

Abstract
Cystinosis is an inherited lysosomal storage disease characterized by defective transport of cystine out of lysosomes. However, the causative gene, CTNS, encodes a seven transmembrane domain lysosomal protein, cystinosin, unrelated to known transporters. To investigate the molecular function of cystinosin, the protein was redirected from lysosomes to the plasma membrane by deletion of its C-terminal GYDQL sorting motif (cystinosin-ΔGYDQL), thereby exposing the intralysosomal side of cystinosin to the extracellular medium. COS cells expressing cystinosin-ΔGYDQL selectively take up l-cystine from the extracellular medium at acidic pH. Disruption of the transmembrane pH gradient or incubation of the cells at neutral pH strongly inhibits the uptake. Cystinosin-ΔGYDQL is directly involved in the observed cystine transport, since this activity is highly reduced when the GYDQL motif is restored and is abolished upon introduction of a point mutation inducing early-onset cystinosis. We conclude that cystinosin represents a novel H+-driven transporter that is responsible for cystine export from lysosomes, and propose that cystinosin homologues, such as mammalian SL15/Lec35 and Saccharomyces cerevisiae ERS1, may perform similar transport processes at other cellular membranes.
Keywords: cystine/cystinosis/H+ symporter/lysosome/transporter
Introduction
Cystinosis is a lysosomal storage disease characterized by an intralysosomal accumulation of cystine, which is due to defective cystine efflux from these organelles. This autosomal recessive disorder comprises three allelic clinical forms, varying in severity and age of onset. The infantile form (MIM 21980) generally appears at 6–8 months of age with a proximal renal tubulopathy, which, in the absence of renal transplantation, can lead to death by 10 years of age due to renal failure (for review see Gahl et al., 1995). Other clinical signs, notably retinal blindness, hypothyroidism, diabetes mellitus, swallowing difficulties and neurological deterioration, eventually appear due to the widespread accumulation of cystine in most tissues. The juvenile form (MIM 219900) is characterized by glomerular renal damage, which manifests at around 10–12 years of age and slowly progresses to glomerular insufficiency and photophobia due to corneal cystine-crystal deposits. Finally, the ocular non-nephropathic form (MIM 219750) is solely characterized by a mild photophobia but no renal anomalies.
Cystine, the disulfide of the amino acid cysteine, is a by-product of lysosomal protein hydrolysis, and is reduced to cysteine in the cytoplasm. As the enzymes involved in cyst(e)ine redox reactions are normal in cystinotic cells, it has been hypothesized that the underlying metabolic defect of cystinosis is a defective lysosomal membrane cystine transport (Gahl et al., 1995). Support for this hypothesis has been provided by the demonstration that cystine is rapidly lost from artificially loaded normal lysosomes, whereas cystine efflux from cystinotic lysosomes is almost non-existent (Gahl et al., 1982b; Jonas et al., 1982a,b; Steinherz et al., 1982). Furthermore, it has been shown that lysosomal cystine transport is carrier mediated due to the observation that the velocity of cystine egress from normal lysosomes shows saturation kinetics (Gahl et al., 1982a) and the demonstration of cystine countertransport across the normal lysosomal membrane (Gahl et al., 1983)—two hallmarks of carrier-mediated transport.
The gene underlying cystinosis, CTNS, was identified using a positional cloning strategy (Town et al., 1998). CTNS encodes a 367 amino acid protein, cystinosin, which comprises seven predicted transmembrane domains, a 128 amino acid N-terminal region bearing seven N-glycosylation sites and a 10 amino acid cytosolic C-terminus containing a tyrosine-based lysosomal sorting motif (GYDQL). We recently demonstrated that cystinosin is indeed a lysosomal membrane protein and that its targeting requires the presence of the GYDQL motif, in particular the Y and L residues, as well as a novel sorting motif localized in the third cytoplasmic loop (Cherqui et al., 2001). Deletion of one or the other of these targeting signals partially redirects cystinosin to the plasma membrane in addition to lysosomes, whereas deletion of both sorting motifs completely redirects cystinosin to the plasma membrane.
Although all forms of cystinosis have been linked to mutations in cystinosin (Shotelersuk et al., 1998; Town et al., 1998; Attard et al., 1999; Thoene et al., 1999), it has not yet been determined whether this protein is directly or indirectly responsible for the defective cystine transport of cystinotic lysosomes. Cystinosin may represent the lysosomal cystine transporter itself. However, neither its sequence nor its predicted topology displays a similarity to currently known transporters. It has thus been alternatively proposed that cystinosin might indirectly influence lysosomal cystine efflux (Attard et al., 1999; Mancini et al., 2000). In this study, we addressed this issue by using a strategy that exploited our recent knowledge of the signals involved in the targeting of cystinosin to lysosomes. As the lysosomal lumen is not easily accessible for transport experiments (Pisoni and Schneider, 1992), the recombinant protein was redirected to the plasma membrane by mutation of its C-terminal tyrosine-based motif (Cherqui et al., 2001), thereby creating a cellular model in which the ability of cystinosin to translocate cystine could be examined using whole cells. This experimental approach allowed us to characterize the molecular function of cystinosin.
Results
Cystinosin is a cystine transporter
We recently characterized several artificial mutations of cystinosin that redirect the recombinant protein to the plasma membrane in different cell lines (Cherqui et al., 2001). To test the potential transport activity of cystinosin, a mutant (ΔGYDQL) in which the C-terminal tyrosine-based motif is deleted was expressed in COS cells, and the ability of transfected cells to take up [35S]l-cystine from the extracellular medium was examined. Such an accumulation is equivalent to an efflux of cystine from lysosomes because the extracellular medium is topologically equivalent to the lysosomal lumen: in both processes, cystine is translocated towards the cytosol (Figure 1).

Fig. 1. Rationale used for the cystine transport assay of cystinosin. In vivo, wild-type cystinosin is localized at the lysosomal membrane (smaller grey circle) and is thought to transport cystine (C-S-S-C) from the lysosomal lumen to the cytosol. In our experimental model, we have deleted the C-terminal lysosomal targeting signal from cystinosin (cystinosin-ΔGYDQL). This results in a partial redirection of this protein to the plasma membrane (larger grey circle) in transfected COS cells. In this model, the extracellular medium is topologically equivalent to the lysosomal lumen, and cystinosin-ΔGYDQL would thus act to transport [35S]cystine from the extracellular medium into the cytosol.
In a standard extracellular medium (buffer A, pH 7.4; Materials and methods), a modest increase in accumulated [35S]cystine was often observed in cells expressing the ΔGYDQL mutant compared with those mock transfected with a plasmid without an insert (‘background’ level) or expressing wild-type cystinosin (Figure 2A). A mean increase of 39 ± 11% (± SEM) over background was obtained from 18 independent experiments. In order to mimic the acidic lumen of lysosomes, the extracellular pH was decreased to 5.6 during the incubation with [35S]cystine (buffer B). This acidification dramatically increased the amount of [35S]cystine accumulated in cystinosin-ΔGYDQL-expressing cells, but not in mock-transfected cells (Figure 2A). A mean value of 635 ± 50% over background was obtained from 45 independent experiments with the ΔGYDQL mutant. This acidification also revealed a low [35S]cystine uptake activity associated with the expression of wild-type cystinosin (131 ± 33% over background; n = 9), in agreement with a faint localization of the wild-type protein at the plasma membrane in addition to lysosomes (see below and Figure 8C).

Fig. 2. Cystine uptake ability of cystinosin-ΔGYDQL-expressing cells. (A) Assay of transfected cells for [35S]cystine uptake in a neutral (pH 7.4, hatched bars) or acidic (pH 5.6, grey bars) extracellular medium. At neutral pH, cells expressing cystinosin-ΔGYDQL show a modest increase in the amount of accumulated [35S]cystine as compared with mock-transfected cells or wild-type cystinosin-expressing cells. At acidic pH, a dramatic increase in accumulated [35S]cystine is observed in cystinosin-ΔGYDQL-expressing cells but not in mock-transfected cells. A small amount of [35S]cystine is also taken up by wild-type cystinosin-expressing cells. Error bars correspond to the SEM for all figures. (B) Cystinosin-ΔGYDQL-mediated [35S]cystine uptake (black squares) remained linear for 10 min. [35S]cystine uptake mediated by mock-transfected cells is indicated by white squares. (C) Amount of accumulated [35S]cystine remaining after a 3 and a 6 min incubation with 20 µM digitonin treatment of mock-transfected (white squares) or cystinosin-ΔGYDQL-expressing (black squares) cells.

Fig. 8. Effect of G308R on the amount of recombinant protein produced or its subcellular localization. (A) Amount of [35S]cystine accumulated by cells expressing GFP or the fusion proteins cystinosin–GFP, cystinosin-ΔGYDQL–GFP and cystinosin-G308R-ΔGYDQL–GFP in a neutral (hatched bars) and acidic (grey bars) uptake medium. (B) Western blot analysis of the same lot of transfected cells using an anti-GFP monoclonal antibody demonstrates that cystinosin-G308R-ΔGYDQL–GFP is not produced at a lower level than cystinosin–GFP or cystinosin-ΔGYDQL–GFP. (C) Immunofluorescence studies on the same lot of transfected cells demonstrate that cystinosin-ΔGYDQL–GFP and cystinosin-G308R-ΔGYDQL–GFP have the same subcellular localization pattern, and that both of these fusion proteins are present at a much higher level at the plasma membrane than cystinosin–GFP. Scale bar 40 µm for all panels.
The cystinosin-ΔGYDQL-mediated [35S]cystine uptake remained linear for 10 min (Figure 2B). We thus used a duration of ≤10 min throughout this study to measure uptake velocities. To determine whether the cystinosin- ΔGYDQL-induced cystine uptake reflected translocation across the plasma membrane or binding to the cell surface, cells exposed to [35S]cystine were subsequently incubated with 20 µM digitonin, a detergent that selectively permeabilizes the plasma membrane (Zuurendonk and Tager, 1974; Fiskum et al., 1980). Digitonin treatment rapidly released the accumulated radioactivity (Figure 2C), which is in agreement with a transport mechanism.
Taken together, these data demonstrate that the expression of cystinosin at the plasma membrane is associated with the induction of a cystine transport activity across this membrane. The simplest interpretation is that cystinosin does transport cystine (see Discussion).
Cystine transport is driven by a proton transmembrane gradient
Cystinosin-ΔGYDQL-mediated cystine transport was stimulated at acidic extracellular pH (Figure 2A). To characterize the pH effect further, cells were incubated with [35S]cystine in buffers ranging from pH 7.4 to 5.6. Cystine uptake by cystinosin-ΔGYDQL increased with decreasing pH, but without reaching a plateau (Figure 3A). This increase may depend exclusively on the pH of the extracellular compartment or, since cells maintain their cytosol at neutral pH, it may have resulted from the artificial transmembrane pH gradient created by the change in extracellular buffer. To discriminate between these hypotheses, the ionophore nigericin was added to the ‘acidic’ uptake buffer (buffer B, pH 5.6). Nigericin exchanges K+ for H+ (Pressman, 1968) and is widely used to dissipate transmembrane pH gradients. As shown in Figure 3B, 5 µM nigericin inhibited [35S]cystine uptake by cystinosin-ΔGYDQL by >85% at pH 5.6, demonstrating that the cystine transport is driven by the pH gradient. This observation suggests that cystinosin operates as a H+ symporter, i.e. that it couples the translocation of cystine to a translocation of H+ in the same direction. With the exception of Figure 7, all the experiments reported hereafter were performed at pH 5.6.

Fig. 3. Effect of a transmembrane pH gradient on cystine uptake. (A) [35S]cystine uptake by mock-transfected (white squares) and cystinosin-ΔGYDQL (black squares) expressing cells was performed in standard uptake buffer (see Materials and methods) adjusted to a pH ranging from 5.6 to 7.4 with 20 mM potassium phosphate. Cystinosin-mediated uptake increased with decreasing pH. (B) Amount of [35S]cystine accumulated by mock-transfected, cystinosin-expressing and cystinosin-ΔGYDQL-expressing cells in an acidic extracellular medium (grey boxes). The addition of 5 µM nigericin to the uptake media (striped boxes) abolished [35S]cystine uptake by cystinosin and cystinosin-ΔGYDQL, demonstrating the dependence of cystine transport on a proton gradient. (C) Effect of the presence of extracellular Na+ and K+ (uptake media containing NaCl as the major osmolyte, buffered with potassium phosphate K+-Pi), solely K+ (sucrose, Pi-K+) and solely Na+ (sucrose, MES-Na+) on [35S]cystine uptake by mock-transfected (white bars) and cystinosin-ΔGYDQL-expressing (black bars) cells. These changes do not significantly alter the amount of [35S]cystine taken up by cystinosin-ΔGYDQL, demonstrating that cystine transport does not require other ions.

Fig. 7. Effect of the cystinotic point mutation G308R on cystine uptake. Amount of [35S]cystine accumulated by mock-transfected, cystinosin-ΔGYDQL-expressing and cystinosin-G308R-ΔGYDQL-expressing cells in a neutral (hatched bars) and acidic (grey bars) uptake medium. The introduction of the G308R point mutation associated with infantile cystinosis abolishes cystine transport by cystinosin-ΔGYDQL.
To examine whether other extracellular ions influence the cystinosin-ΔGYDQL-mediated cystine transport, NaCl in buffer B was replaced by an isotonic concentration of KCl (data not shown) or sucrose (Figure 3C) and the potassium phosphate buffer was replaced by an equivalent amount of 2-(N-morpholino) ethane sulfonate (MES) adjusted to the same pH (Figure 3C). These changes did not significantly alter [35S]cystine uptake by cystinosin- ΔGYDQL; therefore, the transmembrane proton gradient appears to be the sole driving force of cystine transport.
Substrate selectivity of the transport activity
In order to characterize the interaction of cystinosin with cystine, cystinosin-ΔGYDQL-expressing cells were incubated with increasing concentrations of [35S]cystine for 8 min. Cells took up [35S]cystine at a constant rate over this time period at all concentrations tested (data not shown). The uptake velocity increased linearly up to 100 µM (Figure 4A). Above 100 µM, the increase in velocity declined progressively, suggesting a saturation of kinetics. A linear Eadie–Hofstee plot of the cystinosin-ΔGYDQL-dependent data confirmed that the cystine transport is saturable and follows Michaelis–Menten kinetics (Figure 4B). A KM of 278 ± 49 µM was calculated from three independent transfections. The Vmax, which depends on the transfection efficiency, ranged between 108 and 507 pmol/min per well (mean value: 316 ± 116 pmol/min per well).

Fig. 4. Saturation kinetics of cystinosin-mediated cystine uptake. (A) Plot of the velocity (V) of [35S]cystine uptake versus increasing substrate concentration (S) for mock-transfected (white squares) and cystinosin-ΔGYDQL-expressing (black squares) cells over an 8 min uptake period (linear phase). A deduction of background levels demonstrates that cystine uptake by cystinosin-ΔGYDQL is saturable (black triangles). (B) A linear Eadie–Hostee plot of the cystinosin-ΔGYDQL-dependent data demonstrates that cystine transport follows Michaelis–Menten kinetics. KM = 350 µM and Vmax = 507 pmol/min per well for this experiment.
To address the specificity of cystinosin for cystine, we compared the ability of this transporter to take up 40 µM [35S]l-cystine in the presence of 600 µM cold l- or d-cystine (limiting experimental concentration due to cystine insolubility). l-cystine inhibited [35S]l-cystine uptake by >60%, whereas d-cystine did not inhibit cystine uptake significantly (Figure 5).

Fig. 5. Stereoselectivity of cystine uptake. Assay of [35S]cystine uptake by mock-transfected (white bars) or cystinosin-ΔGYDQL (black bars) expressing cells in the absence (control) or presence of 600 µM l- or d-cystine. l-cystine inhibits [35S]cystine uptake by cystinosin-ΔGYDQL by >60%, whereas its stereoisomer d-cystine has no significant effect.
The intralysosomal hydrolysis of proteins generates a variety of amino acids, which may interfere with the cystinosin-mediated cystine efflux. The ability of diverse classes of amino acids to inhibit [35S]l-cystine uptake was therefore tested using a 10 mM concentration of each amino acid (Table I). This concentration greatly exceeds the concentration found in the lysosomal lumen, which occurs in the 10–100 µM range for each amino acid (Vadgama et al., 1991). Therefore, only major effects are expected to be relevant in vivo. l-methionine, l-leucine, l-alanine and l-valine did not inhibit cystinosin-ΔGYDQL-mediated cystine uptake by >40% (Table I). l-serine and l-threonine also inhibited cystine uptake only moderately. Phenylalanine did not significantly inhibit cystine uptake, and l-proline and l-glutamic acid did not inhibit uptake at all. In contrast, l-cysteine, when present at a 10 mM concentration, inhibited cystine uptake by 75%. Therefore, among all amino acids tested, only cysteine appeared as a possible additional substrate.
Table I. Effect of amino acids on [35S]l-cystine accumulation.
| Amino acid (10 mM) | % uninhibited± SEM | Greene et al. (1990) |
|---|---|---|
| l-methionine | 73 ± 12 (n = 4) | 51 ± 4 |
| l-leucine | 75 ± 12 (n = 4) | 61 ± 4 |
| l-alanine | 69 ± 9 (n = 6) | 86 |
| l-valine | 60 ± 7 (n = 6) | 66 |
| l-phenylalanine | 87 ± 7 (n = 6) | 78 |
| l-proline | 102 ± 9 (n = 6) | 87 |
| l-serine | 81 ± 4 (n = 4) | 79 |
| l-threonine | 69 ± 7 (n = 6) | 104 |
| l-cysteine | 25 ± 4 (n = 7) | n.d. |
| l-glutamic acid | 102 ± 11 (n = 4) | n.d. |
Values are expressed as the mean ± SEM of n independent observations, and are compared with those obtained by Greene et al. (1990) for the lysosomal cystine countertransport activity of mouse L-929 fibroblasts.
n.d., not determined.
The effect of increasing concentrations of l-cysteine on cystinosin-ΔGYDQL-mediated cystine uptake was then tested. Half-inhibition was obtained for 1.5 mM cysteine (Figure 6A), a value 5-fold higher than the cystine concentration that half-saturates cystinosin (Figure 4). The fact that 600 µM l-cystine inhibited ∼65% of the [35S]cystine transport, whereas an identical concentration of l-cysteine had no effect, confirmed that cystinosin preferentially recognizes l-cystine (Figure 6B).

Fig. 6. Cysteine uptake ability of cystinosin-ΔGYDQL-expressing cells. (A) [35S]cystine accumulated by cystinosin-ΔGYDQL in the presence of increasing concentrations of l-cysteine (logarithmic scale) is expressed as a percentage of uptake in the absence of cysteine. Half-inhibition of [35S]cystine uptake was obtained for a cysteine concentration of 1.5 mM, a value ∼5-fold higher than the cystine concentration that half-saturates cystinosin (278 ± 49 µM). (B) At equal concentrations, l-cystine inhibits [35S]cystine uptake by cystinosin-ΔGYDQL (black bars), whereas l-cysteine has no effect. [35S]cystine uptake by mock-transfected cells is shown as white bars. (C) At equal substrate occupancy (i.e. in the presence of a 5-fold higher concentration of [35S]cysteine as opposed to [35S]cystine), cystinosin does not translocate cysteine significantly. Bars as for (B).
To examine whether cysteine molecules bound to cystinosin are translocated across the cell membrane, cystinosin-ΔGYDQL-expressing cells were incubated with 200 µM [35S]l-cysteine at pH 5.6. For comparison, a parallel set of cells was incubated with a 5-fold lower concentration of [35S]cystine to compensate for the 5-fold higher affinity of cystine relative to cysteine. In such conditions, if [35S]cysteine and [35S]cystine were translocated by cystinosin with equal molecular turnovers, identical uptake signals (in picomoles) should be observed. This proved not to be the case, as although 201 ± 37 pmol/well [35S]cystine were taken up by cystinosin-ΔGYDQL, identical levels of 383 ± 58 and 378 ± 42 pmol/well [35S]cysteine were detected in cells expressing and not expressing cystinosin-ΔGYDQL, respectively (Figure 6C). In this experiment, background levels of cysteine uptake were decreased by substituting K+ for Na+ in buffer B (Figure 6C) in order to detect a possible cystinosin-mediated translocation of cysteine with more sensitivity. In further experiments, this background level was further reduced ∼2-fold by the addition of 5 mM serine or alanine, in agreement with the existence of an endogenous alanine/serine/cysteine transporter (ASCT) in COS cells (Palacin et al., 1998). However, even under these conditions, which did not significantly inhibit the cystinosin-mediated [35S]cystine uptake (Figure 3C; Table I), no cystinosin-dependent uptake of [35S]cysteine could be detected (data not shown). These experiments show that, at equal substrate occupancy, cystinosin translocates cysteine less efficiently, if at all, than cystine.
Taken together, these data show that cystinosin is highly selective for cystine.
A point mutation causing severe cystinosis abolishes cystine transport activity
Most of the mutations detected in patients with early-onset cystinosis are loss of function mutations, such as a large 57 kb (Touchman et al., 2000) deletion spanning almost the entire length of the gene (Town et al., 1998) and underlying 75% of European cases of infantile cystinosis (Forestier et al., 1999). Missense mutations have also been identified, and these are generally clustered in the C-terminal region of cystinosin (Attard et al., 1999). We tested the effect of one such mutation, G308R, detected in several affected individuals (Shotelersuk et al., 1998; Attard et al., 1999) and which substitutes an arginine for a highly conserved glycine residue in the sixth transmembrane domain, on cystine transport. G308R was introduced into the construct carrying the ΔGYDQL mutant and we found that cystine transport was reduced to background levels (Figure 7).
In order to verify that this effect was not due to an alteration in the amount of cystinosin that reaches the plasma membrane, we used western blot analysis and immunofluorescence studies of cystinosin constructs fused to a C-terminal green fluorescent protein (GFP) tag (Cherqui et al., 2001). Transport studies, western blot analysis and immunofluorescent studies were carried out on the same lot of transfected cells. Expression of the cystinosin-ΔGYDQL–GFP fusion protein in COS cells induced a strong cystine uptake activity at pH 5.6 (Figure 8A), showing that the fusion to GFP did not preclude the transport activity of cystinosin. A more limited activity was detected with the wild-type cystinosin–GFP fusion protein, as observed for the untagged protein (Figure 2A). Although the overall amount of wild-type cystinosin–GFP expressed was higher than that of cystinosin-ΔGYDQL–GFP (Figure 8B), the reduced activity of the wild-type protein could be correlated with a reduced expression at the plasma membrane of COS cells (Figure 8C). In contrast, the introduction of the G308R mutation into the cystinosin-ΔGYDQL–GFP protein abolished the cystine transport activity (Figure 8A), but did not reduce its overall level of expression (Figure 8B), nor did it significantly alter its localization at the plasma membrane (Figure 8C). These data show that the cystinotic point mutation prevents cystinosin from translocating cystine.
Discussion
Cystinosin is the lysosomal cystine transporter
The lysosomal storage disorder cystinosis is characterized by an intralysosomal accumulation of cystine due to a defective cystine efflux from lysosomes (Gahl et al., 1995). The causative gene was identified (Town et al., 1998) and the encoded protein, cystinosin, was recently shown to be a lysosomal membrane protein (Cherqui et al., 2001). However, the molecular function of cystinosin has remained unknown. By deleting the GYDQL lysosomal sorting motif (Cherqui et al., 2001), we were able to partially redirect cystinosin to the plasma membrane, generating a cellular model in which the lysosomal side of cystinosin is easily accessible for transport experiments as it now faces the extracellular medium. Our model provides the first direct evidence that cystinosin transports cystine. Deletion of the lysosomal sorting motif localized in the third cytoplasmic loop was not used because it abolished transport activity (data not shown). It could be argued that cystinosin interacts with and regulates a distinct, as yet unknown, cystine transporter. However, the amount of cystine uptake correlated with the amount of cystinosin at the plasma membrane (Figure 8) and a point mutation inducing infantile cystinosis was sufficient to abolish cystine transport. Therefore, our results strongly support the conclusion that cystinosin is the lysosomal cystine transporter.
The cystine transport mediated by recombinant cystinosin is consistent with previous observations on isolated lysosomes. Cystinosin-mediated cystine transport is saturable and follows Michaelis–Menten kinetics with a mean KM of 278 ± 49 µM. Similarly, countertransport experiments (in which externally added labelled cystine is exchanged for intralysosomal cystine) on lysosomes purified from human leukocytes (Gahl et al., 1983) and mouse L-929 fibroblasts (Greene et al., 1990) resulted in a KM of 500 and 270 µM, respectively. Both the recombinant and native transporters display a high selectivity for l-cystine (see Table I for a comparison). The fact that cystine uptake is not, or only moderately, inhibited by 10 mM l-glutamate on one hand, and 10 mM l-leucine or l-phenylalanine on the other hand, differentiates cystinosin from the two known plasma membrane cystine transporters 4F2hc/xCT (Sato et al., 1999) and rBAT/b°,+AT (Chairoungdua et al., 1999; Feliubadalo et al., 1999; Pfeiffer et al., 1999), respectively (for review see Palacin et al., 1998, 2001). Moreover, the affinity of cystinosin for cystine (KM = 278 µM) is lower than that of recombinant xCT (KM = 81 ± 13 µM) or b°,+AT (KM = 41 ± 20 µM) expressed in mammalian cell lines (Pfeiffer et al., 1999; Shih and Murphy, 2001).
We observed a moderate affinity of cystinosin for l-cysteine (IC50 = 1.5 mM). However, when cysteine and cystine transport were compared at similar levels of occupancy, no cysteine uptake was detected (Figure 6C). Since the background uptake of cysteine was high in this experiment, it may have masked a low level of cystinosin-mediated cysteine uptake. Nevertheless, we may derive from our data that if cystinosin translocated cysteine, the velocity of this translocation would not exceed half that of cystine (considering that a cysteine specific uptake would only be detectable if it exceeds twice the standard error of the background; see Results). Because the affinity for cysteine is also five times weaker, the overall cysteine transport would be at least 10 times slower than cystine transport at identical, non-saturating concentrations [intralysosomal amino acids generally do not exceed 100 µM (Vadgama et al., 1991)]. Therefore, l-cystine appears to be the sole physiological substrate of cystinosin.
The bioenergetics of the cystinosin-ΔGYDQL-mediated and lysosomal uptakes are also similar. We observed a 16-fold stimulation of cystine uptake when the pH of the extracellular medium was decreased from 7.4 to 5.6. This effect was strongly inhibited after disruption of the transmembrane pH gradient by nigericin, suggesting that cystinosin operates as a H+ symporter. Thus, the H+-translocating ATPase that acidifies and positively charges the lysosomal lumen (Nelson and Harvey, 1999) will actively drive the cystinosin-mediated transport in the efflux direction (Figure 9), as proposed for other lysosomal transport processes (Pisoni and Schneider, 1992; Sagné et al., 2001).
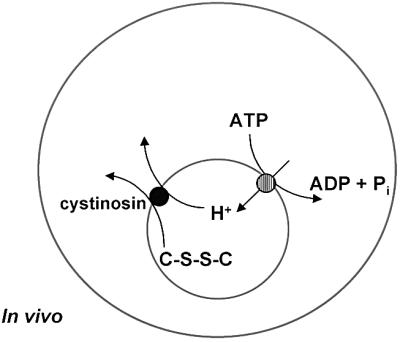
Fig. 9. Chemiosmotic coupling between cystinosin and the lysosomal H+-ATPase. The interior of the lysosome (small grey circle) is acidified by a H+-ATPase present in its membrane. The efflux of cystine (C-S-S-C) from the lysosome by the lysosomal cystine transporter, cystinosin, is coupled to an efflux of H+. Thus, the influx of H+ by the lysosomal H+-ATPase drives cystinosin-mediated cystine transport to the cystosol.
Consistently, previous studies on leukocytes (Gahl et al., 1982a; Greene et al., 1987), lymphoblasts (Jonas et al., 1982b, 1983), fibroblasts (Smith et al., 1987) and rat liver (Jonas, 1986) have shown that cystine efflux from lysosomes is stimulated by Mg-ATP. The use of inhibitors and uncouplers showed that the effect of ATP is mediated by the lysosomal H+-ATPase ( Jonas et al., 1983; Jonas, 1986; Smith et al., 1987). Both the electrical and pH components of the transmembrane electrochemical gradient created by the H+-ATPase were able to drive cystine efflux (Smith et al., 1987). Another study reported that in lysosomes from human leukocytes, cystine efflux was not stimulated by the H+-ATPase (Gahl and Tietze, 1985). However, an excessive lysosomal loading of cystine (artificially performed using a methyl ester derivative) might have masked the transporter-mediated process in these experiments (Greene et al., 1987).
Our characterization of the molecular activity of cystinosin, and previous studies of lysosomal cystine transport, strongly support a model in which cystine efflux is actively driven by the lysosomal H+-ATPase (Figure 9). As noted previously (Gahl and Tietze, 1985; Gahl et al., 1995), this chemiosmotic coupling is somewhat surprising because cystine is believed to be rapidly reduced to cysteine when it enters the cytosol, which in turn should drive cystine out of lysosomes. From a bioenergetical point of view, the H+/cystine symport thus appears to be a waste of energy for the cell. However, such a coupling may have other functions. For example, since intralysosomal acidification also stimulates proteolysis, the H+ symport mechanism may link the production of amino acids to their export from lysosomes, thereby preventing the build-up of high amino acid concentrations in their lumen. In the case of cystine, this coupling would be particularly beneficial due to its low solubility.
Cystinosin defines a novel family of transporters
The existence of diverse transporters involved in the export of lysosomal degradation products to the cytosol has been well documented by biochemical studies (reviewed by Pisoni and Schneider, 1992). Until recently, however, none had been identified. Sialin, the protein defective in sialic acid lysosomal storage diseases (MIM 269920), was identified as a member of the type I Na+/phosphate transporter family (Verheijen et al., 1999). This was in agreement with previous biochemical studies that linked the disease to an impairment of sialic acid and glucuronic acid transport across the lysosomal membrane (Havelaar et al., 1998; Mancini et al., 2000). More recently, a lysosomal amino acid transporter (LYAAT-1) (Sagné et al., 2001) belonging to the amino acid/auxin permease (AAAP) family (Rentsch et al., 1998) was identified. Interestingly, Russnak et al. (2001) demonstrated that, in Saccharomyces cerevisiae, several members of this AAAP family are involved in the transport of amino acids into and/or out of the vacuole, the lysosome equivalent in yeast. Our data demonstrating that cystinosin is the lysosomal cystine transporter thus contribute to the growing list of identified lysosomal transporters and, more generally, of identified intracellular transporters (Van Belle and André, 2001).
The novelty of cystinosin with regard to the two aforementioned transporters is the absence of homology to any known family of transporters, as well as its predicted seven transmembrane domain (TM) topology, which is, to our knowledge, unique among solute transporters. This resemblance to G-protein-coupled receptors suggested that cystinosin may act as part of a complex that binds, rather than actually transports, cystine (Attard et al., 1999). However, the present study and the fact that cystinosis is a monogenic disorder (Town et al., 1998; Anikster et al., 1999) rule out this hypothesis.
A recent study revealed the existence of several distant cystinosin homologues, in addition to the putative orthologues in other species (Zhai et al., 2001). In S.cerevisiae, the family includes five members (YBR147w, YOL092w, YDR352w, YDR090c and YMR101w) in addition to ERS1 (Hardwick and Pelham, 1990), the closest yeast homologue of cystinosin (33% identity over 97 amino acids). In mammals, the protein SL15/Lec35 (Ware and Lehrman, 1996; Anand et al., 2001) also belongs to the cystinosin family. Homology searches using amino acid sequences of the yeast proteins detected additional members in the human genome (B.Gasnier, unpublished data). Proteins from the cystinosin family share similar sizes and predicted topologies (300–400 amino acids, seven TM), as well as a conserved internal repeat, a motif designated PQ as it includes a characteristic PQ dipeptide (Zhai et al., 2001). Interestingly, the glycine residue involved in the cystinotic point mutation G308R, which abolished cystine transport, is one of the residues of the PQ motif that is highly conserved. Finally, the cystinosin family members also show a weaker homology to the family of bacterial rhodopsins, which, like cystinosin, possess seven TM and translocate H+ (Zhai et al., 2001).
To date, among the cystinosin homologues, functional data are available only for yeast ERS1 and mammalian SL15/Lec35. ERS1 has been shown to restore, by an unknown mechanism, the retention of endoplasmic reticulum proteins in yeast erd1 mutants (Pelham et al., 1988; Hardwick and Pelham, 1990). SL15, originally described as a suppressor of the recessive CHO cell glycosylation mutation Lec35 (Ware and Lehrman, 1996), is the gene defective in this mutant (Anand et al., 2001). Several endoplasmic reticulum glycosylation reactions that depend on mannose-P-dolichol (MPD) as a mannose donor are defective in Lec35 cells. Interestingly, MPD is synthesized on the cytoplasmic leaflet of the reticulum membrane and needs to be translocated to the lumenal leaflet (MPD ‘flippase’ activity) to be available for the reactions that are impaired in Lec35 cells (Rush and Waechter, 1995). Moreover, the glycosylation defects of Lec35 cells are corrected by chemical or physical treatments that perturb membranes (Zeng and Lehrman, 1990). It has thus been suggested that SL15/Lec35 acts at a step close to the MPD transbilayer movement (Anand et al., 2001). Since cystinosin operates as a transporter, we propose that SL15/Lec35 is the MPD transporter itself.
In summary, our study establishing that cystinosin is the lysosomal cystine transporter represents the dénouement of our understanding of the molecular basis of cystinosis (Gahl et al., 1995). Furthermore, the introduction of a single cystinotic point mutation situated in the sixth TM provides the first insight into a region of cystinosin critical for cystine transport. A future goal will be to determine what effect the different mutated alleles of the CTNS gene have on cystine transport. This will allow us to delineate the exact regions of cystinosin responsible for this activity as well as to understand the underlying basis of the phenotypic differences between the three forms of cystinosis. Our functional expression assay, which allows the precise quantification of the transport kinetics, will provide an important tool in this respect. Previous assays on lysosomes permitted such measurements only for an exchange reaction (countertransport experiments), which is not the physiological process occurring on lysosomes. In contrast, cells expressing cystinosin-ΔGYDQL, which may be compared with giant inside-out lysosomes, allow quantification for a net (inside-out) ‘efflux’ to the cytosol. The identification of the molecular activity of cystinosin also suggests a novel family of membrane translocators, which, as in the case of SL15/Lec35 for the membrane translocation of MPD, may provide novel insights into the study of eukaryotic cell compartmentalization.
Materials and methods
Generation and mutagenesis of cystinosin-expressing constructs
The CTNS cDNA coding region was amplified using the BamHI-containing forward primer 5′-GGGGATCCTGAGCTCTGCCTCTTCC-3′, situated in the 5′-untranslated region, and the XbaI-containing reverse primer 5′-GCTCTAGAGCCAGCCCTGTGTGCCAG-3′, situated in the 3′-untranslated region. The resultant PCR product was restriction enzyme digested and subcloned into the expression vector pcDNA3.1/Zeo+ (Invitrogen), generating the construct pcDNA-CTNS. Modifications to pcDNA-CTNS to generate constructs carrying a deletion of the C-terminal lysosomal sorting motif GYDQL, ΔGYDQL, and a cystinotic missense mutation, G308R, were carried out using the Stratagene Quikchange site-directed mutagenesis kit according to the manufacturer’s recommendations (primers used are not listed here, but are available upon request). Each construct generated was verified by sequencing. The fusion construct encoding cystinosin bearing a GFP tag at its C-terminus, pCTNS-EGFP, and subsequent modifications to this plasmid were generated as previously described (Cherqui et al., 2001).
Cell culture and transfection
COS-7 cells were grown under 5% CO2 in glucose-rich Dulbecco’s modified Eagle medium (DMEM) containing Glutamax-I (Life Technologies) supplemented with 7.5% fetal calf serum (FCS) and 100 U/ml penicillin/streptomycin. Typically, 2 × 106 cells in 50 µl of ice-cold phosphate-buffered saline (PBS) pH 7.4 were transfected with 5 µg of plasmid by electroporation using a GHT 1287 electropulsator (Jouan). After addition of the plasmid, cells were immediately subjected to eight square pulses (250 V, 3 ms) delivered at 1 Hz by 4-mm-spaced electrodes, and then diluted in 6 ml of culture medium. Cells were subsequently divided into 12 aliquots and cultured in 24-well culture plates (15 mm diameter) for 48 h before being assayed for cystine uptake. As a negative control, cells were mock transfected with the plasmid pcDNA3.1/Zeo+. For immunofluorescence studies, cells were cultured as above on a sterile glass coverslip for 48 h. For western blot analysis, electroporated cells were cultured in a 10 cm cell culture plate for 48 h.
Amino acid uptake studies
The culture wells were washed twice with 500 µl of uptake buffer A (5 mM d-glucose, 140 mM NaCl, 1 mM MgSO4, 20 mM potassium phosphate pH 7.4). Unless stated otherwise, cells were then incubated at room temperature with 200 µl of uptake buffer B (identical to buffer A, but potassium phosphate was adjusted to pH 5.6) supplemented with 40 µM or 1 µCi [35S]l-cystine (50–200 mCi/mmol; Amersham) for 10 min. The reaction was stopped by two brief washes with 500 µl of ice-cold uptake buffer A at pH 7.4. The cells were lysed using 200 µl of 0.1 N NaOH and the radioactivity accumulated by the cells counted by liquid scintillation in Emulsifier-Safe cocktail (Packard) using a Tri-Carb 2100 TR liquid scintillation analyser (Packard). For the substrate selectivity experiments (Table I; Figures 5 and 6), [35S]l-cystine and the unlabelled amino acid were applied to the cells simultaneously. The unlabelled l-cysteine (Merck) solutions were prepared extemporaneously in degassed water to prevent oxidation of the thiol moiety and were verified using thin-layer chromatographic (TLC) analysis of N-ethylmaleimide-conjugated products (States and Segal, 1969). Cysteine uptake measurements were performed with 1 µCi of [35S]l-cysteine (>1000 mCi/µmol; Amersham) and 200 µM unlabelled l-cysteine essentially as above, with any modifications as indicated in the Results. Transport measurements were performed in triplicate and are expressed as mean ± standard error of the mean (SEM). Each experiment was repeated a minimum of three times.
Western blot analysis
Culture plates were placed on ice, rinsed twice with PBS and scraped in 2 ml of PBS containing protease inhibitors (5 µg/ml aprotinin, 5 µg/ml leupeptin, 5 µg/ml pepstatin and 1 mM EGTA). Cells were centrifuged at 18 000 g for 5 min at 4°C, the supernatant removed and the pellet immediately frozen in liquid nitrogen before storing at –20°C. The pellet was resuspended in Laemmli’s sample buffer containing Benzonase (Merck) and the equivalent of 2 × 105 cells was loaded directly onto a 10% SDS–PAGE gel. The separated proteins were electrotransferred to nitrocellulose filters (Bio-Rad) and, after blocking for 1 h in 0.05% Tween/PBS containing 1% polyvinylpyrolidane (Sigma), the membrane was incubated for 1 h at room temperature with a 1/1000 dilution of anti-GFP antibody (Boehringer Mannheim). After three washes in 0.05% Tween/PBS, the filter was incubated for 1 h at room temperature with a 1:200 000 dilution of horseradish peroxidase-conjugated sheep antibody against mouse whole immunoglobulins (Amersham Life Sciences). The final detection was performed using the Lumi-LightPLUS Western Blotting Substrate (Roche Molecular Biochemicals).
Immunofluorescence studies
Transfected cells were washed with PBS containing 100 µM MgCl2 and 100 µM CaCl2, and fixed with 4% paraformaldehyde (Sigma) for 10 min at room temperature. After washing, cells were blocked with 10 mM NH4Cl for 10 min, washed with PBS and permeabilized using 0.5% Triton X-100 (Sigma) for 10 min. Coverslips were then treated with 1 mg/ml RNase for 10 min and the cell nuclei stained with 0.5 µg/ml propidium iodide (Sigma) for 3 min. Finally, coverslips were washed in PBS and mounted using Fluoprep (bioMérieux). Mounted coverslips were visualized using a Leica DMR microscope equipped with Leica QFluoro software for the acquisition and treatment of fluorescence.
Acknowledgments
Acknowledgements
V.K. would like to thank J.P.Henry for the opportunity to work in his laboratory, G.C.Bellenchi, M.Isambert and C.Bedet for their invaluable help and advice, and all the members of CNRS 1929 for their warm welcome. We are indebted to R.Haguenauer-Tsapis and J.P.Henry for critical reading of the manuscript and fruitful discussions. This work was supported by CNRS, INSERM, Vaincre les Maladies Lysosomales, Association Française contre les Myopathies, Association pour l’Utilisation du Rein Artificel and Ministère de l’Education Nationale, de la Recherche et de la Technologie (PhD grant to S.C.).
References
- Anand M., Rush,J.S., Ray,S., Doucey,M.A., Weik,J., Ware,F.E., Hofsteenge,J., Waechter,C.J. and Lehrman,M.A. (2001) Requirement of the Lec35 gene for all known classes of monosaccharide-P-dolichol-dependent glycosyltransferase reactions in mammals. Mol. Biol. Cell, 12, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikster Y., Shotelersuk,V. and Gahl,W.A. (1999) CTNS mutations in patients with cystinosis. Hum. Mutat., 14, 454–458. [DOI] [PubMed] [Google Scholar]
- Attard M., Jean,G., Forestier,L., Cherqui,S., van’t Hoff,W., Broyer,M., Antignac,C. and Town,M. (1999) Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum. Mol. Genet., 8, 2507–2514. [DOI] [PubMed] [Google Scholar]
- Chairoungdua A. et al. (1999) Identification of an amino acid transporter associated with the cystinuria-related type II membrane glycoprotein. J. Biol. Chem., 274, 28845–28848. [DOI] [PubMed] [Google Scholar]
- Cherqui S., Kalatzis,V., Trugnan,G. and Antignac,C. (2001) The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J. Biol. Chem., 276, 13314–13321. [DOI] [PubMed] [Google Scholar]
- Feliubadalo L. et al. (1999) Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (b°,+AT) of rBAT. International Cystinuria Consortium. Nature Genet., 23, 52–57. [DOI] [PubMed] [Google Scholar]
- Fiskum G., Craig,S.W., Decker,G.L. and Lehninger,A.L. (1980) The cytoskeleton of digitonin-treated rat hepatocytes. Proc. Natl Acad. Sci. USA, 77, 3430–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forestier L., Jean,G., Attard,M., Cherqui,S., Lewis,C., van’t Hoff,W., Broyer,M., Town,M. and Antignac,C. (1999) Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. Am. J. Hum. Genet., 65, 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl W.A. and Tietze,F. (1985) pH effects on cystine transport in lysosome-rich leucocyte granular fractions. Biochem. J., 228, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl W.A., Bashan,N., Tietze,F., Bernardini,I. and Schulman,J.D. (1982a) Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science, 217, 1263–1265. [DOI] [PubMed] [Google Scholar]
- Gahl W.A., Tietze,F., Bashan,N., Steinherz,R. and Schulman,J.D. (1982b) Defective cystine exodus from isolated lysosome-rich fractions of cystinotic leucocytes. J. Biol. Chem., 257, 9570–9575. [PubMed] [Google Scholar]
- Gahl W.A., Tietze,F., Bashan,N., Bernardini,I., Raiford,D. and Schulman,J.D. (1983) Characteristics of cystine counter-transport in normal and cystinotic lysosome-rich leucocyte granular fractions. Biochem. J., 216, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl W.A., Schneider,J.A. and Aula,P.P. (1995) Lysosomal transport disorders: cystinosis and sialic acid storage disorders. In Scriver,C.R., Beaudet,A.L., Sly,W.S. and Valle,D. (eds), The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill, New York, NY, pp. 3763–3797.
- Greene A.A., Clark,K.F., Smith,M.L. and Schneider,J.A. (1987) Cystine exodus from normal leucocytes is stimulated by MgATP. Biochem. J., 246, 547–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene A.A., Marcusson,E.G., Morell,G.P. and Schneider,J.A. (1990) Characterization of the lysosomal cystine transport system in mouse L-929 fibroblasts. J. Biol. Chem., 265, 9888–9895. [PubMed] [Google Scholar]
- Hardwick K.G. and Pelham,H.R.B. (1990) ERS1, a seven transmembrane domain protein from Saccharomyces cerevisiae. Nucleic Acids Res., 18, 2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havelaar A.C., Mancini,G.M., Beerens,C.E., Souren,R.M. and Verheijen,F.W. (1998) Purification of the lysosomal sialic acid transporter. Functional characteristics of a monocarboxylate transporter. J. Biol. Chem., 273, 34568–34574. [DOI] [PubMed] [Google Scholar]
- Jonas A.J. (1986) Cystine transport in purified rat liver lysosomes. Biochem. J., 236, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas A.J., Greene,A.A., Smith,M.L. and Schneider,J.A. (1982a) Cystine accumulation and loss in normal, heterozygous and cystinotic fibroblasts. Proc. Natl Acad. Sci. USA, 79, 4442–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas A.J., Smith,M.L. and Schneider,J.A. (1982b) ATP-dependent lysosomal cystine efflux is defective in cystinosis. J. Biol. Chem., 257, 13185–13188. [PubMed] [Google Scholar]
- Jonas A.J., Smith,M.L., Allison,W.S., Laikind,P.K., Greene,A.A. and Schneider,J.A. (1983) Proton-translocating ATPase and lysosomal cystine transport. J. Biol. Chem., 258, 11727–11730. [PubMed] [Google Scholar]
- Mancini G.M., Havelaar,A.C. and Verheijen,F.W. (2000) Lysosomal transport disorders. J. Inherit. Metab. Dis., 23, 278–292. [DOI] [PubMed] [Google Scholar]
- Nelson N. and Harvey,W.R. (1999) Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol. Rev., 79, 361–385. [DOI] [PubMed] [Google Scholar]
- Palacin M., Estevez,R., Bertran,J. and Zorzano,A. (1998) Molecular biology of mammalian plasma membrane amino acid transporters. Physiol. Rev., 78, 969–1054. [DOI] [PubMed] [Google Scholar]
- Palacin M., Borsani,G. and Sebastio,G. (2001) The molecular bases of cystinuria and lysinuric protein intolerance. Curr. Opin. Genet. Dev., 11, 328–335. [DOI] [PubMed] [Google Scholar]
- Pelham H.R., Hardwick,K.G. and Lewis,M.J. (1988) Sorting of soluble ER proteins in yeast. EMBO J., 7, 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer R., Loffing,J., Rossier,G., Bauch,C., Meier,C., Eggermann,T., Loffing-Cueni,D., Kuhn,L.C. and Verrey,F. (1999) Luminal heterodimeric amino acid transporter defective in cystinuria. Mol. Biol. Cell, 10, 4135–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisoni R.L. and Schneider,J.A. (1992) Amino acid transport by lysosomes. In Kilberg,M.S. and Häussinger,D. (eds), Mammalian Amino Acid Transport. Plenum Press, New York, NY, pp. 89–99.
- Pressman B.C. (1968) Ionophorous antibiotics as models for biological transport. Fed. Proc., 27, 1283–1288. [PubMed] [Google Scholar]
- Rentsch D., Boorer,K.J. and Frommer,W.B. (1998) Structure and function of plasma membrane amino acid, oligopeptide and sucrose transporters from higher plants. J. Membr. Biol., 162, 177–190. [DOI] [PubMed] [Google Scholar]
- Rush J.S. and Waechter,C.J. (1995) Transmembrane movement of a water-soluble analogue of mannosylphosphoryldolichol is mediated by an endoplasmic reticulum protein. J. Cell Biol., 130, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russnak R., Konczal,D. and McIntire,S.L. (2001) A family of yeast proteins mediating bidirectional vacuolar amino acid transport. J. Biol. Chem., 276, 23849–23857. [DOI] [PubMed] [Google Scholar]
- Sagné C., Agulhon,C., Ravassard,P., Darmon,M., Hamon,M., El Mestikawy,S., Gasnier,B. and Giros,B. (2001) Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl Acad. Sci. USA, 98, 7206–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H., Tamba,M., Ishii,T. and Bannai,S. (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem., 274, 11455–11458. [DOI] [PubMed] [Google Scholar]
- Shih A.Y. and Murphy,T.H. (2001) xCt cystine transporter expression in HEK293 cells: pharmacology and localization. Biochem. Biophys. Res. Commun., 282, 1132–1137. [DOI] [PubMed] [Google Scholar]
- Shotelersuk V., Larson,D., Anikster,Y., McDowell,G., Lemons,R., Bernardini,I., Guo,J., Thoene,J. and Gahl,W.A. (1998) CTNS mutations in an American-based population of cystinosis patients. Am. J. Hum. Genet., 63, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.L., Greene,A.A., Potashnik,R., Mendoza,S.A. and Schneider,J.A. (1987) Lysosomal cystine transport. Effect of intralysosomal pH and membrane potential. J. Biol. Chem., 262, 1244–1253. [PubMed] [Google Scholar]
- States B. and Segal,S. (1969) Thin-layer chromatographic separation of cystine and the N-ethylmaleimide adducts of cysteine and glutathionen. Anal. Biochem., 27, 323–329. [DOI] [PubMed] [Google Scholar]
- Steinherz R., Tietze,F., Gahl,W.A., Triche,T.J., Chiang,H., Modesti,A. and Schulman,J.D. (1982) Cystine accumulation and clearance by normal and cystinotic leukocytes exposed to cystine dimethyl ester. Proc. Natl Acad. Sci. USA, 79, 4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoene J. et al. (1999) Mutations of CTNS causing intermediate cystinosis. Mol. Genet. Metab., 67, 283–293. [DOI] [PubMed] [Google Scholar]
- Touchman J.W. et al. (2000) The genomic region encompassing the nephropathic cystinosis gene (CTNS): complete sequencing of a 200-kb segment and discovery of a novel gene within the common cystinosis-causing deletion. Genome Res., 10, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town M. et al. (1998) A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nature Genet., 18, 319–324. [DOI] [PubMed] [Google Scholar]
- Vadgama J.V., Chang,K., Kopple,J.D., Idriss,J.M. and Jonas,A.J. (1991) Characteristics of taurine transport in rat liver lysosomes. J. Cell Physiol., 147, 447–454. [DOI] [PubMed] [Google Scholar]
- Van Belle D. and Andre,B. (2001) A genomic view of yeast membrane transporters. Curr. Opin. Cell Biol., 13, 389–398. [DOI] [PubMed] [Google Scholar]
- Verheijen F.W. et al. (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nature Genet., 23, 462–465. [DOI] [PubMed] [Google Scholar]
- Ware F.E. and Lehrman,M.A. (1996) Expression cloning of a novel suppressor of the Lec15 and Lec35 glycosylation mutations of Chinese hamster ovary cells. J. Biol. Chem., 271, 13935–13938. [DOI] [PubMed] [Google Scholar]
- Zeng Y.C. and Lehrman,M.A. (1990) A block at Man5GlcNAc2-pyrophosphoryldolichol in intact but not disrupted castanospermine and swainsonine-resistant Chinese hamster ovary cells. J. Biol. Chem., 265, 2296–2305. [PubMed] [Google Scholar]
- Zhai Y., Heijne,W.H., Smith,D.W. and Saier,M.H. (2001) Homologues of archaeal rhodopsins in plants, animals and fungi: structural and functional predications for a putative fungal chaperone protein. Biochim. Biophys. Acta, 1511, 206–223. [DOI] [PubMed] [Google Scholar]
- Zuurendonk P.F. and Tager,J.M. (1974) Rapid separation of particulate components and soluble cytoplasm of isolated rat-liver cells. Biochim. Biophys. Acta, 333, 393–399. [DOI] [PubMed] [Google Scholar]