

Abstract
The adenovirus E1A protein regulates transcription of cellular genes via its interaction with the transcriptional coactivators p300/CBP. The collagenase promoter activated by the c-Jun protein is repressed by E1A. Here we show that E1A repression is specific for c-Jun, as E1A does not repress the collagenase promoter activated by the homologous transcription factor EB1. Using chimeras of c-Jun and EB1, we demonstrate that a 12 amino acid region in the basic region of the c-Jun DNA-binding domain is essential for repression by E1A. Since repression requires the binding of p300 to E1A, we studied the involvement of p300 acetyltransferase activity in the repression mechanism. We demonstrate that c-Jun is acetylated in vivo, and mutational analysis identified Lys271 in the c-Jun basic region to be essential for repression of the collagenase promoter by E1A. In addition, Lys271 is acetylated both in vitro and in vivo. These results suggest that the specific repression of the collagenase promoter by E1A involves acetylation of c-Jun.
Keywords: acetylation/CBP/c-Jun/E1A/p300
Introduction
The adenovirus E1A oncoprotein regulates a variety of cellular processes such as proliferation and transformation. The interaction of E1A with cellular transcription regulators is essential for adenovirus-induced transformation of cells. The transforming properties of E1A reside in an N-terminal non-conserved region and in CR1 and CR2, two regions conserved among adenovirus serotypes. E1A regulates gene expression by interaction through these regions with cellular proteins such as the transcriptional coactivators p300, the CREB-binding protein (CBP) and the retinoblastoma tumour suppressor protein pRb (reviewed in Hagmeyer et al., 1995).
E1A and p300/CBP frequently have been shown to affect the transcription of the same promoters (Arany et al., 1995). For example, the c-jun promoter is activated by both E1A and p300 independently (Duyndam et al., 1999). On the other hand, the collagenase promoter is inhibited by E1A, a repression dependent on the p300/CBP-binding site of E1A (Dorsman et al., 1995). The binding of E1A to p300/CBP was suggested to be responsible for the effect of E1A on the AP1 family of transcription factors (Lee et al., 1996; Smits et al., 1996), including c-Jun and c-Fos, which play a central role in proliferation and differentiation (Vogt and Bos, 1990; Angel and Karin, 1991). Previous studies have shown that activation of the collagenase promoter by c-Jun and p300/CBP requires the binding of p300/CBP to the transactivation domain (TAD) of c-Jun (Bannister et al., 1995; Lee et al., 1996). However, in apparent contradiction to this finding, E1A represses the c-Jun-activated collagenase promoter via the DNA-binding domain (DBD) of c-Jun (Hagmeyer et al., 1993; Smits et al., 1996). Taken together, the coactivator CBP/p300 appears to mediate the transcription regulation of c-Jun by E1A, but the mechanism remains unclear.
CBP was first identified and cloned in a search of transcriptional coactivators that bound to the phosphorylated CREB transcription factor (Chrivia et al., 1993). The p300 protein, which was cloned originally by virtue of its interaction with E1A (Eckner et al., 1994), was found to have high homology to CBP. p300 and CBP subsequently have been shown to bind to both upstream transcription factors and components of the basal transcription machinery. Given the unique binding mode of these coactivators, a model emerged with p300/CBP serving the function of adaptor proteins, which mediate the association of upstream enhancers with basal transcription factors (reviewed in Giordano et al., 1999). Over the last few years, important insights have been gained into the mechanism of transcriptional regulation by coactivators. It has been demonstrated that many coactivators, including p300, CBP (Bannister and Kouzarides, 1996; Ogryzko et al., 1996), P/CAF (Ogryzko et al., 1996), TAFII250 (Mizzen et al., 1996) and GCN5 (Kuo et al., 1996; Wang et al., 1997), possess histone acetyltransferase (HAT) activity. The HAT activity is important in the regulation of transcription, as hyperacetylation of the N-terminal histone tails correlates with transcriptional activity (Hebbes et al., 1988; Wolffe and Pruss, 1996; Grunstein, 1997; Struhl, 1998). In addition, several of these HAT proteins have also been shown to acetylate non-histone proteins such as transcription factors.
The acetylation of transcription factors and factors related to transcription has been shown to be an important regulatory mechanism of transcription. Acetylation of these factors leads to changes in protein–protein and protein–DNA interactions which subsequently can result in increased or decreased transcription (Sterner and Berger, 2000). An example of impaired transcription is the acetylation by CBP of the high mobility group protein HMGI(Y), a component of the interferon-β (IFN-β) enhanceosome, leading to disruption of the enhanceosome and abrogation of IFN-β transcription (Munshi et al., 1998). In addition, acetylation of the Drosophila transcription factor dTCF by dCBP has been shown to disrupt dTCF interaction with Armadillo and thereby result in repression (Waltzer and Bienz, 1998). In addition, nuclear hormone receptors have been shown to be regulated negatively by the acetylation of the p160 coactivator protein ACTR (Chen et al., 1999).
So far, the mechanism of E1A repression of the collagenase promoter via the c-Jun DBD has been unclear. The aim of this study was to find the mechanism of repression of c-Jun by E1A. We find that E1A is not a general repressor of the collagenase promoter as it does not repress the collagenase promoter activated by the AP1-related factor Epstein–Barr virus transcription factor EB1 (Farrell et al., 1989; Giot et al., 1991), a factor also activated by CBP (Adamson and Kenney, 1999). This study identifies a 12 amino acid motif within the basic region of the c-Jun DBD as being required for the repression by E1A. We investigated the involvement of p300 acetyltransferase activity in this process and found c-Jun to be acetylated in vivo. Mutational analysis of lysine residues identified Lys271 as being essential for the repression by E1A. In addition, Lys271 is acetylated by p300 in vitro, and we found that this residue is also acetylated in vivo. Taken together, our results suggest that repression of the collagenase promoter by E1A occurs via the acetylation of the basic region of c-Jun.
Results
Transcription factor-specific repression by E1A
The E1A protein is considered to be a transcriptional repressor of several cellular genes with 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-responsive element (TRE)-containing promoters (Offringa et al., 1988, 1990; van Dam et al., 1989; Frisch et al., 1990; Hoffman et al., 1993). The collagenase promoter is an example of a promoter induced via its TRE by TPA (Angel et al., 1987) and repressed by E1A (Offringa et al., 1990). c-Jun homodimers and c-Jun–c-Fos heterodimers are transcription factors shown to be important for TRE-dependent activation and repression (Frisch et al., 1990; Offringa et al., 1990). In addition to these cellular transcription factors, the Epstein–Barr virus transcription factor EB1 also activates the collagenase promoter (Farrell et al., 1989; Giot et al., 1991).
To examine the ability of E1A to repress the collagenase promoter activated by c-Jun or EB1, we transiently transfected F9 cells, which do not contain endogenous c-Jun, with c-Jun and EB1. Indeed, both c-Jun and EB1 activated the co-transfected collagenase promoter (Figure 1A). However, there is a striking difference in the capacity of E1A to inhibit the resulting transcription from these two activators. Co-transfection of E1A with c-Jun results in repression of the collagenase promoter while E1A does not significantly inhibit the EB1-activated collagenase promoter (Figure 1A). The small decrease in transcription observed with co-transfection of EB1 and E1A is also observed in the absence of a transcription factor (Figure 1A and C). These results show that E1A is not a general repressor of the collagenase promoter but that repression is specific for the transcription factor that activates the promoter. We used this differential regulation by E1A of c-Jun and EB1 to identify the requirements for repression.

Fig. 1. Transcription factor-specific repression of the collagenase promoter by E1A requires a 12 amino acid region in the c-Jun basic region. (A) E1A specifically represses the c-Jun- and not the EB1-activated collagenase promoter (+517/–63). F9 cells were transfected with collagenase promoter–luciferase reporter and c-Jun or EB1 in the absence (black bars) or presence (white bars) of E1A. The average fold repression ± SD by E1A of six experiments is shown below the graph. (B) Schematic representation of c-Jun–EB1 chimeras. Full-length c-Jun with its functional domains: the N-terminal transactivation domain (TAD) and the C-terminal DNA-binding domain (DBD) consisting of the basic region (BR) and the leucine zipper (LZ). The chimeras of c-Jun (dark grey) and EB1 (light grey) are depicted, showing the basic region (BR) and dimerization domain (DD) enlarged. (C) Luciferase activity after co-transfection of the collagenase promoter–luciferase reporter with the c-Jun/EB1 chimeras in F9 cells. On the left the activity of the chimeras on the collagenase promoter is shown. On the right the fold repression by E1A of the collagenase promoter–luciferase reporter activated with the chimeras is shown. (D) Schematic representation of EB1/c-JunBR-C, which consists of EB1 with the 12 amino acid sequence of c-Jun substituting for the indicated homologous EB1 sequence.
Repression by E1A requires a 12 amino acid region in the c-Jun basic region
The inability of E1A to repress the EB1-activated collagenase promoter allowed us to study transcription factor-specific characteristics required for this repression. We therefore used chimeras of c-Jun and EB1 (Giot et al., 1991; Castellazzi et al., 1993) to identify the region of c-Jun required for repression by E1A more specifically (Figure 1B). The c-Jun/EB1 chimeras used in transient transfections of F9 cells all activate the collagenase promoter (Figure 1C). Repression by E1A is only observed when the collagenase promoter is activated by a hybrid transcription factor that includes the basic region of c-Jun (EB1/c-JunBR, Figure 1C). Further analysis of the c-Jun basic region present in the chimeric EB1/c-JunBR protein demonstrates that transcription by EB1 when the N-terminal half of its basic region is replaced by the c-Jun sequence is not repressed. However, exchanging the C-terminal half of the basic region of EB1 for the c-Jun sequence creates an E1A-repressible transcription factor (EB1/c-JunBR-N/-C, Figure 1C). Thus, a 12 amino acid region within the c-Jun basic region is necessary and sufficient for repression by E1A.
c-Jun is acetylated in vivo
The identification of the region of c-Jun required for repression by E1A allowed us to study the mechanism of repression further. Previously, the interaction of E1A with p300 was shown to be essential for repression of the collagenase promoter (Dorsman et al., 1997). As p300 was shown to bind to the TAD of c-Jun (Lee et al., 1996; Duyndam et al., 1999), this seems contradictory to the finding that the repression by E1A requires the c-Jun DBD. To verify whether under our experimental conditions the interaction of E1A with p300 is necessary in order to repress the collagenase promoter, we studied the repression by the E1A mutant RG2. Indeed, we found that this E1A mutant, which is incapable of interacting with p300/CBP (Duyndam et al., 1999,Lee et al., 1995), does not repress the c-Jun-activated collagenase promoter (Figure 2). A possible model consistent with the involvement of different domains in c-Jun could be that p300, upon binding to the TAD, can acetylate other parts of c-Jun as it acetylates histones when recruited to the promoter.
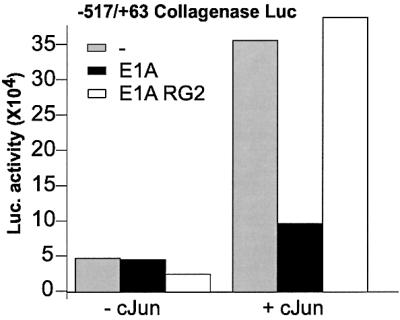
Fig. 2. Repression of the collagenase promoter depends on the p300-binding site of E1A. An E1A mutant, which does not bind p300 does not inhibit c-Jun-activated collagenase promoter. Luciferase activity after co-transfection in F9 cells of collagenase promoter and c-Jun in the absence (grey bar) or presence (black bar) of E1A or in the presence of the E1A RG2 mutant (white bar).
To investigate the involvement of the acetyltransferase activity of p300, we first studied whether c-Jun is acetylated in vivo. Adenovirus type 5-transformed human embryonic retina (Ad5HER) cells were labelled briefly (1 h) with [14C]pyruvate, the biological precursor of acetyl-CoA. Immunoprecipitation with a c-Jun-specific antibody, H79, demonstrated that c-Jun is indeed acetylated in vivo (Figure 3, lane 3). Western analysis with another c-Jun antibody, Ab-1, demonstrates the specificity of this immunoprecipitation for c-Jun (Figure 3, lane 1). The identity of the top band remains to be established, but the molecular weight of the lower band corresponds to that of c-Jun. The labelling of c-Jun is not due to degradation of the pyruvate and incorporation of 14C label into all proteins as E1A immunoprecipated from the same lysates is not labelled (Figure 3, lane 5). The synthesis of both c-Jun and E1A during the labelling time was demonstrated by labelling cells with [35S]methionine followed by immunoprecipitation (Figure 3, lanes 2 and 4). The acetylation of c-Jun in vivo together with the involvement of p300 in the repression of the c-Jun-activated collagenase promoter by E1A suggests that the mechanism of regulation of c-Jun by p300 might include acetylation.

Fig. 3. Acetylation of c-Jun in vivo. c-Jun was immunoprecipitated with the H79 c-Jun antibody from Ad5HER cells. The cells were mock treated or treated with media supplemented with the acetyl-CoA precursor [14C]pyruvate or [35S]methionine for 1 h. The immunoprecipitations were analysed by a western blot with the Ab-1 anti-c-Jun antibody (1) and labelled immunoprecipitations by autoradiography of [35S]methionine (2) and [14C]pyruvate (3). As a control for the incorporation of 14C-label in proteins in general, E1A was immunoprecipitated (5) and no indication for labelling was found. The autoradiogram after SDS–PAGE of the [35S]methionine labelling shows E1A synthesis (4).
c-Jun basic region necessary for repression by E1A is acetylated in vitro
As described above, the repression by E1A involves binding of E1A to the acetyltransferase p300, and the target of repression, c-Jun, is acetylated in vivo. Since repression by E1A occurs via the basic region of c-Jun, we studied whether this region is acetylated by p300. We used bacterially expressed GST fusion proteins of the c-Jun DBD and TAD in in vitro acetylation assays with the GST–HAT domain of p300 (Figure 4A). The acetylation experiments demonstrate that p300 can acetylate the c-Jun DBD in vitro, while the TAD of c-Jun is not acetylated by p300 (Figure 4B). Core histones and p53 were used as positive controls for the acetylation assay (Figure 4B and data not shown).

Fig. 4. Acetylation of c-JunDBD and EB1/c-JunBR-C by p300HAT in vitro. (A) Coomassie stain of SDS–PAGE separation of purified recombinant proteins used in the in vitro acetylation assays. (B) Autoradiogram of the in vitro p300HAT acetylation assay. Histones (Sigma) were used as a positive control. On the right, the acetylation assay with GST–c-JunTAD and GST–c-JunDBD, respectively, is shown. Indicated is the labelled band of GST–c-JunDBD. The top part of this high percentage gel with the autoacetylated p300HAT was cut off. (C) Autoradiogram of an in vitro acetylation assay with GST–p300HAT of GST–EB1 and GST–EB1/c-JunBR-C.
Our transient transfection experiments demonstrated that substitution of only a small part of the DNA-binding basic region of EB1 into the homologous region of c-Jun made the EB1 protein susceptible to E1A repression (Figure 1C). To evaluate whether these data correlated with in vitro acetylation, the DBD of EB1 and the chimera EB1/c-JunBR-C (Figure 4A) were tested in an in vitro acetylation assay. Noticeably, p300 did not acetylate the EB1 DBD while EB1/JunBR-C was acetylated (Figure 4C). The homologous basic regions of both EB1 and c-Jun contain lysine residues (Figure 1D). Therefore, the acetylation requires one or more lysines in the specific amino acid sequence of the basic region of c-Jun. These results show that the region of c-Jun required for repression by E1A corresponds to the region acetylated by p300 in vitro.
Lys271 of c-Jun required for repression by E1A is acetylated in vitro and in vivo
The correlation of repression by E1A of a specific region of c-Jun and acetylation of the same region led us to study the role of the lysine residues in this region as potential targets for the repression by E1A (Figure 1D). The three lysine residues present in the 12 amino acid stretch were replaced by non-acetylatable arginines in order to retain the positive charge while preventing acetylation. The activity of the c-Jun mutants was determined in transient transfections in F9 cells. All mutants activated the collagenase promoter to a level comparable to wild-type c-Jun, indicating that the lysines are not essential for the c-Jun-mediated activation of the collagenase promoter (Figure 5A).

Fig. 5. c-Jun Lys271 is not required for activation but is essential for repression by E1A and is acetylated in vitro and in vivo. (A) Transcription activation of wild-type c-Jun and c-Jun mutants. F9 cells were transiently transfected with a collagenase promoter–luciferase reporter construct and co-transfected with wild-type c-Jun and c-Jun mutants. (B) F9 cells were transiently transfected with the collagenase promoter–luciferase reporter construct and co-transfected with wild-type c-Jun and c-Jun mutants in the presence or absence of co-transfected E1A. The graph shows the fold inhibition by E1A of the different c-Jun mutants. On the right, the expression levels of the c-Jun proteins are shown in the absence or presence of E1A. The expression levels were determined by western analysis of the lysates used to measure luciferase activity, using the 9E10 antibody against a C-terminal c-Myc tag on the c-Jun proteins. (C) Lys271 of c-JunDBD is acetylated in vitro by p300. Acetylation assays were performed with GST–c-Jun proteins shown by Coomassie stain on the right. Autoradiogram of the acetylation reactions after separation by SDS–PAGE for the lysine to arginine mutants is depicted on the left. (D) Immunoprecipitation of transfected myc-tagged wild-type c-Jun, K271R and K3R from [14C]pyruvate-labelled Ad5HER cells. Depicted are the autoradiogram of the 14C-labelled, immunoprecipitated proteins and a direct western analysis, using the 9E10 antibody, of the cell lysates used for the immunoprecipitation.
Subsequently, the repression of the c-Jun mutants by E1A was tested. The collagenase promoter activated by c-JunK268R and c-JunK273R was repressed by E1A to an extent similar to that of the wild-type c-Jun-activated promoter (Figure 5B). Importantly, the collagenase promoter activated by c-JunK271R or c-JunK3R (all three lysines mutated into arginines) was no longer repressed by E1A (Figure 5B). To determine whether this residue is acetylated by p300, the lysine to arginine mutants were tested in in vitro acetylation assays. Mutation of all three lysine residues abolished acetylation of the c-Jun DBD by p300 (c-JunK3R, Figure 5C). Substitution of only Lys273 by arginine did not change the in vitro acetylation of GST–c-Jun, indicating that this is not a target for acetylation. However, substitution of Lys271 abolished acetylation by p300. To a lesser extent, mutation of Lys268 decreased the acetylation by p300 in vitro (Figure 5C), suggesting that it may also be a target for acetylation. Alternatively, mutation of Lys268 may result in decreased acetylation of Lys271. This latter explanation is supported by the relative level of acetylation observed for the c-Jun mutants. Acetylation of c-JunK271R decreases to the same extent as the triple lysine mutant c-JunK3R. The level of acetylation of c-JunK268R, however, remains several fold higher than that of c-JunK271R and c-JunK3R. More important for the repression by E1A is the finding that c-JunK271R harbouring a single point mutation is no longer subject to repression by E1A.
To study whether the in vitro target for acetylation is also acetylated in vivo, we transiently transfected myc-tagged wild-type c-Jun, c-JunK271R and c-JunK3R in the E1A-expressing Ad5HER cells. As seen in Figure 5D, immunopurification of wild-type c-Jun protein showed that it is acetylated, whereas the acetylation of the single and triple point mutants was markedly reduced in vivo. In summary, these results demonstrate that repression of the collagenase promoter by E1A requires Lys271 of c-Jun, a lysine that is acetylated in vitro and in vivo.
TPA induction of collagenase in Ad5HER cells is regained by c-JunK271R expression
Since both the repression by E1A and acetylation of c-Jun require the same residue in the DBD, we compared E1A- and non-E1A-expressing cells for c-Jun acetylation and c-Jun-mediated gene activation. First, we studied whether c-Jun is hypoacetylated in cells that do not express E1A. For this experiment, the E1A-expressing Ad5HER cells were compared with the primary human fibroblast VH10, as this cell line expresses relatively high levels of c-Jun. After TPA stimulation and labelling with [14C]pyruvate, immunoprecipitation of c-Jun revealed the presence of 14C-labelled c-Jun in the Ad5HER cells but not in VH10 cells (Figure 6A). Expression levels of c-Jun were found to be comparable in both cell lines, as seen in the [35S]methionine labelling also shown in Figure 6A. Thus, consistent with the model that E1A can cause acetylation of c-Jun, non-E1A-expressing cells do not show acetylated c-Jun.
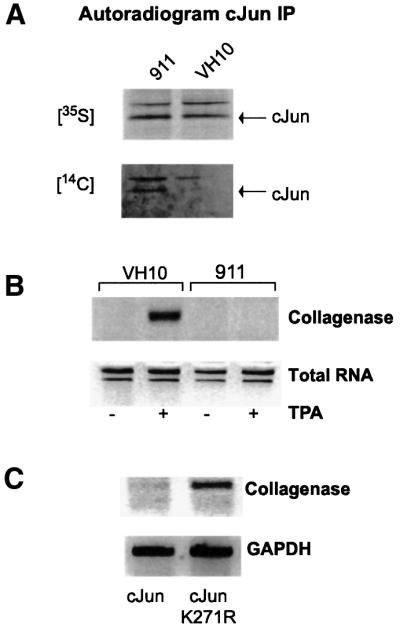
Fig. 6. Differential acetylation and regulation of c-Jun on the collagenase promoter between non-E1A-containing VH10 cells and E1A-expressing Ad5HER cells. (A) Acetylation of c-Jun in VH10 and Ad5HER cells. The cells were pre-treated with TPA 5 h before labelling. Cells were labelled by supplementing the media with the acetyl-CoA precursor [14C]pyruvate or [35S]methionine for 1 h. c-Jun was immunopurified from the extract with the c-Jun-specific H79 antibody and analysed by autoradiography after SDS–PAGE. (B) Endogenous collagenase expression in VH10 and Ad5HER cells. The cells were mock treated or TPA treated 6 h before lysis. Levels of endogenous collagenase mRNA are shown as determined by RT–PCR and visualized by UV after separation on an agarose gel. As a control, 18S and 28S rRNA levels are shown representing the amount of total RNA. (C) Endogenous collagenase expression in Ad5HER cells after transfection with c-Jun or c-JunK271R. Ad5HER cells were transiently transfected with c-Jun or c-JunK271R and stimulated with TPA. Levels of endogenous collagenase mRNA determined by RT–PCR are shown (upper panel). As a control, levels of GAPDH mRNA are depicted (lower panel).
To add in vivo significance to the acetylation and repression via c-Jun Lys271 on the collagenase promoter, expression of the endogenous collagenase gene after TPA stimulation was studied in the Ad5HER and VH10 cells. Treatment of VH10 cells with TPA resulted in expression of collagenase mRNA shown by RT–PCR, as was observed previously in other cell lines (e.g. Angel et al., 1987). In contrast, the E1A-expressing Ad5HER cells did not express detectable levels of collagenase mRNA after TPA stimulation (Figure 6B).
The experiments described above show a difference in the expression levels of a TPA-responsive gene as well as a difference in the acetylation state of c-Jun between an E1A- and a non-E1A-expressing cell line. To understand whether there is indeed a role for the acetylation of Lys271 in this differential regulation of the collagenase gene, we transfected c-Jun or c-JunK271R in the Ad5HER cells. As expected, upon TPA stimulation, collagenase expression was not enhanced in the wild-type c-Jun-transfected cells (Figure 6C). Strikingly, in the cells transfected with the non-acetylatable c-JunK271R, TPA induced expression of the endogenous collagenase gene (Figure 6C). These data show that Lys271 of c-Jun, which is acetylated in vitro and in vivo, is required for the repression by E1A, and mutation of this residue relieves the block on TPA-induced collagenase expression in E1A-expressing cells.
Discussion
E1A has been shown to be a transcriptional regulator for numerous cellular genes; however, little is known about the mechanism of regulation. The present study shows that E1A can inhibit the collagenase promoter in a transcription factor-specific manner. Although the TADs of EB1 and c-Jun are both bound and activated by p300/CBP, only c-Jun activation is repressed by E1A, a repression dependent on the p300/CBP-binding site of E1A. Using chimeras of c-Jun and EB1, we determined that E1A requires a small 12 amino acid stretch in the basic region of c-Jun to repress activation of the promoter. The requirement for the interaction of E1A with p300 for repression of transcription by c-Jun, together with the observation that c-Jun is acetylated in vivo, prompted us to study the role of the lysines present in the 12 amino acid sequence. Comparison of the acetylation of EB1 and EB1 with the 12 amino acids of c-Jun showed that acetylation was specific for the lysine residues of c-Jun. Mutational analysis of the lysine residues showed that Lys271 is not required for activation of c-Jun but is essential for the repression by E1A. In addition, in vitro acetylation experiments identified the c-Jun basic region as an acetylation target of p300. This acetylation occurred on Lys271 both in vitro and in vivo. Importantly, E1A-expressing cells, which cannot be induced by TPA to express endogenous collagenase, can be induced by TPA when transfected with c-JunK271R but not wild-type c-Jun. Therefore, the data suggest that E1A represses the collagenase promoter via acetylation of Lys271 of the c-Jun basic region.
The c-Jun proto-oncogene plays a decisive role in many cellular processes. To accomplish specificity in the regulation of all these processes, c-Jun is tightly regulated by specific dimer formation and phosphorylation (reviewed in Karin et al., 1997) and, as this study demonstrates, also by acetylation. The specificity of repression of c-Jun activation of the collagenase promoter by E1A indicates that the regulation is more complicated than the mere disruption of transcription activator complexes containing p300/CBP. The involvement of p300 in the repression by E1A and the acetylation state of c-Jun in vivo and in vitro suggest that E1A accomplishes this specificity by regulating cellular acetyltransferases. The data concerning the role of E1A in the regulation of activity of HAT enzymes so far have been inconclusive. In vitro inhibition of p300/CBP HAT activity by E1A has been shown previously (Chakravarti et al., 1999; Hamamori et al., 1999; Perissi et al., 1999), while stimulation of CBP HAT activity by E1A has been described in cell cycle progression (Ait-Si-Ali et al., 1998). A more detailed study showed that high levels of E1A inhibited acetylation of histones H3 and H4, while lower levels of E1A stimulated acetylation (Li et al., 1999). These concentrations of E1A also increase acetylation of MyoD, while acetylation of TFIIE, TFIIF (RAP74) and TR-RXR is inhibited. Finally, it was described that E1A has no effect on the histone acetylation activity of p300/CBP (Bannister and Kouzarides, 1996; Kraus et al., 1999). Therefore, E1A may have a differential effect on acetyltransferase activity depending on the substrate or target promoter.
Acetylation of proteins can have both stimulatory and inhibitory effects on transcription (Imhof et al., 1997; Korzus et al., 1998). The acetylation of histones and several transcription factors such as p53 and MyoD enhances transcription of target genes (Sakaguchi et al., 1998; Sartorelli et al., 1999). On the other hand, acetylation of dTCF (Waltzer and Bienz, 1998), HMGI(Y) (Munshi et al., 1998) and ACTR (Chakravarti et al., 1999) results in decreased transcription. Actually, in these examples, acetylation plays a dual role in the regulation of transcription: acetylation of histones stimulates transcription whereas subsequent acetylation of HMGI(Y) and ACTR switches off transcription of the IFN-β promoter (Agalioti et al., 2000) and estrogen receptor (ER) target genes, respectively. A similar model can be implied for the regulation of the collagenase promoter by p300/CBP. Previous studies have shown that both p300 and CBP have a stimulatory effect on transactivation by c-Jun (Bannister et al., 1995; Lee et al., 1996). As both proteins are transcription adaptors and possess acetyltransferase activity, activation is thought to be the result of both complex formation on the promoter and histone acetylation. According to this model, p300/CBP would interact with the TAD of c-Jun and subsequently stimulate the promoter via acetylation of histones, making the chromatin accessible for transcription. In the presence of E1A, the acetylation activity of p300/CBP is targeted towards the DBD of c-Jun, and its acetylation causes the repression of the collagenase promoter. The finding that the mutant K271R does not interfere with c-Jun-mediated activation of the collagenase promoter supports the hypothesis that acetylation of this residue is not involved in the activation process. Whether acetylation of c-Jun is also part of the cellular mechanism to shut off the promoter, analogous to the regulation of the IFN-β promoter and ER target genes, remains to be established.
The crystal structure of the c-Jun–c-Fos–DNA complex (Glover and Harrison, 1995; Chen et al., 1998) shows that Lys271 is not in contact with the DNA, but actually protrudes out away from the DNA (Figure 7). Thus, acetylation of Lys271 is not likely to interfere with DNA binding, and indeed the in vitro DNA-binding affinity of mutant c-Jun was not reduced (data not shown). A possible model is that non-acetylated c-Jun may interact with other factors, resulting in stabilization of the promoter complex on the DNA. Acetylation of c-Jun may compromise this interaction and therefore affect the stability of the complex, resulting in inhibition of transcription.

Fig. 7. Location of Lys271 of c-Jun. Residue 271, acetylated by p300, protrudes away from the c-Jun–c-Fos–DNA complex within a RasMol representation of the structure (Chen et al., 1998). The figure shows the ribbon structure of the basic region and zipper domain of c-Jun and c-Fos bound to double-stranded DNA. Lys271 is shown as space filled. The two pictures differ by 90° rotation of the structure.
Materials and methods
Cell culture and transient transfections
F9 cells were grown in F12–Dulbecco’s modified Eagle’s medium (DMEM) (1:1) supplemented with 10% fetal calf serum (FCS), penicillin (100 µg/ml), streptomycin (100 µg/ml) and 0.1 mM β-mercaptoethanol. F9 cells were transfected on 3 cm dishes by the calcium phosphate method (Graham and van der Eb, 1973). After 6 h, cells were washed in HEPES-buffered saline (HBS) and supplemented with culture media. At 24 or 10 h after transfection, cells were washed in HBS and harvested in 250 µl of lysis buffer [25 mM Tris-phosphate pH 7.8, 2 mM dithiothreitol (DTT), 2 mM 1,2-diaminocyclohexane-N,N,N′N′ tetraacetic acid, 10% glycerol, 1% Triton X-100]. Luciferase activity was determined according to the manufacturer’s protocol (Promega). For the transient transfections, we used 500 ng of reporter plasmid, 500 ng of the Rous sarcoma virus (RSV)-driven transcription factors, 200 ng of the pcDNA constructs of c-Jun and between 125 and 400 ng of E1A plasmid. Experiments shown in Figures 1 and 2 were obtained with RSV-driven constructs (24 h transfection). Data in Figures 5 and 6 were obtained with pcDNA constructs (10 h transfection). The data presented are representative of at least three separate experiments. Results similar to those shown in Figure 5 were obtained with an RSV construct expressing the c-Jun mutants.
Ad5HER cells, human embryonic retina cells transformed with the E1 region of adenovirus type 5 (Fallaux et al., 1996), and human diploid foreskin fibroblasts VH10 (Klein et al., 1990) were grown in DMEM supplemented with 10% FCS, penicillin (100 µg/ml) and streptomycin (100 µg/ml). Ad5HER cells were transfected on 9 cm dishes with the calcium phosphate method described above. The cells were harvested 48 h later, after 1 h 14C labelling or 6 h TPA treatment depending on the experiment.
Plasmids and recombinant proteins
c-Jun, EB1 and EB1/c-Jun chimeras used for transient transfection were cloned into a pUC18-derived vector driven by the RSV promoter. The EB1/c-JunBR chimeras were described previously as ZJ, ZJA and ZJB (Giot et al., 1991). The c-JunEB1 construct consists of amino acids 1–187 of c-Jun and amino acids 140–245 of EB1. c-Jun was also cloned into the EcoRI site of pcDNA3.1(–)/myc-HisB after PCR of the human c-Jun coding sequence from pRSV-c-Jun described in Offringa et al. (1990). c-Jun point mutants were constructed in this pcDNA vector using the QuikChange site-directed mutagenesis method according to the manufacturer’s protocol (Stratagene). The collagenase promoter sequence (–517/+63), as described in Offringa et al. (1990), was cloned in front of the luciferase reporter gene of pGL3 (Promega). Expression constructs for 12S-E1A and 12S-E1A RG2 were obtained from E.Moran (Wang et al., 1993). DNA sequences for recombinant GST proteins were cloned into a derivative of pGEX-2T (Pharmacia), pGEX265 (Angel et al., 1987). GST–p300HAT (amino acids 1195–1673) was made by PCR of the fragment with additional 5′-KpnI and 3′-SpeI sites. GST–c-JunDBD consists of a deletion of amino acids 6–194 out of full-length c-Jun and insertion of the 5′-SalI–3′-EcoRI fragment into the GST vector. GST–c-JunTAD (amino acids 1–166) was created by PCR adding a 5′-SmaI and a 3′-EcoRI site. GST–EB1 (amino acids 141–245) and GST–EB1/c-JunBR-C (amino acids 141–245) were obtained by PCR of the fragments from the full-length sequence. GST–c-Jun point mutants were obtained using the QuikChange site-directed mutagenesis method on the wild-type GST–c-JunBR. All sequences were confirmed by sequence analysis.
Recombinant protein preparation and purification
GST fusion proteins were prepared from Escherichia coli DH5α extracts, except for GST–p300HAT, which was grown in E.coli BL21. Bacterial cultures were grown to OD600 nm 0.6 and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 25°C. After 3 h, cells were spun down and resuspended in lysis buffer (25 mM HEPES–KOH pH 7.6, 0.5 M NaCl, 0.1% NP-40, 0.1 mM EDTA, 12.5 mM MgCl2, 10% glycerol, supplemented with protease inhibitors). Cells were freeze–thawed twice, incubated with 0.5 mg/ml lysozyme for 30 min at 4°C and sonicated for 10 s on ice. The cell lysate was spun down and the supernatant incubated with glutathione beads (Pharmacia) for 2 h at 4°C. After washing four times in lysis buffer, GST fusion proteins were eluted from the beads with 20 mM glutathione for 4 h at 4°C. GST–p300HAT, GST–EB1 and GST–EB1/c-JunBR-C were used directly in acetylation assays. GST–c-JunWT and GST-c-Jun point mutants were purified further on ion exchange columns (SP Sepharose fast flow, Pharmacia). Proteins were eluted with 700 mM KCl in the acetylation reaction buffer described below and dialysed for 16 h at 4°C against the reaction buffer without KCl.
In vitro acetylation assays
In vitro acetylation assays were performed with equal molar amounts of substrate proteins as judged from Coomassie-stained gels after SDS–PAGE. The amounts of GST–p300HAT were also determined by Coomassie staining of gels after SDS–PAGE. Histones were used at a concentration of 25 µg/assay (Sigma). The reaction was performed in a reaction buffer (20 mM Tris–HCl 7.9, 60 mM NaCl, 2 mM EDTA, 0.2% NP-40, 1 mM DTT and protease inhibitors) supplemented with HAT enzyme and substrate in the presence of 0.05 µCi of [1-14C]acetyl-CoA (specific activity 52 mCi/mmol, Amersham). Reactions were performed at 37°C for 30 min and stopped by adding 2× sample buffer [4% SDS, 20% glycerol, 125 mM Tris (pH 6.8), 2% β-mercaptoethanol, bromophenol blue]. The reaction mixture was then subjected to SDS–PAGE and analysed by autoradiography.
In vivo labelling
Ad5HER or VH10 cells were grown in 9 cm dishes. Transfection for the labelling of the c-Jun mutants was carried out 48 h before labelling. Before labelling, the cells were washed in minimal essential medium supplemented with 2% dialysed FCS (Gibco-BRL). For the [35S]methionine labelling, an amino acid mix without methionine was added; for cold c-Jun immunoprecipitation and [14C]pyruvate labelling, all amino acids were added (Gibco-BRL). Cells were then supplemented with the medium described above and 50 µCi of [14C]pyruvate or [35S]methionine were added to 2.5 ml of medium on 5 cm dishes. Cells were incubated for 1 h and then harvested in a buffer containing 25 mM HEPES–KOH (pH 7.0), 250 mM NaCl, 2.5 mM EDTA, 1% NP-40, 1 mM DDT and protease inhibitors. Extracts were used for immunoprecipitation.
Antibodies and immunoprecipitations
c-Jun rabbit polyclonal antibodies H79 and Ab-1 were purchased from Santa Cruz and Calbiochem, respectively. The c-Myc mouse monoclonal antibody, 9E10, was described before by Evan et al. (1985). E1A precipitation was performed with monoclonal mouse antibody M73, a gift from E.Harlow (Massachusetts General Hospital Cancer Center, Charleston, MA). Immunoprecipitation assays were performed by binding the antibodies to protein A–Sepharose beads. Subsequently, the lysates were incubated with the antibody-coupled beads for 2 h. Samples were washed and separated on 10% gels by SDS–PAGE. The signal was enhanced by incubation of the gels in PPO/DMSO for 2 h. The proteins were then visualized by autoradiography.
RT–PCR
RNA was isolated using the NP-40 method (Sambrook et al., 1989). cDNA was then prepared with random hexamers and Superscript Reverse TranscriptaseII (Gibco-BRL). PCR was performed using 100 pmol of primer, 250 µM dNTPs and 1.25 U of Pfu (Stratagene). Thirty cycles of 1 min at 94, 60 and 72°C were carried out, and samples were separated on agarose gels and visualized by UV light. Primers: for collagenase, primers directed against two different exons (exons 2 and 3) of the genomic sequence were used so that possible DNA contamination of the sample would not interfere with the mRNA signal: Coll-Exon2pr, GGCATGGTCCACATCTGCTC; and Coll-Exon3pr, CTCACGGACTACACCGAGTC. The primers for the GAPDH control were AATCCCATCACCATCTTCC and ATGAGTCCTTCCACGATACC.
Acknowledgments
Acknowledgements
We thank E.Harlow for the M73 antibody, M.Castellazzi and A.Sergeant for the plasmids of c-JunEB1 chimeras, E.Moran for the E1A and mutant E1A plasmids, N.de Ruiter for the GST–c-JunK268R and K271R plasmids, M.Duyndam and Paul H.Smits for their assistance and initial experiments, and H.van Dam, T.Mahmoudi, A.J.van der Eb and C.P.Verrijzer for useful discussions. This work was funded by the Chemical Sciences Council of the Netherlands Organization for Scientific Research (NWO-CW), with support of EC grants (TMR CT96-0044 and Biomed CT97-2567). and E.K. was supported by the Dutch Cancer Society (KWF).
References
- Adamson A.L. and Kenney,S. (1999) The Epstein–Barr virus BZLF1 protein interacts physically and functionally with the histone acetylase CREB-binding protein. J. Virol., 73, 6551–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agalioti, T., Lomvardas,S., Parekh,B., Yie,J., Maniatis,T., and Thanos,D. (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell, 103, 667–678. [DOI] [PubMed] [Google Scholar]
- Ait-Si-Ali S. et al. (1998) Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature, 396, 184–186. [DOI] [PubMed] [Google Scholar]
- Angel, P. and Karin,M. (1991) The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta, 1072, 129–157. [DOI] [PubMed] [Google Scholar]
- Angel P., Baumann,I., Stein,B., Delius,H., Rahmsdorf,H.J. and Herrlich,P. (1987) 12-O-tetradecanoyl-phorbol-13-acetate induction of the human collagenase gene is mediated by an inducible enhancer element located in the 5′-flanking region. Mol. Cell. Biol., 7, 2256–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z., Newsome,D., Oldread,E., Livingston,D.M. and Eckner,R. (1995) A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature, 374, 81–84. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Oehler,T., Wilhelm,D., Angel,P. and Kouzarides,T. (1995) Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene, 11, 2509–2514. [PubMed] [Google Scholar]
- Castellazzi M., Loiseau,L., Piu,F. and Sergeant,A. (1993) Chimeric c-Jun containing an heterologous homodimerization domain transforms primary chick embryo fibroblasts. Oncogene, 8, 1149–1160. [PubMed] [Google Scholar]
- Chakravarti D., Ogryzko,V., Kao,H.Y., Nash,A., Chen,H., Nakatani,Y. and Evans,R.M. (1999) A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell, 96, 393–403. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Xie,W., Wilpitz,D. and Evans,R.M. (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell, 98, 675–686. [DOI] [PubMed] [Google Scholar]
- Chen L., Glover,J.N., Hogan,P.G., Rao,A. and Harrison,S.C. (1998) Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature, 392, 42–48. [DOI] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok,R.P., Lamb,N., Hagiwara,M., Montminy,M.R. and Goodman,R.H. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Dorsman J.C., Hagmeyer,B.M., Veenstra,J., Elfferich,P., Nabben,N., Zantema,A. and van der Eb,A.J. (1995) The N-terminal region of the adenovirus type 5 E1A proteins can repress expression of cellular genes via two distinct but overlapping domains. J. Virol., 69, 2962–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsman J.C., Teunisse,A.F., Zantema,A. and van der Eb,A.J. (1997) The adenovirus 12 E1A proteins can bind directly to proteins of the p300 transcription co-activator family, including the CREB-binding protein CBP and p300. J. Gen. Virol., 78, 423–426. [DOI] [PubMed] [Google Scholar]
- Duyndam M.C., van Dam,H., Smits,P.H., Verlaan,M., van der Eb,A. and Zantema,A. (1999) The N-terminal transactivation domain of ATF2 is a target for the co-operative activation of the c-jun promoter by p300 and 12S E1A. Oncogene, 18, 2311–2321. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Evan, I.E., Lewis,G.K., Ramsay,G. and Bishop,J.M. (1985) Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol., 5, 3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallaux F.J., Kranenburg,O., Cramer,S.J., Houweling,A., van Ormondt,H., Hoeben,R.C. and van der Eb,A.J. (1996) Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther., 7, 215–222. [DOI] [PubMed] [Google Scholar]
- Farrell P.J., Rowe,D.T., Rooney,C.M. and Kouzarides,T. (1989) Epstein–Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. EMBO J., 8, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Reich,R., Collier,I.E., Genrich,L.T., Martin,G. and Goldberg,G.I. (1990) Adenovirus E1A represses protease gene expression and inhibits metastasis of human tumor cells. Oncogene, 5, 75–83. [PubMed] [Google Scholar]
- Giordano A. and Avantaggiati,M.L. (1999) p300 and CBP: partners for life and death. J. Cell. Physiol., 181, 218–230. [DOI] [PubMed] [Google Scholar]
- Giot J.F., Mikaelian,I., Buisson,M., Manet,E., Joab,I., Nicolas,J.C. and Sergeant,A. (1991) Transcriptional interference between the EBV transcription factors EB1 and R: both DNA-binding and activation domains of EB1 are required. Nucleic Acids Res., 19, 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.N. and Harrison,S.C. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos–c-Jun bound to DNA. Nature, 373, 257–261. [DOI] [PubMed] [Google Scholar]
- Graham ,F.L. and van der Eb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology, 52, 456–467 [DOI] [PubMed] [Google Scholar]
- Grunstein M. (1997) Histone acetylation in chromatin structure and transcription. Nature, 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Hagmeyer B.M., Konig,H., Herr,I., Offringa,R., Zantema,A., van der Eb,A., Herrlich,P. and Angel,P. (1993) Adenovirus E1A negatively and positively modulates transcription of AP-1 dependent genes by dimer-specific regulation of the DNA binding and transactivation activities of Jun. EMBO J., 12, 3559–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmeyer B.M., Angel,P. and van Dam,H. (1995) Modulation of AP-1/ATF transcription factor activity by the adenovirus-E1A oncogene products. BioEssays, 17, 621–629. [DOI] [PubMed] [Google Scholar]
- Hamamori Y., Sartorelli,V., Ogryzko,V., Puri,P.L., Wu,H.Y., Wang,J.Y., Nakatani,Y. and Kedes,L. (1999) Regulation of histone acetylCell, 96, 405–413. [DOI] [PubMed] [Google Scholar]
- Hebbes T.R., Thorne,A.W. and Crane-Robinson,C. (1988) A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J., 7, 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof A., Yang,X.J., Ogryzko,V.V., Nakatani,Y., Wolffe,A.P. and Ge,H. (1997) Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol., 7, 689–692. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu,Z. and Zandi,E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol., 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Klein B., Pastink,A., Odijk,H., Westerveld,A. and van der Eb,A.J. (1990) Transformation and immortalization of diploid xeroderma pigmentosum fibroblasts. Exp. Cell Res., 191, 256–262. [DOI] [PubMed] [Google Scholar]
- Korzus E., Torchia,J., Rose,D.W., Xu,L., Kurokawa,R., McInerney,E.M., Mullen,T.M., Glass,C.K. and Rosenfeld,M.G. (1998) Transcrip tion factor-specific requirements for coactivators and their acetyltransferase functions. Science, 279, 703–707. [DOI] [PubMed] [Google Scholar]
- Kraus, W.L., Manning,E.T., and Kadonaga,J.T. (1999) Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol. Cell. Biol., 19, 8123–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo M.H., Brownell,J.E., Sobel,R.E., Ranalli,T.A., Cook,R.G., Edmondson,D.G., Roth,S.Y. and Allis,C.D. (1996) Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature, 383, 269–272. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Galvin,K.M., See,R.H., Eckner,R., Livingston,D., Moran,E. and Shi,Y. (1995) Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev., 9, 1188–1198. [DOI] [PubMed] [Google Scholar]
- Lee J.S., See,R.H., Deng,T. and Shi,Y. (1996) Adenovirus E1A downregulates c-Jun- and JunB-mediated transcription by targeting their coactivator p300. Mol. Cell. Biol., 16, 4312–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Imhof,A., Collingwood,T.N., Urnov,F.D. and Wolffe,A.P. (1999) p300 stimulates transcription instigated by ligand-bound thyroid hormone receptor at a step subsequent to chromatin disruption. EMBO J., 18, 5634–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizzen C.A. et al. (1996) The TAF(II)250 subunit of TFIID has histone acetyltransferase activity. Cell, 87, 1261–1270. [DOI] [PubMed] [Google Scholar]
- Munshi N., Merika,M., Yie,J., Senger,K., Chen,G. and Thanos,D. (1998) Acetylation of HMG I(Y) by CBP turns off IFN β expression by disrupting the enhanceosome. Mol. Cell, 2, 457–467. [DOI] [PubMed] [Google Scholar]
- Offringa ,R., Smits,A.M., Houweling,A., Bos,J.L. and van der Eb,A.J. (1988) Similar effects of adenovirus E1A and glucocorticoid hormones on the expression of the metalloprotease stromelysin. Nucleic Acids Res., 16, 10973–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offringa R. et al. (1990) A novel function of the transforming domain of E1a: repression of AP-1 activity. Cell, 62, 527–538. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Perissi V., Dasen,J.S., Kurokawa,R., Wang,Z., Korzus,E., Rose,D.W., Glass,C.K. and Rosenfeld,M.G. (1999) Factor-specific modulation of CREB-binding protein acetyltransferase activity. Proc. Natl Acad. Sci. USA, 96, 3652–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sartorelli V., Puri,P.L., Hamamori,Y., Ogryzko,V., Chung,G., Nakatani,Y., Wang,J.Y. and Kedes,L. (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell, 4, 725–734. [DOI] [PubMed] [Google Scholar]
- Smits P.H., de Wit,L., van der Eb,A.J. and Zantema,A. (1996) The adenovirus E1A-associated 300 kDa adaptor protein counteracts the inhibition of the collagenase promoter by E1A and represses transformation. Oncogene, 12, 1529–1535. [PubMed] [Google Scholar]
- Sterner D.E. and Berger,S.L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- van Dam, H., Offringa,R., Smits,A.M., Bos,J.L., Jones,N.C. and van der Eb,A.J. (1989) The repression of the growth factor-inducible genes JE, c-myc and stromelysin by adenovirus E1A is mediated by conserved region 1. Oncogene, 4, 1207–1212. [PubMed] [Google Scholar]
- Vogt P.K. and Bos,T.J. (1990) Jun: oncogene and transcription factor. Adv. Cancer Res., 55, 1–35. [DOI] [PubMed] [Google Scholar]
- Waltzer L. and Bienz,M. (1998) Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature, 395, 521–525. [DOI] [PubMed] [Google Scholar]
- Wang H.G., Rikitake,Y., Carter,M.C., Yaciuk,P., Abraham,S.E., Zerler,B. and Moran,E. (1993) Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol., 67, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Mizzen,C., Ying,C., Candau,R., Barlev,N., Brownell,J., Allis,C.D. and Berger,S.L. (1997) Histone acetyltransferase activity is conserved between yeast and human GCN5 and is required for complementation of growth and transcriptional activation. Mol. Cell. Biol., 17, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe A.P. and Pruss,D. (1996) Targeting chromatin disruption: transcription regulators that acetylate histones. Cell, 84, 817–819. [DOI] [PubMed] [Google Scholar]