

Abstract
The T and natural killer (NK) cell-specific gene SAP (SH2D1A) encodes a ‘free SH2 domain’ that binds a specific tyrosine motif in the cytoplasmic tail of SLAM (CD150) and related cell surface proteins. Mutations in SH2D1A cause the X-linked lymphoproliferative disease, a primary immunodeficiency. Here we report that a second gene encoding a free SH2 domain, EAT-2, is expressed in macrophages and B lympho cytes. The EAT-2 structure in complex with a phosphotyrosine peptide containing a sequence motif with Tyr281 of the cytoplasmic tail of CD150 is very similar to the structure of SH2D1A complexed with the same peptide. This explains the high affinity of EAT-2 for the pTyr motif in the cytoplasmic tail of CD150 but, unlike SH2D1A, EAT-2 does not bind to non-phosphorylated CD150. EAT-2 binds to the phosphorylated receptors CD84, CD150, CD229 and CD244, and acts as a natural inhibitor, which interferes with the recruitment of the tyrosine phosphatase SHP-2. We conclude that EAT-2 plays a role in controlling signal transduction through at least four receptors expressed on the surface of professional antigen-presenting cells.
Keywords: antigen-presenting cells/crystal structure/EAT-2/SAP/XLP
Introduction
The SH2D1A (or SAP) gene encodes a 15 kDa protein whose absence or mutation causes X-lymphoproliferative (XLP) primary immunodeficiency (Coffey et al., 1998; Nichols et al., 1998; Sayos et al., 1998), a disease characterized by an extreme sensitivity to infection with Epstein–Barr virus (EBV) (Purtilo et al., 1975; Hamilton et al., 1980; Seemayer et al., 1995; Sullivan, 1999; Howie et al., 2000; Morra et al., 2001a). Both T and natural killer (NK) cell dysfunctions have been observed in XLP patients (Sullivan et al., 1980; Lanier, 1998; Benoit et al., 2000; Parolini et al., 2000). Uniquely, the SH2D1A protein comprises only a single SH2 domain with a 26 C-terminal amino acid tail (Coffey et al., 1998; Nichols et al., 1998; Sayos et al., 1998). SH2D1A, which is expressed in T and NK cells (Nagy et al., 2000), binds to a motif [TIpYxx(V/I)] in the cytoplasmic tail of SLAM (CD150) (Sayos et al., 1998), 2B4 (CD244) (Lanier, 1998; Tangye et al., 1999; Parolini et al., 2000; Sayos et al., 2000), Ly-9 (CD229) and CD84 (Sayos et al., 2001) via its SH2 domain. Classically, SH2 domain binding depends upon phosphorylation of the tyrosine in the ligand and requires additional contacts C-terminal to the pTyr, usually at the +3 position. Characteristically, SH2D1A uses a ‘three-pronged’ modality of binding to the Tyr281 motif of CD150 (Sayos et al., 1998; Li et al., 1999; Poy et al., 1999), where residues N-terminal to the phosphotyrosine, Ile (–1) and Thr (–2), interact in a specific manner with the β-pleated sheet βD and with the tyrosine pocket of SH2D1A, respectively (see Figure 3A and B for SH2 domain nomenclature). SH2D1A can bind to the unphosphorylated cytoplasmic tail of CD150 (Sayos et al., 1998), and it blocks recruitment of the SHP-2 phosphatase to the tail of phosphorylated CD150 (Sayos et al., 1998), CD244 (Tangye et al., 1999; Sayos et al., 2000), CD84 and CD229 (Sayos et al., 2001). Recently, SH2D1A has been shown to bind to a 62 kDa phosphoprotein adaptor (p62dok) (Sylla et al., 2000).





Fig. 3. Structure of mouse EAT-2–CD150 phosphopeptide complex. (A) Structure-based sequence comparison of EAT-2 with other SH2 domains. Upper panel: the human and mouse EAT-2 and SH2D1A protein sequences are compared. Yellow areas, identical residues; green areas, blocks of similarity; blue areas, conserved positions. Exon boundaries are indicated (Ex, exon). Elements of secondary structure are indicated at the top and labeled using the standard SH2 domain nomeclature (Eck et al., 1993). Key residues for the peptide–SH2 domain interactions are indicated by the symbol ‘+’. Amino acid substitutions found in XLP patients are indicated at the bottom. An arrow indicates Cys15 of EAT-2 and Gly16 of SH2D1A. Lower panel: the mouse EAT-2 SH2 domain is compared with the SH2 domain of the human inositol polyphosphate-5-phosphatase (h SHIP), viral Abelson leukemia oncogene (v abl), Rous sarcoma virus oncogene (v src), human tyrosine kinase lck (h lck) and human tyrosine phosphatase SHP-2 (h SHP-2). Blocks of color indicate similarity or identity as indicated in the above panel. (B) Ribbon diagram showing the EAT-2 SH2 domain in complex with the CD150 phosphopeptide. The bound phosphopeptide is shown in a stick representation (yellow). Selected EAT-2 residues that form the binding site are shown in blue. The N-terminal residues of the peptide make a parallel β-sheet interaction with strand βD; the side chains of these residues make hydrophobic contacts with Leu49 and Tyr51 in strand βD (see text, and D). Interestingly, R12 (at position αA2), which is conserved in most SH2 domains and generally contributes to phosphotyrosine coordination, does not participate in phosphate binding in the EAT-2 complex. Instead, Arg54 (βD6) hydrogen-bonds with the phosphate group. Similar coordination was described for the SH2D1A SH2 domain–CD150 phosphotyrosine peptide complex (Poy et al., 1999). Interactions C-terminal to the phosphotyrosine are dominated by Val +3pY, which binds in a hydrophobic cleft. (C) Surface representation of the EAT-2 SH2 domain with the bound CD150 pTyr281 peptide. Hydrophobic residues at the –1 and –3 positions of the peptide (in a stick representation) intercalate with hydrophobic and aromatic residues on the surface of the SH2 domain. Thr2 (Thr 279 of SLAM) hydrogen-bonds with Glu16. C-terminal to the phosphotyrosine, Val +3 is buried in a mostly hydrophobic groove. Key residues for the SH2 domain–peptide interaction are represented in light green. (D) Stereo view showing the details of CD150 coordination. Residues 278–286 of the CD150 phosphopeptide are shown in green; EAT-2 residues surrounding the bound peptide are colored yellow. White lines indicate hydrogen bond interactions; red spheres represent ordered water molecules that bridge between the peptide and the SH2 domain. EAT-2 residues are labeled in white; N and C indicate the respective termini of the CD150 peptide. Green arrows indicate the phosphotyrosine and ‘+3’ binding pockets. Note the β-sheet hydrogen bonding pattern between the main chain of residues 49–53 of EAT-2 and the N-terminal residues of the CD150 phosphopeptide. The peptide essentially forms an additional strand in the central β-sheet of EAT-2. (E) Superimposition of the mouse EAT-2 and human SH2D1A structures bound to the CD150 pTyr281 peptide. Mouse EAT-2 (gray) and human SH2D1A (blue) α-carbon traces are superimposed. Note that the peptides are bound in essentially identical conformations. (F) Stereo view of the electron density map for the CD150 phosphopeptide bound to the EAT-2 SH2 domain. The 2Fo – Fc annealed omit map was calculated at 2.15 Å resolution and contoured at 1.2σ.
CD150 is found not only on cells of T and NK lineage, but also on resting B cells, dendritic cells and macrophages (Sidorenko and Clark et al., 1993; Cocks et al., 1995; Wang et al., 2001). Because CD150 is a self-ligand, it is involved bi-directionally in antigen-presenting cell (APC)–T cell interactions (Punnonen et al., 1997; Sayos et al., 1998; Mavaddat et al., 2000). CD244 is expressed on APCs such as macrophages or monocytes, and on NK cells and a subset of CD8+ T cells (Nakajima and Colonna, 2000). CD229 and CD84 are expressed on myeloid cells, macrophages, B cells and cells of the T lineage (Sandrin et al., 1992; De la Fuente et al., 1997). Thus, all four receptors, which interact with SH2D1A, are expressed on the surface of professional APCs, where SH2D1A is absent.
Because of the importance of SH2D1A in T and NK cell signaling, we reasoned that APCs must contain a regulator with properties similar to SH2D1A. We focused on a previously reported cDNA, termed EAT-2, which encodes a 132 amino acid single SH2 domain protein with unknown functions (Thompson et al., 1996). Here we show that EAT-2 is the SH2D1A equivalent in B lymphocytes and macrophages as it binds to CD84, CD150, CD244 and CD229 through its SH2 domain. The structure of a complex of EAT-2 with a phosphotyrosine peptide (pTyr281) derived from the CD150 cytoplasmic tail is very similar to that of SH2D1A with the same peptide. Thus, EAT-2 and SH2D1A are free SH2 domains that define a new class of proteins that play a role either in T cells or in APCs.
Results
The human EAT-2 gene
A cDNA library made with RNA from human splenocytes was used to clone a cDNA encoding human EAT-2. The human EAT-2 cDNA has a coding region of 399 nucleotides (DDBJ/EMBL/GenBank accession No. AF256653) (Figure 1A). Its nucleotide sequence is 83% identical to the mouse cDNA (Figure 1A). The complete genomic organization of human EAT-2 was obtained using BLAST analysis (Altschul et al., 1990) of the High Throughput Genomic (HTG) database and the human EAT-2 cDNA sequence. Using seven different GenBank sequences of pBACs containing the EAT-2 exons, but in particular pBAC AL359699 and AC068536, the EAT-2 gene was shown to have an exon–intron organization similar to that of mouse and human SH2D1A (Coffey et al., 1998; Wu et al., 2000) (Figure 1B). Like the SH2D1A gene, EAT-2 consists of four exons spanning ∼14 kb. The coding region of the first and second exons is highly conserved between human and mouse (87 and 90% identity, respectively), while exon 3 is slightly less conserved (81%) (Figure 1A). Two additional sequences highly homologous to the first and third exon are located in the same chromosomal area (exon IA, which is located ∼30 kbp upstream of the first exon, and exon IIIA) (Figure 1B). Interestingly, the sequence of the coding region of the fourth exon is extremely conserved between species (Figure 1A). Exon 1, exon 2 and approximately the first two-thirds of exon 3 code for the EAT-2 SH2 domain (Figure 3A), while the terminal portion of the third exon and exon 4 account for the EAT-2 tail (Figure 3A). A human expressed sequence tag (EST) sequence that is 99% identical to human EAT-2 and derived from a lung cDNA library has been found recently (#BG569733).

Fig. 1. The human EAT-2 gene. (A) Alignment of the human and mouse EAT-2 nucleotide sequences. The coding region sequences of the human (hEAT-2) and mouse (mEAT-2) EAT-2 cDNAs are compared. Exon boundaries are indicated (bold font, identity of nucleotides; regular font, difference of nucleotides). (B) Genomic organization of the human EAT-2 gene. The human EAT-2 gene consists of four exons that present an overall organization similar to that of the SH2D1A gene. The putative exon IIIA represents part of exon III (see text).
The nucleotide region 5′ to the ATG contains a canonical TATA box at 335 nucleotides upstream of the ATG. The length of the 3′-untranslated region (UTR) was determined by comparing the genomic DNA sequence downstream of the stop codon with three ESTs (#BE896279, #BF375549 and #AW613569). We therefore predict that the major human EAT-2 mRNA will be ∼2400 nucleotides, similar to a major 2.5 kb cDNA encoding murine EAT-2 (Thompson et al., 1996). The 3′-UTR of EAT-2 contains three ARE recognition sites, which indicates that EAT-2 mRNA levels may be controlled post-transcriptionally by triggering cell surface receptors, as is the case with SH2D1A (Wu et al., 2000).
EAT-2 is expressed in B lymphocytes and macrophages
SH2D1A is expressed mostly in T lymphocytes and NK cells (Nagy et al., 2000). To establish whether EAT-2 was expressed in cells of the immune system that are SH2D1A-negative, several populations of immunocytes were tested. EAT-2 is highly expressed in organs such as spleen, lymph nodes, lung and small intestine (Thompson et al., 1996; M.Morra, data not shown). To enrich for B cells and other APCs, splenocytes from an immunodeficient mouse, tgε26 (Wang et al., 1994), which lacks NK and T cells, were used. EAT-2 but not SH2D1A is expressed in tgε26 spleen and lymph nodes (Figure 2A). No murine EAT-2 transcript was detected in the thymus (Figure 2B). The murine B-cell leukemia lines K46 and M12 tested positive for the EAT-2 transcript, while the T-leukemia line EL-4 is negative (Figure 2B). Human EAT-2 nucleotide sequences were amplified using RNA from five out of the six B-lymphoma or lymphoblastoid cell lines tested (Cess, Daudi, Namalwa, Raji and RPMI1888) (M.Morra, data not shown). Expression of mouse EAT-2 in highly purified cell-sorted B lymphocytes (B220+ cells) and macrophages (CD11b+ cells) was confirmed by TaqMan analysis (Figure 2C). Peritoneal exudate macrophages isolated from RAG-2 null mice that lack T and B cells also tested positive for EAT-2 expression (M.Morra, data not shown). Taken together, these results show that within the hematopoietic cell lineage, EAT-2 is expressed in APCs such as B lymphocytes and macrophages.
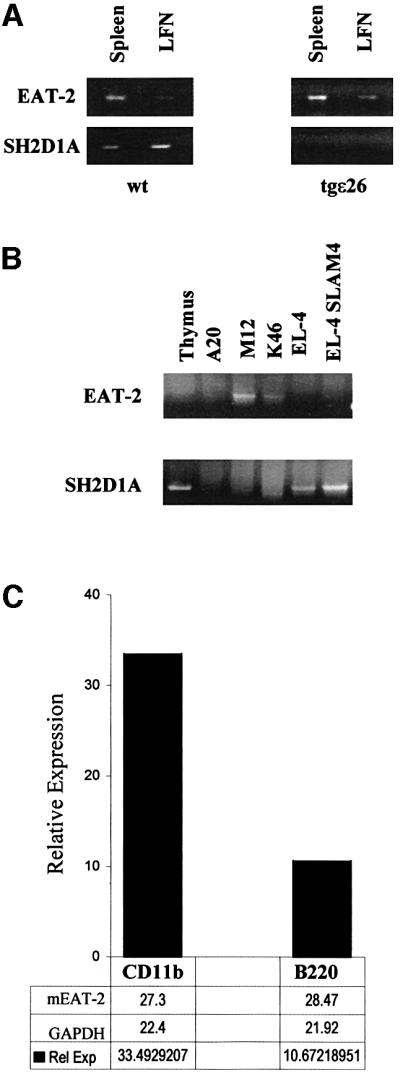
Fig. 2. EAT-2 is expressed in B lymphocytes and macrophages. RT–PCR and TaqMan analysis of murine EAT-2 and SH2D1A expression. (A) EAT-2 and SH2D1A expression in spleen and lymph nodes (LFN) of wild-type mice and T and NK cell-deficient tgε26 mice (Wang et al., 1994). (B) Expression of the mouse EAT-2 transcript in the murine B leukemia M12 and K46, but not in the T-cell line EL-4. (C) Purified B lymphocytes (B220+ cells) and macrophages (CD11b+ cells) from wild-type mice are positive for EAT-2 transcripts by TaqMan analysis (see Materials and methods).
The structure of EAT-2 is similar to that of SH2D1A
The amino acid sequences of the SH2 domains of human and mouse EAT-2 share sequence homologies with all other SH2 domains (Figure 3A, lower panel). The EAT-2 SH2 domain is homologous to the SH2 domain of Grb2 (35%), Csk (30%), Lck (30%) and Syk (30%). However, the highest homology of the mouse and human EAT-2 SH2 domain is with the SH2 domain of mouse and human SH2D1A (47 and 40%, respectively) (Figure 3A, upper panel). To enable a comparison of the structures of EAT-2 and SH2D1A, an attempt was made to grow crystals of mouse EAT-2 with a short (14mer) peptide segment of the cytoplasmic tail of CD150 including Tyr281. Although both the unphosphorylated (Tyr281) and the phosphorylated (pTyr281) peptide had co-crystallized with human SH2D1A (Li et al., 1999; Poy et al., 1999), only a crystal of the mouse EAT-2 SH2 domain complexed with the pTyr281 peptide was obtained.
We expressed and purified a fragment of EAT-2 (residues 1–103) lacking the C-terminal tail and crystallized it in complex with a 14 residue CD150 phosphopeptide (residues 273–286, with the Tyr281 phosphorylated). The structure was solved by molecular replacement with the SH2D1A SH2 domain and refined to an R-value of 21.6% at 2.15 Å resolution (see Materials and methods) (Figure 3F). The final model included all 103 residues of EAT-2, residues 277–286 of CD150 and 86 water molecules.
EAT-2 has a characteristic SH2 fold (Kuriyan and Cowburn, 1997), which includes a central β-sheet with α-helices packed against either side (Figure 3B). Canonical SH2 domains bind phosphopeptides in a ‘two-pronged’ fashion: the phosphotyrosine residue binds in a pocket on one side of the central sheet, and the three to five residues C-terminal to it bind in a pocket on the opposite side (Kuriyan and Cowburn, 1997). The CD150 phosphotyrosine peptide retains these general binding features in the complex with EAT-2 (Figure 3B). Further, EAT-2, like SH2D1A (Poy et al., 1999), forms additional interactions with the three amino acids N-terminal to CD150 Tyr281 (Ile –1, Thr –2 and Leu –3; Figure 3B–D). The CD150 peptide makes a parallel β-sheet interaction with the βD strand of the domain, and the side chains of residues Leu –3 and Ile –1 of the CD150 peptide pack with Leu49 and Tyr51 in strand βD of EAT-2 (Figure 3B and D). Thr –2 (Thr279 of CD150) hydrogen-bonds with Glu16 of EAT-2, and with a buried water molecule that is also coordinated by Arg31 (Figure 3D). Corresponding interactions are also observed in SH2D1A–CD150 complexes (Poy et al., 1999).
The phosphorylated Tyr281 is coordinated in a manner similar to that observed in other SH2 domain complexes; the conserved Arg31 forms the expected bi-dentate hydrogen bonds with phosphate oxygens (Figure 3D). The five residues following Tyr281 are coordinated by interactions with the EF and BG loops, and the βD strand of the central sheet (Figure 3D). These C-terminal interactions are similar to those seen in other SH2 domain– phosphopeptide complexes. Val284 (pY +3) inserts into a hydrophobic cleft in a manner analogous to that of an isoleucine at this position in Src family SH2 complexes (Eck et al., 1993; Waksman et al., 1993) (Figure 3D). We conclude that the interactions between the CD150 peptide and EAT-2 are very similar to those of SH2D1A with the same peptide and utilize the same unusual three-pronged binding mode (Li et al., 1999; Poy et al., 1999). Although similar to other SH2 domains in their phosphotyrosine and C-terminal (+3) recognition, they are different in their ability to recognize specifically residues N-terminal to the phosphotyrosine. Thus, SH2D1A and EAT-2 represent a distinct class of SH2 domains. These results validate previous data obtained by screening of a library of random peptides with the EAT-2 SH2 domain (Poy et al., 1999).
In spite of only 40% identical residues between mouse EAT-2 and human SH2D1A, their structures are very similar. This point is illustrated clearly by superimposing the EAT-2 SH2 domain onto the SH2D1A SH2 domain. The two structures superimpose with an r.m.s. deviation of 0.69 Å for Cα atoms (Figure 3E). Moreover, amino acid residues that are substituted in the SH2D1A protein of XLP patients, and which severely affect the functions of SH2D1A (Morra et al., 2001b), are conserved in mouse and human EAT-2 as well as in mouse and human SH2D1A (Figure 3A, upper panel). These residues are located mostly in positions that are key for the interaction between the EAT-2 SH2 domain and the CD150 phosphopeptide (indicated by the symbol ‘+’ in Figure 3A, upper panel), or in positions critical for the stability of the SH2D1A protein (Morra et al., 2001b).
EAT-2 binds to the phosphorylated cytoplasmic tail of CD84, CD150, CD244 and CD229
The structural analysis revealed that EAT-2 binds to a phosphorylated peptide derived from the cytoplasmic tail of CD150 in a three-pronged fashion. To measure the affinity of binding between EAT-2 and the CD150 Tyr281 peptide, a fluorescence polarization assay was used. An 11mer peptide encompassing the CD150 cytoplasmic region 276–286 was labeled with fluorescein isothiocyanate (FITC) in its α-amino group. The affinity of binding of this peptide, either with or without phosphorylation of Tyr281, was determined in a polarimeter using varying concentrations of GST–EAT-2 and GST– SH2D1A. GST–EAT-2 (Figure 4, upper panel) binds the phosphorylated pTyr281 peptide with an affinity comparable with GST–SH2D1A (Figure 4, middle and lower panel) (Kd = 131 nM for EAT-2/pTyr281; Kd = 127 nM for SH2D1A/pTyr281). However, in contrast to SH2D1A, EAT-2 fails to bind the Tyr281 peptide in the absence of phosphorylation (Figure 4, upper and lower panel). Taken together, these in vitro binding studies distinguish between the SH2 domains of EAT-2 and SH2D1A in that only SH2D1A can bind to the peptide in the absence of phosphorylation.

Fig. 4. EAT-2 binds exclusively to a phosphorylated peptide (pY281) derived from the cytoplasmic tail of CD150. (A) Fluorescence polarization analysis of the EAT-2 binding to a phosphorylated pY281 peptide. Different concentrations of GST–mouse EAT-2 (or GST–human SH2D1A) and an 11mer synthetic peptide identical to amino acid residues 276–287 of human CD150 (Sayos et al., 1998), tyrosine phosphorylated or not, were used. Top panel: binding of GST–mouse EAT-2 to the pY281 (filled triangles and continuous line) or the Y281 peptide (open squares and dashed line). Bottom panel: binding of GST–human SH2D1A to the pY281 (filled triangles and continuous line) or the Y281 peptide (open squares and dashed line). x-axis: protein concentration (nM); y-axis: polarization units (mP). The table summarizes the apparent dissociation constant (kD). (B) Hybrid system analysis of the interaction between EAT-2 and the cytoplasmic tail of CD150 in the presence or absence of fyn. Dashed bars indicate the interaction between the EAT-2 (or SH2D1A) full-length protein fused to a GAL4 DNA-binding domain and the GAL4 DNA activation domain fused to the cytoplasmic tail of the CD150 receptor. An empty pGAD424 vector was used as a control (solid bars). The test was conducted in either the presence or absence of fyn420,531Y–F. y-axis = β-galactosidase (U/ml).
Next, we determined whether EAT-2 binds to the complete cytoplasmic tails of CD150 and of the related receptors CD84, CD229 and CD244, as does SH2D1A (Sayos et al., 2000, 2001). An altered yeast two-hybrid system was used. We chose the yeast two-hybrid system because there is no tyrosine phosphorylation in yeast cells. Moreover, we previously had developed an altered yeast two-hybrid system in which phosphorylation of tyrosine could be established without interfering with the read-out of the assay (Sayos et al., 2001). Briefly, yeast cells were co-transformed with two vectors: (i) the first is a bi-cistronic vector, containing coding sequences for either the EAT-2 sequence alone (two-hybrid system) or EAT-2 and mutant tyrosine kinase fyn420,531Y–F (modified two-hybrid system); and (ii) the second vector contains the coding sequences for the cytoplasmic tail of human CD84, CD150, CD229 or mouse CD244.
In line with the fluorescence polarization results, EAT-2 interacted with CD150 only in the presence of fyn, while no reporter activity was detected without fyn (Figure 4B). SH2D1A was used as a control for its ability to interact with CD150 in the absence of fyn (Sayos et al., 1998) (Figure 4B).
Next, we expanded the analysis to the CD150-related receptors CD84, CD229 and CD244. As in the case of CD150, a reporter activity was evident only in the presence of fyn (Figure 5). Thus, this assay demonstrated the ability of EAT-2 to interact with CD150, CD244, CD229 or CD84. This interaction requires the presence of fyn420,531Y–F (Figure 5) and is dependent upon tyrosine phosphorylation of the receptors.

Fig. 5. EAT-2 binds only to the phosphorylated cytoplasmic tail of CD150, CD84, CD229 and CD244 in a modified yeast hybrid system. (A) Interactions between EAT-2 and CD150, CD244, CD229 or CD84 require the presence of the tyrosine kinase fyn. Yeast cells (strain Y187) were co-transformed with full-length mouse EAT-2 and mutant fyn420,531Y–F in pBRIDGE, and with vector pGAD424 containing the cytoplasmic tail of CD150, CD244, CD229 or CD84. An empty pGAD424 vector was used as a control. Protein interactions were detected by measuring activation of β-galactosidase (U/ml) in the yeast lysate. Interactions in the presence of the protein tyrosine kinase fyn420,531Y–F are indicated by dashed bars. In control experiments with the same transformed yeast cells, fyn420,531Y–F expression was repressed specifically by adding l-methionine to the culture medium (solid bars). (B) Schematic representation of the putative EAT-2-binding motifs in the cytoplasmic tail of the CD150 family members. The motifs in the cytoplasmic tail of CD150 and CD229 that have been shown to bind SH2D1A by mutational analyses are indicated by an asterisk (Sayos et al., 2001; Howie et al., 2001).
EAT-2 partially blocks recruitment of SHP-2 to the APC receptors
The apparent high affinity of the interaction between EAT-2 and the pTyr281 peptide predicted that EAT-2, like SH2D1A, potentially could block recruitment of signal transduction molecules to the cytoplasmic tails of the four APC cell surface receptors. To avoid interference by endogenous EAT-2 or receptor molecules, we utilized a previously published COS-7 cell assay. Cells were transiently transfected with different combinations of plasmids encoding EAT-2, fyn and either CD84, CD150, CD229 or CD244. As expected, EAT-2 binds to all four proteins in COS-7 cells (Figure 6). As shown in Figure 6, EAT-2 binding blocks SHP-2 recruitment to the CD150, CD84, CD229 and CD244 receptors, albeit less efficiently than SH2D1A. This difference might be explained by the absence of the non-phospho binding in the case of EAT-2, which could affect the transient triple transfection assay.

Fig. 6. In vivo binding of EAT-2 to CD150 family members. Interactions were examined after co-transfection of combinations of mouse EAT-2 (in pCMV-FLAG) with CD150, CD244, CD229 or CD84, and fyn into COS-7 cells (plasmid combinations are indicated above each set of panels). All cells were surface biotinylated prior to being subjected to detergent lysis. Immunoprecipitates (I.P.) made with an antibody againsT cD150-related receptors (panels on the left of each figure) are analyzed by western blotting. Anti-phosphotyrosine = W.B. α-PY; avidin = W.B. AV-HRP; EAT-2 = W.B. α-FLAG; SHP-2 = W.B. α-SHP-2. In the panels on the right of each figure, lysates of the same transfections are analyzed by western blotting. (A) EAT-2 and CD150. (B) EAT-2 and CD244. (C) EAT-2 and CD84. (D) EAT-2 and CD229.
Interestingly, as previously observed for SH2D1A (Sayos et al., 1998, 2001), overexpression of EAT-2 apparently induces tyrosine phosphorylation of the CD150-related receptors. This explains the binding of EAT-2 to the CD150 family of glycoproteins in the absence of fyn (Figure 6), whilst they fail to do so in the phosphorylation-free environment of the yeasT cell. Recent findings (Latour et al., 2001; A.Lanyi, unpublished data) indicate that SH2D1A selectively activates/recruits fyn to CD150. Thus, phosphorylation of CD150-related receptors by SH2D1A would derive by both inhibition of SHP-2 recruitment and activation of fyn. According to our phosphorylation results (Figure 6), we predict that EAT-2 might play a role similar to that of SH2D1A in activat ing fyn- or src-related kinases in B lymphocytes and macrophages.
Next, we tested EAT-2 binding in the presence of CD150 and CD229 receptors where the cytoplasmic tyrosines were mutated to phenylalanine. Using a direct interaction hybrid system in the presence of fyn, we observed that mutation CD150 Y1F (CD150 Y281F) (Figure 7A) or the double mutation CD229 Y23F (CD229 Y558F, Y581F) (Figure 7B) totally ablated EAT-2 binding. These findings perfectly parallel results with CD150–SH2D1A (Howie et al., 2001) and CD229– SH2D1A (Sayos et al., 2001). In vivo binding experiments supported and extended these findings. EAT-2, like SH2D1A (Howie et al., 2001), binds to both phosphorylated Tyr281 and Tyr327 in CD150 (Figure 7C). Combined mutation of these binding sites totally ablated EAT-2 binding to the CD150 and CD229 receptors (Figure 7). These results unambiguously indicate that EAT-2 binding to the CD150-related receptors is dependent upon phosphorylation of the motif in the receptors.

Fig. 7. Analysis of EAT-2 binding to Y–F CD150 and CD229 mutant receptors. (A) EAT-2 binding to CD150 Y–F mutants in a direct interaction hybrid system. Hybrid system direct interaction analysis of the binding between EAT-2 in pBRIDGE-Fyn420,531Y–F (dashed bars) and different cD150 Y–F mutations in pGAD424. As control, vector pBRIDGE was used (solid bars). In particular, mutants CD150 Y1F (Tyr281 to phenylalanine), Y3F (Tyr327 to phenylalanine) or Y13F (Tyr281 and Tyr327 to phenylalanine) were used in this study. The lower part of the figures depicts the mutations in the CD150 cytoplasmic tail. (B) EAT-2 binding to CD229 Y–F mutants in a direct interaction hybrid system. Hybrid system direct interaction analysis of the binding between EAT-2 in pBRIDGE-Fyn420,531Y–F (dashed bars) and different cD229 Y–F mutations in pGAD424. As control, vector pBRIDGE was used (solid bars). In particular, mutants CD229 Y2F (Tyr558 to phenylalanine), Y3F (Tyr581 to phenylalanine) and Y23F (Tyr558 and Tyr581 to phenylalanine) were used in this study. The lower part of the figure depicts the mutations in the CD229 cytoplasmic tail. (C) EAT-2 binding to CD150 Y–F mutants in an in vivo system. Full-length wild-type CD150, CD150 Y1F (Tyr281 to phenylalanine), CD150 Y2F (Tyr307 to phenylalanine), CD150 Y3F (Tyr327 to phenylalanine) and CD150 Y123F (Tyr281, Tyr307 and Tyr327 to phenylalanine) were co-transfected with EAT-2 and fyn in a COS-7 cell system. Mutant cD150 cDNAs used in the transfections are indicated above each set of panels. Immunoprecipitates (I.P.) were made with an anti-CD150 antibody and analyzed by western blotting. Anti-phosphotyrosine = W.B. α-PY; avidin = W.B. AV-HRP; EAT-2 = W.B. α-FLAG. The bottom panel shows the lysates of the same cell extracts used for the precipitation.
Fyn phosphorylation of mutants CD150 Y1F, Y2F or Y123F is much lower compared with phosphorylation of wild-type CD150 (Figure 7C). Similar results were obtained using SH2D1A (Howie et al., 2001). Both Tyr315 and Tyr335 in the cytoplasmic tail of mouse CD150 (equivalent to Tyr307 and Tyr327 of human CD150) are required for fyn binding to CD150 (Latour et al., 2001). We speculate that the lower phosphorylation of CD150 of Y–F mutants might be because of an inefficient ternary complex formation among the CD150, EAT-2 and fyn molecules. Because a similar pattern of phosphorylation of CD150 mutants and EAT-2 binding to CD150 in COS-7 cells was also detected in the absence of fyn (M.Morra, data not shown), EAT-2 might activate src-related kinases other than fyn.
Discussion
Here we show that EAT-2 is an SH2D1A-like molecule in professional APCs such as B lymphocytes and macrophages. Although the SH2D1A–CD150 interaction is unique in its binding in the absence of tyrosine phosphorylation (Sayos et al., 1998; Poy et al., 1999), this distinction between CD150 and the other cell surface proteins cannot be explained easily. The presence of a Cys15 in EAT-2 instead of a Gly16 in SH2D1A might be of significance. Residue 15 lies behind Arg31 (Arg32 in SH2D1A) in the phosphotyrosine-binding pocket; substitution with cysteine eliminates a buried water molecule, which in SH2D1A hydrogen-bonds to the arginine. It is possible that the more hydrophobic environment surrounding Arg31 of EAT-2 makes binding of a non-phosphorylated tyrosine residue energetically unfavorable. Thus, while EAT-2 and SH2D1A SH2 domains share a ‘three-pronged’ mode of interaction, the interaction with non-phosphorylated tyrosines may be unique to the interaction of SH2D1A with Tyr281 in CD150.
The interactions of EAT-2 and SH2D1A with the CD150 family members are summarized in Figure 8. We speculate that SH2D1A and EAT-2 are introduced into the immune synapse via CD150 and CD229, CD84 or CD244 to ensure their presence at the T cell–APC interface. It is likely that following T-cell receptor (TCR) triggering, CD244, CD229 and CD84 are tyrosine phosphorylated rapidly, thus recruiting EAT-2 and SH2D1A. In this fashion, SH2D1A and EAT-2 could function indirectly to prolong phosphorylation of important substrates during TCR triggering. Because our results indicate that overexpression of EAT-2 induces tyrosine phosphorylation of the CD150-related receptors as does SH2D1A (Sayos et al., 1998; Latour et al., 2001), EAT-2 might activate fyn or other src-related kinases in B lymphocytes and macrophages.

Fig. 8. A possible model for the interactions between EAT-2, SH2D1A and the four cell surface receptors at the interface of B lymphocytes, macrophages (Mφ) and T cells. The receptor–ligand interactions of CD48/CD244 (Brown et al., 1998) and CD150/CD150 (Punnonen et al., 1997; Mavaddat et al., 2000) have been reported.
The difference between EAT-2 and SH2D1A binding to CD150 could partly account for the differences in CD150 signaling in T and B lymphocytes (Cocks et al., 1995; Punnonen et al., 1997), which are SH2D1A- and EAT-2-expressing cells, respectively. Preliminary binding experiments using peptides with an amino acid sequence surrounding EAT-2 Tyr127 indicated that the phospholipase-C-γ enzyme might bind the EAT-2 tail if phosphorylated (M.Morra, data not shown). The fact that EAT-2 may act as an adaptor articulating interactions with SH2 domain-containing molecules through its tail might also account for differences in CD150 signaling in different lymphocyte populations.
EAT-2 and SH2D1A have a similar exon–intron organization, which suggests their origin by duplication from a common ancestor gene. The EAT-2 gene maps closely to the CD150 cluster, which includes CD48, CD84, CD229, CD244 and 19A (Morra et al., 2001a; Wang et al, 2001), and which is a located in a 260 kb fragment on chromosome 1q22. Thus, a relationship between genomic localization and function of these genes and EAT-2 is suggested. The SH2D1A gene deletion leads to multiple defects in cell signaling (Wu et al., 2001). In addition to typical XLP patients, the SH2D1A gene has been found to be altered in some patients affected by lymphomas (Brandau et al., 1999) or common variable immunodeficiency syndrome (CVID) (Gilmour et al., 2000; Morra et al., 2001c). Patients characterized by a chronic infection by EBV and XLP-like patients with a negative family history tested negative for SH2D1A mutations (Sumegi et al., 2000; M.Morra, unpublished data). Searching for mutations in EAT-2 in these patients may be necessary. In particular, because EAT-2 is expressed mostly in B lymphocytes and macrophages, it is plausible that the gene may be involved in lymphomagenesis or other proliferative diseases. Because EAT-2 can be found in ESTs derived from a variety of human tumors, it is likely that the gene plays an important role in signal transduction events in non-hematopoietic cells. Furthermore, the EAT-2 protein may interact with novel members of the CD150 family that are expressed in a variety of different tissues. The creation of a mouse with a disrupted EAT-2 gene or overexpression of the gene will permit a deeper understanding of the function of EAT-2 in different cell types and of its role in cell signal transduction.
Materials and methods
Cells and antibodies
The A20, M12, K46, EL-4 and COS-7 cells (ATCC) were grown as previously described (Sayos et al., 1998). Anti-human CD150 2E7 monoclonal antibody (mAb) was a gift from DNAX (Cocks et al., 1995). Anti-mouse CD244 mAb was purchased from BD Pharmingen. Anti-human CD229 (clone HCD229.1.84) and CD84 (clone CD84.1.2.21 and CD84.1.7) antibodies were produced as previously described (Sayos et al., 2001). For western blotting, anti-FLAG M5 mAb (Kodak) and polyclonal rabbit anti-SHP-2 were used (Santa Cruz). The detection system consisted of anti-mouse or anti-rabbit IgG horseradish peroxidase (HRP)-conjugated polyclonal antibodies (Santa Cruz). For antiphosphotyrosine blotting, we used a directly conjugated HRP–antibody cocktail (Zymed).
Plasmid construction
Mouse EAT-2 cDNA was cloned in the vector pCMV-2/FLAG (Kodak). The CD150 construct was generated by subcloning the human CD150 cDNA in vector pJFE14-SRα (a gift from DNAX Research Institute). The mouse CD244, human CD229 and human CD84 cDNAs were subcloned in the pCDNA3.1 vector.
Human CD150 mutants (Y281F or Y1F; Y307F or Y2F; Y327F or Y3F; Y281,327F or Y13F; Y281,307,327F or Y123F) were generated by PCR-mediated mutagenesis (Howie et al., 2001).
Immunoprecipitation and western blotting
COS-7 cells (10 × 106) were transfected by the DEAE-dextran method (Ausubel et al., 1995). Proteins were immunoprecipitated as previously described (Sayos et al., 1998) and precipitates were separated by SDS–PAGE and transferred to a PVDF Immobilon membrane (Millipore Corp.). Filters were blocked with 3% bovine serum albumin (BSA) and then probed with the antibodies indicated.
Cloning of the human EAT-2 cDNA
A cDNA containing the complete human EAT-2 sequence was obtained by PCR using spleen cDNA as template (Marathon library, Clontech). Primers recognizing the first exon were paired with primers recognizing sequences surrounding Y120 and Y127 of the mouse EAT-2 sequence. Thus, a fragment of ∼400 bp was generated using the pair of primers recognizing Y127. Amplified fragments were subcloned in a TA cloning vector and the inserts were sequenced.
RT–PCR and TaqMan analysis
Total RNA was isolated by TRIzol Reagent (BRL, Gaithersburg, MA). One step RT–PCR (Access RT–PCR kit, Invitrogen) was performed using the mouse EAT-2 primer combinations F5′-GACCAACCGAGAGTGTGA-3′ and R5′-TTATTGGGTTTGAAAGGTGAA-3′.
For the TaqMan analysis, spleens were mashed and depleted of red blood cells. Individual cell populations (CD11b and B220) were isolated using antibodies conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA). cDNA was synthesized by a SuperScript First-Strand synthesis system (Invitrogen).
VIC-labeled rodent GAPDH control primers and probe were purchased from Applied Biosystems (Foster City, CA). The following mouse EAT-2 primer combination was used: F5′-gctgccacatctgcaagtgt-3′ and R5′- gaacagatcttgctatcccaatca-3′.
The EAT-2 probe (5′-tgccaatttctagtgagccactgagaccc-3′) was labeled using 6-carboxyfluorescein (Applied Biosystems). Reactions were conducted in TaqMan Universal PCR Master Mix (Applied Biosystems) using an ABI PRISM 7700 Sequence Detection System (Applied Biosystems).
A modified yeast two-hybrid system
The sequence encoding mouse EAT-2 was cloned in the multiple cloning site 1 of the bi-cistronic vector pBRIDGE (Clontech). A mutant of human fyn420,531Y–F was subcloned in the second multiple cloning site of pBRIDGE, as described previously (Sayos et al., 2001).
Sequences encoding the cytoplasmic tail of CD84, CD150, CD229 or CD244 were cloned in the GAL4 DNA activation domain site of vector pGAD424 (Clontech). Human CD150 fragment was obtained from the pGBT9–CD150 construct (Sayos et al., 2001). Mouse CD244 was amplified by PCR using the CD244 5′ sense primer 5′-CCGGAATTCAAGAAGAGGAAGCAGTTACAGTTC-3′ and the CD244 3′ antisense primer 5′-GGAAGATCTCTAGGAGTAGACATCAAAGTTCTC-3′. Human CD84 was amplified by PCR using the CD84 5′ sense primer 5′-ATCGAATTCTTCCGTTTGTTCAAGAGAAGA-3′ and the CD84 3′ antisense primer 5′-ATCGGATCCCTAGATCACAATTTCATAGCT-3′. Human CD229 and CD229 Y–F mutants (mutants Ly-9 558 Y–F or CD229 Y2FY; Ly-9 581 Y–F or CD229 Y3F; and Ly-9558, 581 Y–F or CD229 Y23F) were generated as previously described (Sayos et al., 2001).
Plasmids pBRIDGE and pGAL4 were co-transformed and plated on SD agar supplemented with a –Trp, –Leu, –Met dropout. The β-galactosidase colony lift filter assay and liquid culture assay using o-nitrophenyl-β-d-thiogalactopyranoside (ONPG) as a substrate were carried out as described in the Clontech Yeast Protocols Handbook.
Expression, purification and crystallization of the mouse EAT-2 molecule
Mouse EAT-2 (residues 1–103) was expressed in Escherichia coli strain BL21 (DE3) using the pRSET plasmid (Invitrogen). Cell pellets were lysed by sonication, and the EAT-2 protein was purified to homogeneity from clarified lysates using cation exchange chromatography (S-Sepharose FastFlow, Pharmacia) followed by phosphotyrosine affinity chromatography essentially as described for SH2D1A (Poy et al., 1999).
EAT-2 crystals were grown in hanging drops at 22°C. The EAT-2 protein (20 mg/ml) was combined with a 3-fold excess of the phosphorylated CD150 pY281 peptide (VEKKSLTIpYAQVQK). The complex was crystallized over a well solution containing 30% PEG 8000, 100 mM sodium citrate pH 5.6, 25 mM ammonium sulfate and 10 mM dithiothreitol (DTT). All diffraction data were recorded at –165°C. Crystals were ‘dunked’ briefly in a buffer containing well solution plus 20% glycerol prior to flash freezing in liquid nitrogen. The EAT-2/pY281 data were recorded using a Mar Research image plate detector mounted on a Rigaku rotating anode source with mirror optics at –165°C. All diffraction data were integrated and scaled using the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997) (Table I).
Table I. Data collection and refinement statistics.
| Resolution (Å) | 2.1 |
| Space group | P21212 |
| Unit cell (Å) | 58.9, 59.5, 34.9 |
| Molecules/asymmetric unit | 1 |
| Rsym (% overall/2.16–2.1 Å shell) | 5.4/33.0 |
| Reflections (total/unique) | 24 668/7460 |
| Completeness (%) |
97.0 |
| Refinement statistics | |
| Resolution range (Å) | 20.0–2.15 |
| Protein atoms | 908 |
| Water molecules | 86 |
| Rcryst/Rfree (%, F > 2σ) | 21.6/27.9 |
| Rcryst (%, all data) | 23.1 |
| R.m.s.d. bond length/angles | 0.016 Å/2.151° |
The atomic structure of the EAT-2–CD150 complex was then determined by molecular replacement with the program AmoRe (Navaza, 1992). The SAP/pY281 SH2 domain structure, with the phosphopeptide removed, was used as a search model (PDB code ID4W) (Poy et al., 1999). The rotation and translation searches were unambiguous. Difference electron density maps calculated after rigid body refinement of the properly positioned model yielded clear electron density for the bound CD150 phosphopeptide. The peptide was built and the Eat-2 SH2 domain was refitted manually using the program O (Jones and Kjeldgaard, 1997). Ordered solvent was built with the aid of the program ARP/wARP (Lamzin and Wilson, 1997) and the structure was refined with data extending to 2.1 Å resolution using REFMAC in the CCP4 program suite (CCP4, 1994). Refinement statistics are presented in Table I. Figure 3 was prepared using the programs MOLSCRIPT, O and GRASP. Coordinates have been deposited with the Brookhaven Protein Data Bank (ID code 1I3Z).
Measurement of peptide binding affinity using fluorescence polarization
Fluorescent polarization assays were performed according to Danliker et al. (1981) as previously described (Morra et al., 2001b). A Beacon system was used in these experiments. Polarization values are expressed in millipolarization units (mP). The curves were fit by non-linear regression using the Prizm curve-fitting software (Graphpad Software, San Diego, CA).
Acknowledgments
Acknowledgements
This work was supported by grants from the NIH (PO1-AI-35714 to CT) and the National Foundation of Dimes (1FY00-382 to C.T.). M.M. is supported by an American–Italian Cancer Foundation Fellowship.
References
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1995) Current Protocols in Molecular Biology. Suppl. 17. Green Publishing Associates/Wiley-Interscience, New York, NY, p. 16.13.1.
- Benoit L., Wang,X., Pabst,H.F., Dutz,J. and Tan,R. (2000) Defective natural killer cell activation in X-linked lymphoproliferative disease. J. Immunol., 165, 3549–3553. [DOI] [PubMed] [Google Scholar]
- Brandau O. et al. (1999) Epstein–Barr virus-negative boys with non-Hodgkin lymphoma are mutated in the SH2D1A gene, as are patients with X-linked lymphoproliferative disease (XLP). Hum. Mol. Genet., 8, 2407–2413. [DOI] [PubMed] [Google Scholar]
- Brown M.H., Boles,K., van der Merwe,P.A., Kumar,V., Mathew,P.A. and Barclay,A.N. (1998) 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med., 188, 2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks B.G., Chang,C.C., Carballido,J.M., Yssel,H., de Vries,J.E. and Aversa,G. (1995) A novel receptor involved in T-cell activation. Nature, 376, 260–263. [DOI] [PubMed] [Google Scholar]
- Coffey A.J. et al. (1998) Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nature Genet., 20, 129–135. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–776. [DOI] [PubMed] [Google Scholar]
- Danliker W.B., Hsu,M.L., Levin,J. and Rao,B.R. (1981) Equilibrium and kinetic inhibition assays based upon fluorescence polarization. Methods Enzymol., 74, 3–28. [DOI] [PubMed] [Google Scholar]
- De la Fuente M.A., Pizcueta,P., Nadal,M., Bosch,J. and Engel,P. (1997) CD84 leukocyte antigen is a new member of the Ig superfamily. Blood, 90, 2398–2405. [PubMed] [Google Scholar]
- Eck M.J., Shoelson,S.E. and Harrison,S.C. (1993) Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of p56lck. Nature, 362, 87–91. [DOI] [PubMed] [Google Scholar]
- Gilmour K.C., Cranston,T., Jones,A., Davies,E.G., Goldblatt,D., Thrasher,A., Kinnon,C., Nichols,K.E. and Gaspar,H.B. (2000) Diagnosis of X-linked lymphoproliferative disease by analysis of SLAM-associated protein expression. Eur. J. Immunol., 30, 1691–1697. [DOI] [PubMed] [Google Scholar]
- Hamilton J.K. et al. (1980) X-linked lymphoproliferative syndrome registry report. J. Pediatr., 96, 669–673. [DOI] [PubMed] [Google Scholar]
- Howie D., Sayos,J., Terhorst,C. and Morra,M. (2000) The gene defective in X-linked lymphoproliferative disease controls T cell dependent immune surveillance against Epstein–Barr virus. Curr. Opin. Immunol., 12, 474–478. [DOI] [PubMed] [Google Scholar]
- Howie D., Sayos,J., Guirado,M., Simarro,M., Sancho,J. and Terhorst,C. (2001) Molecular dissection of the signaling and co-stimulatory functions of CD150 (SLAM). Blood, in press. [DOI] [PubMed] [Google Scholar]
- Jones T.A. and Kjeldgaard,M. (1997) Electron-density map interpretation. Methods Enzymol., 277, 173–208. [DOI] [PubMed] [Google Scholar]
- Kuriyan J. and Cowburn,D. (1997) Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomol. Struct., 26, 259–288. [DOI] [PubMed] [Google Scholar]
- Lamzin V.S. and Wilson,K.S. (1997) Automated refinement for protein crystallography. Methods Enzymol., 277, 269–305. [DOI] [PubMed] [Google Scholar]
- Lanier L.L. (1998) NK cell receptors. Annu. Rev. Immunol., 16, 359–393. [DOI] [PubMed] [Google Scholar]
- Latour S., Gish,G., Helgason,C.D., Humphries,R.K., Pawson,T. and Veillette,A. (2001) Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nature Immunol., 2, 681–690. [DOI] [PubMed] [Google Scholar]
- Li S.C., Gish,G., Yang,D., Coffey,A.J., Forman-Kay,J.D., Ernberg,I., Kay,L.E. and Pawson,T. (1999) Novel mode of ligand binding by the SH2 domain of the human XLP disease gene product SAP/SH2D1A. Curr. Biol., 9, 1355–1362. [DOI] [PubMed] [Google Scholar]
- Mavaddat N. et al. (2000) Signaling lymphocytic activation molecule (SLAM, CDw150) is homophilic but self-associates with very low affinity. J. Biol. Chem., 275, 28100–28109. [DOI] [PubMed] [Google Scholar]
- Morra M., Howie,D., Simarro Grande,M., Sayos,J., Wang,N., Wu,C., Engel,P. and Terhorst,C. (2001a) X-linked lymphoproliferative disease, a progressive immunodeficiency. Annu. Rev. Immunol., 19, 657–682. [DOI] [PubMed] [Google Scholar]
- Morra M. et al. (2001b) Characterization of SH2D1A missense mutations identified in X-linked lymphoproliferative disease patients. J. Biol. Chem., 276, 36809–36816. [DOI] [PubMed] [Google Scholar]
- Morra M., Silander,O., Calpe,S., Choi,M., Oettgen,H., Myers,L., Etzioni,A., Buckley,R. and Terhorst,C. (2001c) Alterations of the X-linked lymphoproliferative disease gene SH2D1A in common variable immunodeficiency syndrome. Blood, 98, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Nagy N. et al. (2000) SH2D1A and SLAM expression in human lymphocytes and derived cell lines. Int. J. Cancer, 88, 439–447. [PubMed] [Google Scholar]
- Nakajima H. and Colonna,M. (2000) 2B4: an NK cell activating receptor with unique specificity and signal transduction mechanism. Hum. Immunol., 61, 39–43. [DOI] [PubMed] [Google Scholar]
- Navaza J. (1992) AMoRe: a new package for molecular replacement. SERC Daresbury Laboratory, Warrington, UK, pp. 87–90.
- Nichols K.E. et al. (1998) Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc. Natl Acad. Sci. USA, 95, 13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Parolini S. et al. (2000) X-linked lymphoproliferative disease: 2B4 molecules displaying inhibitory rather than activating function killer cells to kill Epstein–Barr virus-infected cells. J. Exp. Med., 192, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy F., Yaffe,M.B., Sayos,J., Saxena,K., Morra,M., Sumegi,J., Cantley,L.C., Terhorst,C. and Eck,M.J. (1999) Crystal structures of the XLP protein SAP reveal a class of SH2 domains with extended, phosphotyrosine-independent sequence recognition. Mol. Cell, 4, 555–561. [DOI] [PubMed] [Google Scholar]
- Punnonen J., Cocks,B.G., Carballido,J.M., Bennett,B., Peterson,D., Aversa,G. and de Vries,J.E. (1997) Soluble and membrane-bound forms of signaling lymphocytic activation molecule (SLAM) induce proliferation and Ig synthesis by activated human B lymphocytes. J. Exp. Med., 185, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtilo D.T., Cassel,C.K., Yang,J.P. and Harper,R. (1975) X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease). Lancet, 1, 935–940. [DOI] [PubMed] [Google Scholar]
- Sandrin M.S., Gumley,T.P., Henning,M.M., Vaughan,H.A., Gonez,L.J., Trapani,J.A. and McKenzie,I.F. (1992) Isolation and characterization of cDNA clones for mouse Ly-9. J. Immunol., 149, 1636–1641. [PubMed] [Google Scholar]
- Sayos J. et al. (1998) The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature, 395, 462–469. [DOI] [PubMed] [Google Scholar]
- Sayos J., Nguyen,K.B., Wu,C., Stepp,S.E., Howie,D., Schatzle,J.D., Kumar,V., Biron,C.A. and Terhorst,C. (2000) Potential pathways for regulation of NK and T cell responses: differential X-linked lymphoproliferative syndrome gene product SAP interactions with SLAM and 2B4. Int. Immunol., 12, 1749–1757. [DOI] [PubMed] [Google Scholar]
- Sayos J., Martin,M., Chen,A., Simarro,M., Howie,D., Morra,M., Engel,P. and Terhorst,C. (2001) The cell surface receptors Ly-9 and CD84 recruit the XLP gene product SAP. Blood, 97, 3867–3874. [DOI] [PubMed] [Google Scholar]
- Seemayer T.A., Gross,T.G., Egeler,R.M., Pirruccello,S.J., Davis,J.R., Kelly,C.M., Okano,M., Lanyi,A. and Sumegi,J. (1995) X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatr. Res., 38, 471–478. [DOI] [PubMed] [Google Scholar]
- Sidorenko S.P. and Clark,E.A. (1993) Characterization of a cell surface glycoprotein IPO-3, expressed on activated human B and T lymphocytes. J. Immunol., 151, 4614–4624. [PubMed] [Google Scholar]
- Sullivan J.L. (1999) The abnormal gene in X-linked lymphoproliferative syndrome. Curr. Opin. Immunol., 11, 431–434. [DOI] [PubMed] [Google Scholar]
- Sullivan J.L., Byron,K.S., Brewster,F.E. and Purtilo,D.T. (1980) Deficient natural killer cell activity in X-linked lymphoproliferative syndrome. Science, 210, 543–545. [DOI] [PubMed] [Google Scholar]
- Sumegi J. et al. (2000) Correlation of mutations of the SH2D1A gene and Epstein–Barr virus (EBV) infection with clinical phenotype and outcome in X-linked lymphoproliferative disease (XLP). Blood, 96, 3118–3125. [PubMed] [Google Scholar]
- Sylla B.S., Murphy,K., Cahir-McFarland,E., Lane,W.S., Mosialos,G. and Kieff,E. (2000) The X-linked lymphoproliferative syndrome gene product SH2D1A associates with p62dok (Dok1) and activates NF-κB. Proc. Natl Acad. Sci. USA, 97, 7470–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye S.G., Lazetic,S., Woollatt,E., Sutherland,G.R., Lanier,L.L. and Phillips,J.H. (1999) Human 2B4, an activating NK cell receptor, recruits the protein tyrosine phosphatase SHP-2 and the adaptor signaling protein SAP. J. Immunol., 162, 6981–6985. [PubMed] [Google Scholar]
- Thompson A.D., Braun,B.S., Arvand,A., Stewart,S.D., May,W.A., Chen,E., Korenberg,J. and Denny,C. (1996) EAT-2 is a novel SH2 domain containing protein that is up regulated by Ewing’s sarcoma EWS/FLI1 fusion gene. Oncogene, 13, 2649–2658. [PubMed] [Google Scholar]
- Waksman G., Shoelson,S.E., Pant,N., Cowburn,D. and Kuriyan,J. (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell, 72, 779–790. [DOI] [PubMed] [Google Scholar]
- Wang B., Biron,C., She,J., Higgins,K., Sunshine,M.J., Lacy,E., Lonberg,N. and Terhorst,C. (1994) A block in both early T lymphocyte and natural killer cell development in transgenic mice with high-copy numbers of the human CD3E gene. Proc. Natl Acad. Sci. USA, 91, 9402–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Morra,M., Wu,C., Gullo,C., Howie,D., Coyle,T., Engel,P. and Terhorst,C. (2001) CD150 is a member of a family of genes that encode glycoproteins on the surface of hematopoietic cells. Immunogenetics, 53, 382–394. [DOI] [PubMed] [Google Scholar]
- Wu C., Sayos,J., Wang,N., Howie,D., Coyle,A. and Terhorst,C. (2000) Genomic organization and characterization of murine SAP: the gene coding for X-linked lymphoproliferative disease. Immunogenetics, 51, 805–815. [DOI] [PubMed] [Google Scholar]
- Wu C. et al. (2001) The SAP gene controls CD8 T cell responses to virus and terminal differentiation of T helper 2 cells. Nature Immunol., 2, 410–414. [DOI] [PubMed] [Google Scholar]