involved, we measured the ability of a fusion protein containing glutathione S-transferase (GST) and the Grb2 SH2 domain to bind in vitro to endogenous PTPα in cell lysates from unsynchronized or mitotic cells (Figure 4, lanes 7 and 8). Only ∼6% of the total PTPα was affinity-precipitated from unsynchronized cell lysates, possibly due to competition with endogenous Grb2. In contrast, <0.75% of the mitotic PTPα was affinity-precipitated from mitotic cell lysates (a >8-fold reduction). This excluded the possibility that changes in cellular Grb2 or intracellular sequestration were responsible for the decreased binding. It also showed that the mitotic reduction in PTPα-Grb2 affinity could be observed in the absence of the two Grb2 SH3 domains normally present in the full-length protein.
The phosphotyrosine displacement model (Zheng et al., 2000) predicts that reduced PTPα-Grb2 binding will increase Src pTyr527 dephosphorylation if the PTPα-Src SH2 binding is not similarly reduced. The (weaker) PTPα-Src SH2 domain binding is difficult to detect in non-overexpressor cells, so the mitotic changes in the binding of PTPα to both the Grb2 and Src SH2 domains were examined in PTPα-overexpressor cells: GST–Grb2 SH2 and GST–Src SH2 fusion proteins were used to affinity-precipitate PTPα-HA from overexpressor cell lysates (Figure 5A). About 4% of the overexpressed PTPα-HA was affinity-precipitated from unsynchronized lysates by GST–Grb2 SH2 domain fusion protein (Figure 5A, lanes 1 and 5). (Again, this amount is probably reduced because of competition from endogenous Grb2.) This amount was reduced 2.2 ± 0.3-fold (two experiments) when mitotic lysate was used (Figure 5A, lanes 5 and 6). Because of its weaker binding, only about 1% of the PTPα-HA was affinity-precipitated from unsynchronized cell lysates by GST–Src SH2 domain fusion protein (Figure 5A, lanes 1 and 7). However, in contrast with Grb2 SH2 domain binding, there was no decrease in this binding during mitosis (Figure 5A, lanes 7 and 8).

Fig. 5. In vitro binding of unsynchronized and mitotic PTPα to the Grb2 and Src SH2 domains. (A) Lysates from unsynchronized (U, odd lanes) or mitotic (M, even lanes) PTPα-HA overexpressor cells were affinity-precipitated by incubation with GST (lanes 3 and 4), a GST–Grb2 SH2 domain fusion protein (lanes 5 and 6) or a GST–Src SH2 domain fusion protein (lanes 7 and 8) bound to Sepharose beads. The washed beads were then analyzed by 9% SDS–PAGE and anti-PTPα immunoblotting. For comparison, lanes 1 and 2 (WCL) contained 0.033 times the amount of complete whole cell lysate. (B) PTPα-HA was immunopurified from unsynchronized or mitotic overexpressor cells and affinity-precipitated by GST or the GST–SH2 domain fusion proteins used above, analyzed by 9% SDS–PAGE and immunoblotted with anti-PTPα antibody (lanes 3–8). For comparison, lanes 1 and 2 (Total) contain 0.3 times the amount of immunopurified PTPα used in the affinity precipitations. The positions of molecular weight standards are indicated in kDa.
To test the possibility that interference by a third protein blocked PTPα binding to Grb2 during mitosis, we also compared the ability of the GST–Grb2 SH2 and GST–Src SH2 fusion proteins to affinity-precipitate PTPα-HA that had been immunopurified from unsynchronized and mitotic cells by a procedure that removed any associated Grb2 and, presumably, other non-covalently associated proteins (see Materials and methods). About 28% of the PTPα-HA immunopurified from unsynchronized cells was affinity-precipitated by the GST–Grb2 SH2 domain fusion protein (Figure 5B, lanes 1 and 5). This probably represents much of the PTPα-HA that was phosphorylated at Tyr789, since the concentration of Grb2 SH2 domain in the binding reactions (0.6 µM) was high compared with typical high-affinity SH2-pTyr binding constants of this type. The mitotic reduction in the binding of the Grb2 SH2 domain observed in these experiments (2.4 ± 0.1-fold; Figure 5B, lanes 5 and 6) was essentially the same as that observed in the direct affinity-precipitation experiments. This indicates that the reduction in Grb2 SH2-PTPα affinity does not result from effects of other associated proteins. Again, no mitotic change in the affinity of the Src SH2 for PTPα was observed.
We conclude that there is an ∼2.3-fold differential reduction in binding of PTPα to the Grb2 SH2 domain relative to binding to the Src SH2 domain during mitosis, and predict that this will result in reduced inhibition of Src-directed PTPα activity by Grb2 in vivo.
Increased PTPα-Src association during mitosis
The reduced inhibition of PTPα by Grb2 during mitosis is predicted to result in increased transient co-association between PTPα and Src. To test this prediction, anti-Src immune complexes from unsynchronized and mitotic PTPα overexpressor cells were made with monoclonal antibody mAb 2–17 (which binds to an epitope at the N-terminus of Src) and the immunoprecipitates were immunoblotted with anti-PTPα antibody (Figure 6A). This showed that the amount of PTPα co-associated with Src increased ∼2.5-fold in mitotic cells (Figure 6A, lanes 1 and 2 in panel a) even though there was no change in the amount of immunoprecipitated Src (panel b) or the total amount of PTPα in the cells (Figure 6A, panel c).
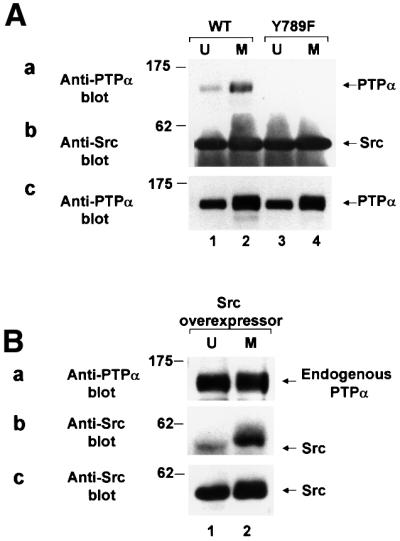
Fig. 6. Increased co-association in vivo of Src and PTPα during mitosis. (A) Immunoprecipitates made with anti-Src antibody from lysates (containing 1.5 mg total cell protein) from unsynchronized (U, odd lanes) or mitotic (M, even lanes) cells overexpressing wt PTPα-HA (lanes 1–2) or PTPα(Y789F)-HA (lanes 3–4) were analyzed by 9% SDS–PAGE and immunoblotted with anti-PTPα or -Src antibody. The positions of molecular weight standards are indicated in kDa. (a) Anti-PTPα immunoblot of anti-Src immunoprecipitate. (b) Anti-Src immunoblot of anti-Src immunoprecipitate. (c) Anti-PTPα immunoblot of cell lysates containing 10 µg total cell protein. (B) Immunoprecipitates made with anti-PTPα antibody from lysates (containing 1.5 mg total cell protein) from unsynchronized or mitotic Src-overexpressor cells were analyzed by 10% SDS–PAGE and immunoblotted with anti-Src or -PTPα antibody. The positions of molecular weight standards are indicated in kDa. (a) Anti-PTPα immunoblot of anti-PTPα immunoprecipitate. (b) Anti-Src immunoblot of anti-PTPα immunoprecipitate. (c) Anti-Src immunoblot of cell lysates containing 5 µg total cell protein.
The converse experiment, in which anti-PTPα immunoprecipitates from unsynchronized and mitotic Src overexpressor cells were immunoblotted with anti-Src antibody was also performed (Figure 6B). In agreement, this showed that the amount of Src co-associated with PTPα increase ∼2.6-fold in mitotic cells (Figure 6B, lanes 1 and 2 in panel a) with no change in the amount of immunoprecipitated PTPα (Figure 6B, panel b) or the total amount of Src in the cells (Figure 6B, panel c).
Cells overexpressing either PTPα or Src were used in these experiments because, as previously reported (Zheng et al., 2000), we are not able to detect co-immunoprecipitation of endogenous PTPα with endogenous Src—probably because of the low signal-to-noise ratio.
PTPα activity and Grb2 binding return to interphase levels near the time of cell division
To determine the duration of the mitotic changes in PTPα activity and Grb2 binding, nocodazole-arrested mitotic non-overexpressor cells were collected and then incubated in medium without nocodazole for varying times (i.e. mitotic release). PTPα phosphatase activity and Grb2 binding were then measured using the methods of Figures 1 and 5A. As seen in Figure 7A, PTPα activity decreased halfway to its interphase level by about 40 min after release; the decrease was essentially complete by 80 min after release. PTPα-Grb2 binding and electrophoretic mobility returned to their interphase levels with about the same kinetics (Figure 7A–C). This was slightly slower than the temporal progression of the cells to cytokinesis, which occurred roughly 30–40 min after release (data not shown). Similar results were obtained with overexpressed PTPα-HA (data not shown).
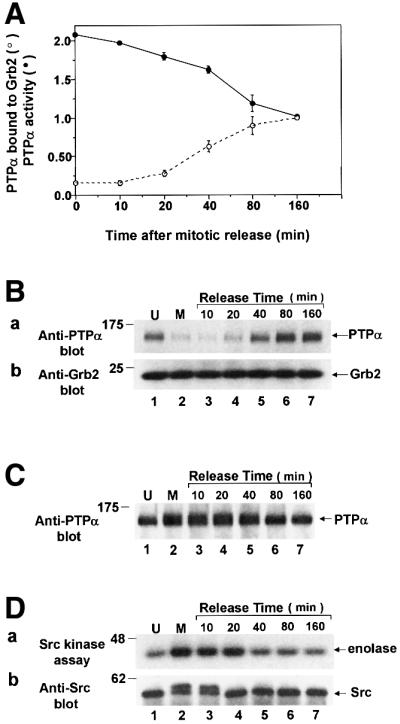
Fig. 7. PTPα tyrosine phosphatase activity and Grb2 binding following release from mitotic arrest. Mitotic non-overexpressor cells were collected after arrest with nocodazole and were then incubated for the indicated amounts of time in normal medium without nocodazole. Cell lysates were then prepared and analyzed for PTPα activity, binding to Grb2 in vivo, electrophoretic mobility and Src activity. (A) The ability of immunoprecipitated endogenous PTPα to dephosphorylate [32P]pTyr-containing MBP was measured as described in Figure 1. The amount of 32P released per molecule of PTPα normalized to the amount released by overexpressed PTPα after 160 min is shown (filled circles). The binding of PTPα to Grb2 was measured in experiments like the one shown in Figure 7B. The amount of PTPα bound normalized to the amount bound after 160 min release is shown (open circles). Error bars indicate the SEM (two experiments). (B) Grb2 was immunoprecipitated from the cell lysates and the amount of co-immunoprecipitated endogenous PTPα was revealed by anti-PTPα immunoblotting (panel a). Immunoblotting of the lower portion of the blot with anti-Grb2 antibody showed that there was no change in the level of Grb2 throughout the release period (panel b). Experimental procedures were as in Figure 4. (C) Anti-PTPα immunoblot of immunoprecipitated endogenous PTPα. (D) Endogenous Src was immunoprecipitated from the cell lysates and its ability to phosphorylate enolase was measured as described in Figure 3A. Aliquots of each immunoprecipitate were immunoblotted with anti-Src antibody (panel b). The positions of molecular weight standards are indicated in kDa.
Measurement of the Src activity time-course, using aliquots of the cell lysates that were used in Figure 7A–C, showed that Src activity returned to normal by 40 min after release (Figure 7D, panel a). Interestingly, the mitotic electrophoretic mobility retardation of Src disappeared about 20 min sooner (Figure 7D, panel b). This retardation results from Cdc2-mediated serine/threonine phosphorylations of Src (Shenoy et al., 1989), and the return to interphase mobility reflects their dephosphorylation. This suggests that the activation of Src persists for about 20 min after Cdc2 is deactivated (in anaphase) and that Src deactivation is roughly cotemporal with the slower decrease in PTPα activity and increase in Grb2 binding.
Mitotic activation of PTPα depends on mitotic serine hyperphosphorylation
The observed electrophoretic mobility retardation of PTPα suggests that it is hyperphosphorylated during mitosis and that this could be the cause of the altered PTPα specific activity and Grb2 binding. If so, this is probably at serine since: (i) phosphoamino acid analysis of immunoprecipitates of radiolabelled PTPα showed that only serine and tyrosine residues were phosphorylated, both during interphase and mitosis (data not shown); (ii) the anti-phosphotyrosine immunoblots indicate that there is no mitotic change in the overall level of tyrosine phosphorylation in PTPα (Figure 1); and (iii) the absence of detectable phosphotyrosine in PTPα(Y789F) even during mitosis (Figure 1B) strongly suggests that there are no new mitotic tyrosine phosphorylation sites in PTPα.
To test this hypothesis, PTPα immunoprecipitated from unsynchronized and mitotic cells was treated with the serine/threonine phosphatase PP2A and analyzed for PTP activity, electrophoretic mobility and Grb2 binding using the methods described above. PP2A treatment of mitotic PTPα restored to interphase levels PTPα electrophoretic mobility (Figure 8B), phosphatase activity (Figure 8A), and binding to the GST–Grb2 SH2 domain fusion protein (Figure 8E). PP2A treatment also eliminated the mitotic increase in PTPα’s Src-activating activity (Figure 8D, lanes 3 and 4). It also reduced the ability of PTPα from unsynchronized cells to dephosphorylate Src (Figure 8D, lanes 1 and 2).

Fig. 8. Effect of serine-dephosphorylation on phosphatase activity and Grb2 binding of PTPα from unsynchronized and mitotic cells. Wild-type PTPα-HA was immunoprecipitated with anti-HA antibody from unsynchronized (U) or mitotic (M) overexpressor cells, either treated (+) or not treated (–) with serine/threonine phosphatase PP2A, and then assayed. (A) The ability of the immunoprecipitates to dephosphorylate [32P]pTyr-containing MBP was assayed as in Figure 1A. Error bars indicate the SEM. (B) Anti-PTPα immunoblot in 10% SDS–PAGE of the immunoprecipitates. (C) Anti-pTyr immunoblot in 10% SDS–PAGE of the immunoprecipitates. The slightly decreased level of tyrosine phosphorylation in unsynchronized untreated cells was not routinely reproducible. (D) The treated and untreated PTPα was eluted and assayed for its ability to dephosphorylate and active immunoprecipitated overexpressed Src as in Figure 2. (E) The treated and untreated PTPα was eluted and affinity-precipitated by GST–Grb2 SH2 domain fusion protein, analyzed by 9% SDS–PAGE and immunoblotted with anti-PTPα antibody (lanes 5–8) as in Figure 5B. For comparison, lanes 1–4 (Total) each contain 40% of the amount of eluant which was affinity-precipitated. The positions of molecular weight standards are indicated in kDa.
Anti-phosphotyrosine immunoblots (Figure 8C) showed that, as expected, PP2A did not dephosphorylate phosphotyrosine. (The apparent increase in phosphotyrosine in this figure probably results from condensation of the gel band following dephosphorylation.) Moreover, control experiments showed that addition of okadaic acid (a specific inhibitor of PP2A serine/threonine phosphatase activity) to the reaction mixture completely blocked the PP2A reduction of mitotic PTPα specific activity (data not shown). This indicates that the mitotic electrophoretic mobility retardation results from hyperphosphorylation at serine(s) in PTPα and that this is required for the mitotic changes in PTPα activity and Grb2 binding. The indication that PP2A treatment caused a small reduction in the specific activity of PTPα from unsynchronized cells could result from mitotic cells within the population or if these serine(s) are also phosphorylated to a small extent during interphase.
Src is not activated during mitosis in PTPα–/– cells
To determine if PTPα is required for the mitotic activation of Src, we measured the kinase activity of Src from unsynchronized and mitotic PTPα-knockout mouse cells (Figure 9). In agreement with Su et al. (1999) and Ponniah et al. (1999), we found that Src had lower specific activity in unsynchronized PTPα–/– cells than in a control PTPα+/+ line derived by reintroducing PTPα back into the PTPα–/– line (Figure 9, lanes 1 and 3; Table I). Interestingly, there was very little, if any, mitotic increase in Src activity in the PTPα–/– cells (Figure 9, lanes 3 and 4), indicating that PTPα plays a unique role in mitotic activation of Src.
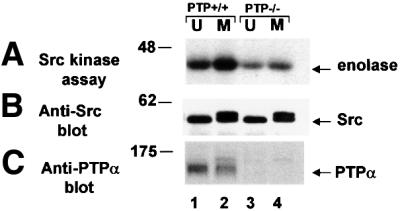
Fig. 9. Effect of PTPα knockout on Src kinase activity in unsynchronized and mitotic cells. Src was immunoprecipitated from unsynchronized (U) or mitotic (M) cells and (A) assayed for in vitro kinase activity by incubating it with enolase substrate and [γ-32P]ATP in phosphorylation buffer, separating the reaction products by 9% SDS–PAGE and autoradiography. (B) Anti-Src immunoblot of the immunoprecipitates. (C) Anti-PTPα immunoblot of cell lysates containing 50 µg total cell protein. Lanes: 1 and 2, mouse E3 PTPα-knockout cells into which PTPα gene had been reintroduced; lanes 3 and 4, E3 PTPα-knockout cells (Su et al., 1999). The positions of molecular weight markers are indicated in kDa.
Discussion
We discovered that two separate effects increase the ability of PTPα to dephosphorylate pTyr527 in Src during mitosis: (i) its intrinsic specific activity increases ∼2-fold, and (ii) there is a 2.9- to 3.7-fold reduction in the fraction of PTPα bound by Grb2, a specific inhibitor of PTPα dephosphorylation of pTyr527 in Src. The increase in intrinsic specific activity was also observed in PTPα(Y789F) (which does not bind Grb2) and in PTPα that was stripped of noncovalently-associated proteins, indicating that the binding of Grb2 or other proteins does not account for the altered intrinsic specific activity. Moreover, experiments with immunopurified PTPα and recombinant GST–Grb2 SH2 fusion proteins showed that the in vivo mitotic decrease in PTPα-Grb2 binding resulted from an intrinsic change in PTPα, not from changes in cellular Grb2, sequestration or the effects of other proteins. Both the changes in PTPα specific activity and Grb2 binding correlated with increased serine phosphorylation of PTPα during mitosis and were removed by treatment with the serine/threonine phosphatase PP2A.
It is important to note that these changes occur without reduced phosphorylation of Tyr789 in PTPα (Figure 1), which is required for dephosphorylation of pTyr527 in Src (Zheng et al., 2000). Moreover, there is no mitotic change in the affinity of PTPα for the Src SH2 domain (Figure 5). This binding, although of lower affinity than the PTPα-Grb2 SH2 domain binding, plays a key role in the displacement of Src pTyr527 from the Src SH2 domain prior to its dephosphorylation (Zheng et al., 2000). The net ∼2.3-fold decrease in the ratio between the affinities of pTyr789 for the Grb2 SH2 and Src SH2 domains suggests that the competitive inhibition of PTPα by Grb2 is reduced during mitosis. Findings by den Hertog et al. (1994) and our unpublished immunodepletion experiments indicate that essentially all of the PTPα that contains phosphorylated pTyr789 is bound by Grb2 during interphase. Thus, very little PTPα can act on Src during interphase and the mitotic reduction of Grb2 inhibition is predicted to be significant in vivo. Consistent with this, we found that the binding of Src and overexpressed PTPα increases ∼2.5-fold during mitosis (Figure 6).
The mitotic reduction in Grb2 inhibition is predicted to act in multiplicative combination with the increased PTPα specific activity to cause an even greater increase in total pTyr527-directed PTPα activity. Assuming that the mitotic increase in PTPα-Src binding provides a measure of the reduction in Grb2 inhibition, we estimate the combined increase in pTyr527-directed PTPα activity is ∼2.5 × 2 = 5-fold. We have not measured this composite increase directly in vitro because the procedure required for the pTyr527 dephosphorylation assay strips associated Grb2 from PTPα. On the other hand, quantitative analysis of the in vivo mitotic changes in Tyr527 phosphorylation and Src kinase activity is consistent with this hypothesis: a previous analysis that accounted for the strongly nonlinear relationship between the level of Tyr527 phosphorylation and the rate constants of Tyr527 phosphorylation (k+) and dephosphorylation (k–) showed that the ∼2.3-fold mitotic increase in wild-type Src kinase activity results from a ∼14-fold increase in k–/k+ (Shenoy et al. 1992). Since the mitotic activation of Src is blocked by orthovanadate, a PTP inhibitor, we believe that most of this increase is in k– (Bagrodia et al., 1993). This increase results from the combination of two effects: (i) decreased intramolecular association between the Src SH2 domain and pTyr527 (caused by Cdc2-mediated amino-proximal phosphorylations of Src) that deprotects pTyr527 and makes it ∼3.3-fold more accessible to PTPs (Bagrodia et al., 1994; Stover et al., 1994), and (ii) a ∼4.3-fold increase in pTyr527-directed phosphatase activity (Shenoy et al., 1992). This ∼4.3-fold increase is, within the accuracy of the experiments, consistent with the ∼5-fold increase in pTyr527-directed PTPα phosphatase activity predicted to result from the combination of increased specific activity and decreased Grb2 inhibition.
The hypothesis that PTPα is the mitotic activator of Src is consistent with previous studies (Bagrodia et al., 1993; Kaech et al., 1993), which have shown that the mitotic activation of Src is blocked by treatment with orthovanadate and in Src mutants that lack the N-terminal myristate which is required for its membrane localization. Combined with the observation (Chackalaparampil et al., 1994) that Tyr527 phosphorylation is decreased and Src is activated when cells are treated with okadaic acid, a specific inhibitor of PP2A, these studies have suggested that Src is regulated by a membrane-associated PTP whose activity can be increased by serine/threonine phosphorylation. PTPα satisfies all these criteria. In direct support of this hypothesis, we found that pTyr527 dephosphorylation and Src kinase activity were increased even more during mitosis in PTPα overexpressor cells (Figure 3). Moreover, since there is no mitotic activation of Src in PTPα-knockout cells (Figure 9), we conclude that PTPα is required for the mitotic activation of Src. PTPα also acts on the Tyr527 homologues found in the Src family members Fyn and Yes (Bhandari et al., 1998; Harder et al., 1998), and it is possible that it also participates in the mitotic activation of these Src-family members (Roche et al., 1995).
The fact that almost all mitotic activation of Src is eliminated by PTPα knockout is somewhat surprising since some evidence suggests that other PTPs also participate in regulating Src family members: whereas microinjecting a neutralizing antibody specific for Src, Fyn and Yes blocks progression of fibroblasts from G2 to M (Roche et al., 1995), almost complete depletion of PTPα by antisense methods does not block cell cycle progression (Arnott et al., 1999). Moreover, while transgenic mice that lack all three Src family members are not viable (Klinghoffer et al., 1999), PTPα–/– transgenic mice are viable and Src and Fyn still retain about 30–50% of their normal activity (Ponniah et al., 1999; Su et al., 1999; Table I). Indeed, at least three other PTPs, SHP1, SHP2 (Syp) and PTP1B (Peng and Cartwright, 1995; Lorenz et al., 1996; Somani et al., 1997; Cheng et al., 2001), have been implicated in Src regulation. It will be interesting to test whether these or other PTPs are activated during mitosis.
The mitotic retardation of PTPα electrophoretic mobility was eliminated by treatment with PP2A, a highly specific phosphoserine/phosphothreonine phosphatase. Since PTPα is only phosphorylated at serine and tyrosine during mitosis (data not shown), we conclude that PTPα is hyperphosphorylated at one or more serines during mitosis. The simplest hypothesis is that these phosphorylations are sufficient to alter PTPα catalytic activity and Grb2 binding. However, the possibility that serine hyperphosphorylation is required but not sufficient, and that some other modification to PTPα itself is also involved, is not excluded.
PKC is a serine kinase that could be responsible for the mitotic activation, as there is evidence that it can phosphorylate and activate PTPα in other situations: PTPα serine phosphorylation and activity increases in vivo following treatment of quiescent NIH 3T3 cells with phorbol ester, an activator of PKC (Tracy et al., 1995). In addition, PTPα can be phosphorylated and activated by PKC in vitro at Ser180 and Ser204, which are located near the plasma membrane on the cytoplasmic side (den Hertog et al., 1995; Tracy et al., 1995). PKC has been implicated in both negative and positive control of the G2→M transition (see reviews by Livneh and Fishman, 1997; Black, 2000) and it is possible that Ser180 and Ser204 are the residues that are hyperphosphorylated during mitosis. The fact that these residues are phosphorylated to some extent during interphase (Tracy et al., 1995) could explain the observation that PP2A treatment caused some decrease even in the activity of PTPα from unsynchronized cells (Figure 8). Thus, the possibility that these sites and/or PKC are involved in the mitotic activation of PTPα merits further examination.
PTPα also contains a serine within a Cdc2 phosphorylation consensus sequence (Pearson and Kemp, 1991) in its membrane-proximal cytoplasmic region (Krueger et al., 1990) and a potential casein kinase II phosphorylation site (Ser787) near Tyr789. These sites and kinases must also be considered.
The fact that the binding of PTPα to the isolated Grb2 SH2 domain decreased in mitosis (Figures 4 and 5) means that the mitotic change must affect PTPα in the region surrounding Tyr789 near its C-terminus, and that the binding between the Grb2 SH3 domain and other regions of PTPα (den Hertog and Hunter, 1996; Su et al., 1996) need not be affected. Indeed, a decrease in PTPα-Grb2 SH2 domain affinity without change in PTPα-Grb2 SH3 binding might explain the fact that a larger relative decrease (>8-fold) was observed in the Grb2 SH2 domain affinity-precipitations than in the co-immunoprecipitations with full-length Grb2 (Figure 4). If the SH3-PTPα binding were unchanged in mitosis, the cellular Grb2, which binds PTPα using both its SH2 and COOH SH3 domain, would be even more effective during mitosis than during interphase at competing binding from the exogenous Grb2 SH2 domain alone (because of the increased relative importance of the SH3-PTPα binding during mitosis).
These biochemical observations pose two structural puzzles. How are both the specific activity and SH2 domain binding of PTPα simultaneously modulated? How is the high affinity of PTPα for the Grb2 SH2 domain reduced without affecting its moderate affinity for the Src SH2 domain? The first question arises because the change in specific activity is likely to be caused by changes affecting the PTPα’s D1 domain, which is responsible for most catalytic activity (Wang and Pallen, 1991; den Hertog et al., 1993; Wu et al., 1997; Harder et al., 1998). This region is removed in sequence from Tyr789, so the manner by which both the SH2-binding region and D1 are simultaneously affected is an interesting question. The answer to the second question may be related to the observation that the Grb2 SH2 domain has the unusual property of binding peptide ligands in a β-turn conformation (Rahuel et al., 1996; Ogura et al., 1999). In contrast, the Src SH2 domain binds its ligands in the more common extended conformation (Waksman et al., 1993; Gilmer et al., 1994). The differential reduction in SH2 domain binding could result if the mitotic serine phosphorylation(s) prevented the PTPα C-terminus from forming a β-turn.
Dimerization and/or inhibitory interactions between PTPα’s D2 and D1 domains can completely inhibit PTPα specific activity (Jiang et al., 1999), and might be capable of affecting Grb2 binding as well. However, it seems unlikely that they are involved in the mitotic changes observed here since the high-salt immunoprecipitation and immunopurification procedures used for measuring specific activity and SH2 domain binding would be expected to dissociate dimers. Indeed, non-denaturing gel electrophoresis and cross-linking assays have not detected the presence of any PTPα dimers in the cell lysates used in these experiments (data not shown).
Because of technical limitations, we have not been able to study the time course of the Src-related biochemical events during the G2→M transition, but indirect evidence suggests that the mitotic activation of Src can occur at earliest shortly before nuclear envelope breakdown (Chackalaparampil and Shalloway, 1988). It is likely that this occurs after the mitotic Cdc2-cyclin B complex is activated by Cdc25-mediated dephosphorylation (see, for example, the review by Kishimoto and Okumura, 1997) and phosphorylate Src in its amino-proximal region (Shenoy et al., 1989). On the other hand, the mitotic release experiments (Figure 7) more precisely determined the temporal relationships between Cdc2-mediated phosphorylation of Src, Src activity (and by inference, Tyr527 phosphorylation) and the serine hyperphosphorylation and activation of PTPα after metaphase. It is noteworthy that the mobility-retarding Cdc2-mediated phosphorylations in Src disappeared about 20 min before Src specific activity, PTPα specific activity and PTPα-Grb2 binding returned to their interphase levels. This is consistent with the prior hypothesis (Taylor and Shalloway, 1993) that the Cdc2-mediated phosphorylations act by promoting dephosphorylation of pTyr527, and that its dephosphorylation by a PTP (which we now suggest to be PTPα) is the proximal cause of activation.
It is evident in Figure 7D and in previously published mitotic release experiments (Chackalaparampil and Shalloway, 1988) that Src activity returns to normal by 30–40 min after release from nocodazole arrest, which is close to the time of cytokinesis. But the PTPα modifications decrease somewhat more gradually and do not return to their interphase levels until ∼80 min after release. This temporal divergence may just reflect the strongly nonlinear relationship between the pTyr527-dephosphorylation rate and the level of Tyr527 phosphorylation (see Materials and methods). However, other events—possibly associated with cytoskeletal reorganization during cytokinesis—may also be involved in Src deactivation. For example, down-regulation of Src by Csk (Okada and Nakagawa, 1989) which phosphorylates Tyr527, appears to be regulated by colocalization of Csk and Src to focal adhesion plaques (,Howell et al., 1994; Sabe et al., 1994). The adhesion plaques partially dissipate during cell rounding in mitosis, and their reorganization as cells flatten after cytokinesis with concomitant colocalization of Csk and Src may contribute to abrupt Src down-regulation.
Figure 10 summarizes the functional and temporal sequence of events that we believe to be involved in the activation and subsequent deactivation of Src by the coordinated activities of the Cdc2-cyclin B complex, PTPα, and the Csk tyrosine kinase (see figure legend for references): (i) activation of the Cdc2-cyclin B complex and activation of PTPα (the temporal relationship of these two events is unknown); (ii) amino-proximal phosphorylation of Src by Cdc2, which weakens the intramolecular association between the Src SH2 domain and pTyr527; (iii) dephosphorylation of pTyr527 and activation of Src; (iv) degradation of cyclin B and deactivation of Cdc2 by APC/cyclosome; (4) amino-proximal dephosphorylation of Src (pTyr527 remains dephosphorylated since PTPα activity remains high and Grb2 inhibition remains low); and (v) serine dephosphorylation and deactivation of PTPα and return of inhibition by Grb2 binding (these result in deactivation of Src by phosphorylation of Tyr527, probably by Csk).

Fig. 10. Temporal model for activation of PTPα and Src during mitosis. Hypothetical time lines for phosphorylation and activation of the Cdc2–Cyclin B complex, Src and PTPα are shown which consolidate the multiple known functional and temporal relationships: (1) Cdc 25 activates the Cdc2-Cyclin B complex by dephosphorylating Thr14 and Tyr15 in Cdc2 (Kishimoto and Okumura, 1997); (2) the complex initiates nuclear envelope breakdown (Kishimoto and Okumura, 1997) and (3) phosphorylates mammalian Src at Thr36 and Ser74 (Shenoy et al., 1989; Lin, 1994); (4) these phosphorylations reduce Src SH2-pTyr527 affinity (Stover et al, 1994), thus (5) making pTyr527 more accessible for dephosphorylation (Bagrodia et al., 1991, 1994; Stover et al., 1994); (6) PTPα is activated by Ser hyperphosphorylation (this study) and (7) dephosphorylates pTyr527 by a phosphotyrosine displacement mechanism involving PTPα pTyr789 (Zheng et al., 2000); (8) both PTPα and Src are activated during metaphase (this study and Chackalaparampil and Shalloway, 1988); (9) APC/cyclosome deactivates the Cdc2–Cyclin B complex leading to anaphase (Page and Hieter, 1999); (10) the mitotic Ser/Thr Src phosphorylations are removed and Src is subsequently deactivated by rephosphorylation at Tyr527 at about the time of cytokinesis (Chackalaparampil and Shalloway, 1988), (11) probably by Csk (Sabe et al., 1994); (12) the mitotic modifications of PTPα (i.e. serine decreased Grb2 binding and enhanced specific activity) are removed during an extended period surrounding cytokinesis (this stydy). Dark colors indicate the functionally activated state. Small circles surrounding S, T or Y indicate phosphorylation of the respective amino acid types: in Cdc2, the inhibitory sites, Thr14 and Tyr15, and the stimulatory site Thr161; in mammalian Src, the mitosis-specific targets of Cdc2 phosphorylation, Thr36 and Ser74, and Tyr527; and in PTPα, Tyr789 and at least one serine site. The relative timing of all events is established except for that of activation of PTPα relative to activation of the Cdc2–Cyclin B complex, which is unknown. The absolute times of the phosphorylation of Src by Cdc2–Cyclin B and its dephosphorylation by PTPα are not known, except that they must occur sometime within an interval beginning shortly before nuclear envelope breakdown and ending at metaphase.
In contrast with the mitotic increase in PTPα-specific activity, which probably enhances its dephosphorylation of all substrates, the reduction in Grb2 binding may specifically increase the dephosphorylation of the negative-regulatory phosphotyrosines of Src family members (Zheng et al., 2000). If so, this component of the mitotic regulation provides an elegant mechanism that nature may use to solve the intrinsic problem posed by the use of a protein tyrosine phosphatase as an activator of a protein tyrosine kinase. Alternatively, this reduction might increase PTPα dephosphorylation of a broader range of SH2-domain containing targets, a possibility requiring further investigation. In either case, it provides an additional mechanism by which PTPα activity can be regulated, and one that could be used in other physiological contexts.
Materials and methods
Cell lines, nocodazole arrest of mitotic cells and induction of PTPα expression
The following inducible PTPα-overexpressor and non-overexpressor (control) NIH 3T3-derived cell lines were used (see Table I; Zheng et al., 2000): wt PTPα, NIH(pNTPTPα/cos)1; HA-tagged wt PTPα, NIH(pTPTPα/cos)1; PTPα(Y789F), NIH(pNTPTPα/Y789F/cos)1; HA-tagged PTPα(Y789F), NIH(pTPTPα/Y789F/cos)1; control (no PTPα transgene), NIH(pTet-SPLICE/cos)1. The Src overexpressor cell line was NIH(pMcsrc/foc)B1 (Johnson et al., 1985). The PTPα–/– line, a generous gift of J.Sap, was clone 3BP2 which had been derived from E3 PTPα-knockout cells by the 3T3 protocol (J.Sap, personal communication; Ponniah et al., 1999); the control PTPα+/+ line was clone 3αP9 derived by reintroducing PTPα into E3.
Unless otherwise specified, overexpressor cells were maintained in the presence of 5 ng/ml doxycycline to suppress PTPα overexpression prior to experimentation. Because most genes are only slowly expressed during mitosis, a special induction procedure was used to achieve approximately equal levels of PTPα overexpression in both the unsynchronized and mitotic cell populations: cells were induced by removal of doxycyline for 8 h, during which time PTPα levels steadily increased. Doxycycline was then added to a concentration of 5 ng/ml and the cells were incubated for 10 h either in the presence (mitotic arrest) or the absence (unsynchronized cells) of 0.1 µg/ml nocodazole. Mitotic cells were collected by manual shake-off. The half-life of overexpressed PTPα protein in these cells is ∼16 h (data not shown), so substantial (and equal) levels of PTPα overexpression (∼5–9× endogenous) resulted.
For time course experiments, mitotic cells were washed in media without nocodazole at 4°C (cells remain in mitosis at this temperature), replated in normal media and then incubated for the indicated times at 37°C.
Antibodies and immunoblotting
Antibody against the GST–PTPα(B20) fusion protein, which contains GST and residues 165–793 from mouse PTPα (Zheng et al., 2000), was used for all PTPα immunoblots. Antibody against the GST–PTPα(D2) fusion protein, which contains GST and the human PTPα D2 domain (residues 505–793), was prepared in the same way as described for GST–PTPα(B20) (Zheng et al., 2000). Anti-Grb2 monoclonal antibody (G16720, Signal Transduction Laboratories) was used for immunoblots in 1:5000 dilution and anti-Grb2 polyclonal antibody (C23SC-255, lot D190, Santa Cruz Biotechnology) was used for Grb2 immunoprecipitations. Anti-HA, -Src antibodies, -pTyr antibodies, cross-linking of antibodies to Sepharose beads and immunoblotting procedures were as previously described (Zheng et al., 2000) with the addition of mAb 2-17, a monoclonal antibody made against Src residues 2-17 which was produced from hybridoma 203-7D10 (Viromed Biosafety Laboratories, Camden, NY) and purified from cell culture medium using Thiofilic-Uniflow resin (Clontech) according to the manufacturer’s protocol.
Immunoprecipitation and immunopurification of PTPα
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in 1 ml of lysis buffer for 20 min at 4°C with rocking. Lysis buffer for samples that were to be used in PTP assays was 1% NP-40, 50 mM HEPES pH 7.2, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 10 µg/ml aprotinin, 10 µg/ml leupeptin and 1 mM phenylmethylsulfonyl fluoride. For other samples, lysis buffer also contained 1 mM Na3VO4. Cell lysates were clarified by centrifugation for 30 min at 20 000 g at 4°C and whole-cell protein concentrations were determined using the Bio-Rad DC protein assay.
For immunoprecipitations, unless specified otherwise, lysates were adjusted to contain either 1.5 mg (for endogenous PTPα) or 0.3 mg (for overexpressed PTPα) total cell protein in 300 µl of lysis buffer. This resulted in approximately equal concentrations of overexpressed and endogenous PTPα. Immunoprecipitations were performed by incubating the lysate with the specified antibody for 2 h at 4°C, then incubating with 20 µl of a 50% suspension of protein A-Sepharose beads (Pharmacia) for 1 h at 4°C. Prior to immunoprecipitate phosphatase assays, immunoprecipitates were washed once with lysis buffer plus 500 mM NaCl (to remove any non-covalently bound proteins) and then twice with lysis buffer.
Immunopurification of HA-tagged PTPα proteins with anti-HA conjugated protein A–Sepharose beads was as described by Zheng et al. (2000) from cell lysates containing 1 mg total cell protein. The Sepharose bead–antibody–protein complexes were washed once with lysis buffer plus 500 mM NaCl and then twice with lysis buffer prior to elution of PTPα.
Dephosphorylation and kinase assays and quantitative analysis
In vitro dephosphorylation assays of MBP were performed as described by Zheng et al. (2000), except that they used 10% of the PTPα immunoprecipitated from cell lysates containing 1.5 mg total cell protein and the incubation time was 15 min. The remainder of the immunoprecipitate was used for immunoblotting.
The Src pTyr527 dephosphorylation and kinase assays were performed as described by Zheng et al., (2000). These assays had to be analyzed carefully because roughly half of the pTyr527 was dephosphorylated in some cases, and so changes in PTPα activity were non-linearly related to changes in Tyr527 phosphorylation: We expect the amount of pTyr to depend on the specific activity (σ) of the phosphatase and incubation time (t) according to pTyr(t)/pTyr(0) = exp(-c σ t), where c depends on the experimental conditions. Thus, σ is proportional to –log[pTyr(t)/pTyr(0)]. Using this relationship and the data from multiple experiments like that shown in Figure 2, we computed the mitotic/unsynchronized pTyr527-directed phosphatase ratio, σM/σU = log(pTyrM/pTyr–)/ log(pTyrU/pTyr–), where pTyr–, pTyrU and pTyrM are the amounts of pTyr527 remaining after dephosphorylation with mock-immunopurified protein, PTPα from unsynchronized cells or PTPα from mitotic cells, respectively. A similar analysis was applied using the Src activity data and the previously determined relationship between the fractional Src Tyr527 phosphorylation level (P) and Src specific kinase activity (SKA), where SKA = 1–10.5 P (Shenoy et al., 1992). Both these analyses implied that σM/σU is ∼1.8–2.2.
A simple first-order kinetic model implies that P is related to the Tyr527 phosphorylation and dephosphorylation rate constants, k+ and k–, by P = 1/(1 + k–/k+), which is strongly nonlinear in the physiological situation where P ∼0.85–0.95. [See Shenoy et al. (1992) for some small adjustments to this model, but note that there is a typographical error in the formula for P in the legend to figure 4.]
Co-immunoprecipitation assays
Either Src, Grb2 or PTPα were immunoprecipitated as specified above except that, following den Hertog et al. (1994) and Harder et al. (1998), the lysis buffer was supplemented with 10% glycerol. In addition, immunoprecipitates were washed three times with lysis buffer plus 10% glycerol (and not with a high-salt buffer). For co-immunoprecipitation experiments, Src was immunoprecipitated using 2 µg mAb 2–17.
SH2 affinity-precipitation of PTPα
PTPα was affinity-precipitated from cell lysates as described in Zheng et al. (2000) using 10 µg GST, GST–Grb2 SH2 domain or GST–Src SH2 domain in a binding volume of 300 µl. The lysates used in the affinity precipitations shown in Figures 4 and 5A contained 1 mg and 0.3 mg total cell protein, respectively. The affinity precipitations in Figure 5B used PTPα-HA immunopurified from lysates containing 1 mg total cell protein in a binding volume of 500 µl.
Serine dephosphorylation and subsequent assays of PTPα
For the tyrosine phosphatase assay, HA-tagged PTPα was immunoprecipitated from cell lysates containing 300 µg total cell protein using anti-HA conjugated protein A–Sepharose beads as described above. Beads were washed twice with lysis buffer and then once with PP2A buffer [20 mM HEPES pH 7.0, 1 mM dithiothreitol (DTT), 1 mM MnCl2, 100 µg/ml bovine serum albumin (BSA)], and then resuspended in 100 µl PP2A buffer. The suspension was split into two aliquots and incubated with or without 0.5 U (∼0.25 µg) purified PP2A (Upstate Biotechnology, #14-111) at 30°C for 30 min. For control experiments, 0.05 µM okadaic acid (LC Laboratories, Woburn, MA) was added to inhibit PP2A activity. Beads were then washed twice with lysis buffer, once with PTPα phosphatase buffer (50 mM imidazole pH 7.2, 5 mM DTT) and then resuspended in 100 µl phosphatase buffer. One tenth of the resuspension was assayed for tyrosine phosphatase activity with MBP as substrate as described (Zheng et al., 2000); the remainder was used for immunoblotting.
An analogous procedure was used for Grb2 SH2 domain affinity assay except that the cell lysates contained 1 mg total cell protein and, following incubation of the aliquots with or without PP2A, the beads were washed twice with lysis buffer and PTPα was eluted in 0.1 ml of 0.1 M glycine pH 2.5 followed by neutralization to pH 7.2–7.4 with 5 µl 1 M Tris–HCl pH 8.0. The eluate was then analyzed for affinity-precipitation by GST–Grb2 SH2 domain and by anti-PTPα immunoblotting as described above.
Acknowledgments
Acknowledgements
We thank Ross J.Resnick for invaluable technical assistance, unpublished data, many helpful discussions and critical review of the manuscript. We thank Jan Sap, Jing Su and Madhavi Muranjan for the generous gift of PTPα-knockout cells. We also thank an anonymous reviewer who made a number of excellent suggestions. This work was supported by a National Institutes of Health grant (CA32317) to D.S.
References
- Arnott C.H., Sale,E.M., Miller,J. and Sale,G.J. (1999) Use of an antisense strategy to dissect the signaling role of protein-tyrosine phosphatase α. J. Biol. Chem., 274, 26105–26112. [DOI] [PubMed] [Google Scholar]
- Bagrodia S.,I. Chackalaparampil,T.E. Kmiecik and D. Shalloway (1991) Altered tyrosine 527 phosphorylation and mitotic activation of p60c–src. Nature, 349, 172–175. [DOI] [PubMed] [Google Scholar]
- Bagrodia S., Laudano,A.P. and Shalloway,D. (1994) Accessibility of the c-src SH2-domain for binding is increased during mitosis. J. Biol. Chem., 269, 10247–10251. [PubMed] [Google Scholar]
- Bagrodia S., Taylor,S.J. and Shalloway,D. (1993) Myristylation is required for Tyr-527 dephosphorylation and activation of pp60c–src in mitosis. Mol. Cell. Biol., 13, 1464–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari B., Lim,K.L. and Pallen,C.J. (1998) Physical and functional interactions between receptor-like protein-tyrosine phosphatase α and p59fyn. J. Biol. Chem., 173, 8691–8698. [DOI] [PubMed] [Google Scholar]
- Bilwes A.M., den Hertog,J., Hunter,T. and Noel,J.P. (1996) Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization. Nature, 382, 555–559. [DOI] [PubMed] [Google Scholar]
- Black J.D. (2000) Protein kinase C-mediated regulation of the cell cycle. Front. Biosci., 5, 406–423. [DOI] [PubMed] [Google Scholar]
- Blanchetot C. and den Hertog,J. (2000) Multiple interactions between receptor protein-tyrosine phosphatase (RPTP)α and membrane-distal protein-tyrosine phosphatase domains of various RPTPs. J. Biol. Chem., 275, 12446–12452. [DOI] [PubMed] [Google Scholar]
- Brown M.T. and Cooper,J.A. (1996) Regulation, substrates and functions of src. Biochim. Biophys. Acta, 1287, 121–149. [DOI] [PubMed] [Google Scholar]
- Chackalaparampil I. and Shalloway,D. (1988) Altered phosphorylation and activation of pp60c–src during fibroblast mitosis. Cell, 52, 801–810. [DOI] [PubMed] [Google Scholar]
- Chackalaparampil I., Bagrodia,S. and Shalloway,D. (1994) Tyrosine dephosphorylation of pp60c–src is stimulated by a serine/threonine phosphatase inhibitor. Oncogene, 9, 1947–1955. [PubMed] [Google Scholar]
- Cheng A., Bal,G.S., Kennedy,B.P. and Tremblay,M.L. (2001) Attenuation of adhesion-dependent signaling and cell spreading in transformed fibroblasts lacking protein tyrosine phosphatase-1B. J. Biol. Chem., 276, 25848–25855. [DOI] [PubMed] [Google Scholar]
- den Hertog J., Pals,C.E.G.M., Peppelenbosch,M.P., Tertoolen,L.G.J., de Laat,S.W. and Kruijer,W. (1993) Receptor protein tyrosine phosphatase α activates pp60c–src and is involved in neuronal differentiation. EMBO J., 12, 3789–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J., Tracy,S. and Hunter,T. (1994) Phosphorylation of receptor protein-tyrosine phosphatase α on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J., 13, 3020–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J., Sap,J., Pals,C.E., Schlessinger,J. and Kruijer,W. (1995) Stimulation of receptor protein-tyrosine phosphatase α activity and phosphorylation by phorbol ester. Cell Growth Differ., 6, 303–307. [PubMed] [Google Scholar]
- Gilmer T. et al. (1994) Peptide inhibitors of src SH2-SH3-phosphoprotein interactions. J. Biol. Chem., 269, 31711–31719. [PubMed] [Google Scholar]
- Harder K.W., Moller,N.P.H., Peacock,J.W. and Jirik,F.R. (1998) Protein-tyrosine phosphatase α regulates Src family kinases and alters cell-substratum adhesion. J. Biol. Chem., 273, 31890–31900. [DOI] [PubMed] [Google Scholar]
- Howell B.W. and Cooper,J.A. (1994) Csk suppression of Src involves movement of Csk to sites of Src activity. Mol. Cell. Biol., 14, 5402–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G., den Hertog,J., Su,J., Noel,J., Sap,J. and Hunter,T. (1999) Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-α. Nature, 401, 606–610. [DOI] [PubMed] [Google Scholar]
- Jiang G., den Hertog,J. and Hunter,T. (2000) Receptor-like protein tyrosine phosphatase α homodimerizes on the cell surface. Mol. Cell. Biol., 20, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PJ., Coussens,P.M., Danko,P.M. and Shalloway,D. (1985) Overexpressed pp60c-src can induce focus formation without complete transformation of NIH 3T3 cells. Mol. Cell Biol., 5, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S., Covic,L., Wyss,A. and Ballmer-Hofer,K. (1991) Association of pp60c–src with polyoma virus middle-T antigen abrogating mitosis-specific activation. Nature, 350, 431–433. [DOI] [PubMed] [Google Scholar]
- Kaech S., Schnierle,B., Wyss,A. and Ballmer-Hofer,K. (1993) Myristylation and amino-terminal phosphorylation are required for activation of pp60c–src during mitosis. Oncogene, 8, 575–581. [PubMed] [Google Scholar]
- Kaplan R., Morse,B., Huebner,K., Croce,C., Howk,R., Ravera,M., Ricca,G., Jaye,M. and Schlessinger,J. (1990) Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl Acad. Sci. USA, 87, 7000–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. and Okumura,E. (1997) In vivo regulation of the entry into M-phase: initial activation and nuclear translocation of cyclin B/Cdc2. Prog. Cell Cycle Res., 3, 241–249. [DOI] [PubMed] [Google Scholar]
- Klinghoffer R.A., Sachsenmaier,C. Cooper,J.A. and Soriano,P. (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J., 18, 2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik T.E. and Shalloway,D. (1987) Activation and suppression of pp60c–src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell, 49, 65–73. [DOI] [PubMed] [Google Scholar]
- Krueger N.X., Streuli,M. and Saito,H. (1990) Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J., 9, 3241–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.-H. (1994) Regulation of c-Src by modifications in its amino-proximal region. PhD Thesis, The Pennsylvania State University.
- Livneh E. and Fishman,D.D. (1997) Linking protein kinase C to cell-cycle control. Eur. J. Biochem., 248, 1–9. [DOI] [PubMed] [Google Scholar]
- Lorenz U., Ravichandran,K.S., Burakoff,S.J. and Neel,B.G. (1996) Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc. Natl Acad. Sci. USA, 93, 9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R., Bilwes,A.M., Noel,J.P., Hunter,T. and Weiss,A. (1998) Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science, 279, 88–91. [DOI] [PubMed] [Google Scholar]
- Neel B.G. and Tonks,N.K. (1997) Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell. Biol., 9, 193–204. [DOI] [PubMed] [Google Scholar]
- Ogura K., Tsuchiya,S., Terasawa,H., Yuzawa,S., Hatanaka,H., Mandiyan,V., Schlessinger,J. and Inagaki,F. (1999) Solution structure of the SH2 domain of Grb2 complexed with the Shc-derived phosphotyrosine-containing peptide. J. Mol. Biol., 289, 439–445. [DOI] [PubMed] [Google Scholar]
- Okada M. and Nakagawa,H. (1989) A protein tyrosine kinase involved in regulation of pp60c–src function. J. Biol. Chem., 264, 20886–20893. [PubMed] [Google Scholar]
- Page A.M. and Hieter,P. (1999) The anaphase-promoting complex: new subunits and regulators. Annu. Rev. Biochem., 68, 583–609. [DOI] [PubMed] [Google Scholar]
- Pearson R.B. and Kemp,B.E. (1991) Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol., 200, 62–81. [DOI] [PubMed] [Google Scholar]
- Peng Z.Y. and Cartwright,C.A. (1995) Regulation of the Src tyrosine kinase and Syp tyrosine phosphatase by their cellular association. Oncogene, 11, 1955–1962. [PubMed] [Google Scholar]
- Petrone A. and Sap,J. (2000) Emerging issues in receptor protein tyrosine phosphatase function: lifting fog or simply shifting? J. Cell Sci., 113, 2345–2354. [DOI] [PubMed] [Google Scholar]
- Ponniah S., Wang,D.Z., Lim,K.L. and Pallen,C.J. (1999) Targeted disruption of the tyrosine phosphatase PTPα leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol., 9, 535–538. [DOI] [PubMed] [Google Scholar]
- Rahuel J. et al. (1996) Structural basis for specificity of Grb2-SH2 revealed by a novel ligand binding mode. Nature Struct. Biol., 3, 586–589. [DOI] [PubMed] [Google Scholar]
- Roche S., Fumagalli,S. and Courtneidge,S.A. (1995) Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science, 269, 1567–1569. [DOI] [PubMed] [Google Scholar]
- Sabe H., Hata,A., Okada,M., Nakagawa,H. and Hanafusa,H. (1994) Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc. Natl Acad. Sci. USA, 91, 3984–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaapveld R., Wieringa,B. and Hendriks,W. (1997) Receptor-like protein tyrosine phosphatases: alike and yet so different. Mol. Biol. Rep., 24, 247–262. [DOI] [PubMed] [Google Scholar]
- Shenoy S., Choi,J.-K., Bagrodia,S., Copeland,T.D., Maller,J.L. and Shalloway,D. (1989) Purified maturation promoting factor phosphorylates pp60c–src at the sites phosphorylated during fibroblast mitosis. Cell, 57, 763–774. [DOI] [PubMed] [Google Scholar]
- Shenoy S., Chackalaparampil,I., Bagrodia,S., Lin,P.-H. and Shalloway,D. (1992) Role of p34cdc2-mediated phosphorylations in two-step activation of pp60c–src during mitosis. Proc. Natl Acad. Sci. USA, 89, 7237–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somani A.K., Bignon,J.S., Mills,G.B., Siminovitch,K.A. and Branch,D.R. (1997) Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem., 272, 21113–21119. [DOI] [PubMed] [Google Scholar]
- Stover D.R., Liebetanz,J. and Lydon,N.B. (1994) Cdc2-mediated modulation of pp60c–src activity. J. Biol. Chem., 269, 26885–26889. [PubMed] [Google Scholar]
- Su J., Batzer,A. and Sap,J. (1994) Receptor tyrosine phosphatase R-PTP-α is tyrosine phosphorylated and associated with the adaptor protein Grb2. J. Biol. Chem., 269, 18731–18734. [PubMed] [Google Scholar]
- Su J., Yang,L.-T and Sap,J. (1996) Association between receptor protein-tyrosine phosphatase RPTPα and the Grb2 adaptor. J. Biol. Chem., 271, 28086–28096. [DOI] [PubMed] [Google Scholar]
- Su J., Muranjan,M. and Sap,J. (1999) Receptor protein tyrosine phosphatase α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol., 9, 505–511. [DOI] [PubMed] [Google Scholar]
- Taylor S.J. and Shalloway,D. (1993) The cell cycle and c-Src. Curr. Opin. Genet. Dev., 3, 26–34. [DOI] [PubMed] [Google Scholar]
- Taylor S.J. and Shalloway,D. (1996) Src and the control of cell division. BioEssays, 18, 9–11. [DOI] [PubMed] [Google Scholar]
- Tertoolen L.G.J., Blanchetot,C., Jiang,G., Overvoorde,J., Gadella Jr.,T.W.J., Hunter,T. and den Hertog,J. (2001) Dimerization of receptor protein-tyrosine phosphatase α in living cells. BMC Cell Biol., 2, article 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.M. and Brugge,J.S. (1997) Cellular functions regulated by Src family kinases. Annu. Rev. Cell. Biol., 13, 513–609. [DOI] [PubMed] [Google Scholar]
- Tracy S., van der Geer,P. and Hunter,T. (1995) The receptor-like protein-tyrosine phosphatase, RPTPα, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J. Biol. Chem., 270, 10587–10594. [DOI] [PubMed] [Google Scholar]
- Waksman G., Shoelson,S.E., Pant,N., Cowburn,D. and Kuriyan,J. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell, 72, 767–778. [DOI] [PubMed] [Google Scholar]
- Wang Y. and Pallen,C.J. (1991) The receptor-like protein tyrosine phosphatase HPTPα has two active catalytic domains with distinct substrate specificities. EMBO J., 10, 3231–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Buist,A., den Hertog,J. and Zhang,Z.-Y. (1997) Comparative kinetic analysis and substrate specificity of the tandem catalytic domains of the receptor-like protein-tyrosine phosphatase α. J. Biol. Chem., 272, 6994–7002. [DOI] [PubMed] [Google Scholar]
- Zeng L., D’Allesandri,L., Kalousek,M.B., Vaughan,L. and Pallen,C.J. (1999) Protein tyrosine phosphatase α (PTPα) and contactin form a novel neuronal receptor complex linked to the intracellular tyrosine kinase fyn. J. Cell Biol., 147, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.M., Wang,Y. and Pallen,C.J. (1992) Cell transformation and activation of pp60c–src by overexpression of a protein tyrosine phosphatase. Nature, 359, 336–339. [DOI] [PubMed] [Google Scholar]
- Zheng X.M., Resnick,R.J. and Shalloway,D. (2000) A phosphotyrosine displacement mechanism for activation of Src by PTPα. EMBO J., 19, 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]