

Abstract
The tumor suppressor protein p53 is a transcription factor that is frequently mutated in human cancers. In response to DNA damage, p53 protein is stabilized and activated by post-translational modifications that enable it to induce either apoptosis or cell cycle arrest. Using a novel yeast p53 dissociator assay, we identify hADA3, a part of histone acetyltransferase complexes, as an important cofactor for p53 activity. p53 and hADA3 physically interact in human cells. This interaction is enhanced dramatically after DNA damage due to phosphorylation event(s) in the p53 N-terminus. Proper hADA3 function is essential for full transcriptional activity of p53 and p53-mediated apoptosis.
Keywords: apoptosis/DNA damage/hADA3/histone acetyltransferase/p53
Introduction
The human tumor suppressor protein p53 induces cell cycle arrest or apoptosis in response to stress conditions, such as DNA damage, hypoxia or ribonucleotide depletion. It does so, in large part, as a homo-tetrameric transcription factor that activates downstream effector genes, such as p21WAF1/CIP1/SDI1 for the mediation of G1 arrest (Ko and Prives, 1996; Levine, 1997; Giaccia and Kastan, 1998; Oren, 1999).
p53 has a very short half-life in unstressed cells that is due primarily to a negative feedback mechanism with MDM2. MDM2 is a downstream target gene of p53; the protein binds to the p53 N-terminus and induces ubiquitin-mediated degradation of p53 (Haupt et al., 1997; Kubbutat et al., 1997). DNA damage activates kinases, such as ATM, ATR, DNA-PK, hCHK1 and Chk2/hCds1, that phosphorylate serine residues in the N-terminus of p53, thus stabilizing p53 by preventing MDM2 from binding to p53 (Giaccia and Kastan, 1998; Ashcroft and Vousden, 1999; Chehab et al., 2000; Hirao et al., 2000; Shieh et al., 2000). While the relative importance of these and other p53 modifications is still being worked out, it has become quite clear that a cascade of post-translational modifications is required to stabilize p53 and fully activate it as a transcription factor (Giaccia and Kastan, 1998; Ashcroft and Vousden, 1999).
In this context, the effect of histone acetyltransferases (HATs) on p53 activity is particularly interesting. HATs are coactivators for many transcription factors. This activity initially was attributed entirely to their ability to relax chromatin at promoters by acetylating histones, but it quickly became clear that HATs regulate many other factors through acetylation, including transcription factors. The mechanisms that govern the timely and appropriate acetylation of transcription factors currently are largely unknown (Giles et al., 1998; Struhl, 1998; Kouzarides, 2000; Schiltz and Nakatani, 2000; Sterner and Berger, 2000).
Two homologous HATs, CBP (CREB-binding protein) and p300, function as coactivators of p53 (Avantaggiati et al., 1997; Gu et al., 1997; Lill et al., 1997; Scolnick et al., 1997). p53 can be acetylated at specific lysine residues in the C-terminus by p300/CBP and PCAF (p300/CBP-associated factor), another HAT that frequently is associated with p300 or CBP (Yang et al., 1996; Gu and Roeder, 1997; Sakaguchi et al., 1998; Liu et al., 1999). This leads to a markedly increased affinity of p53 for p53 DNA-binding sites in in vitro assays. N-terminally phosphorylated p53 is a much better substrate for PCAF acetylation than non-phosphorylated p53 (Sakaguchi et al., 1998). p53 acetylation occurs after many different types of cellular stress (Ito et al., 2001). Thus, p53 acetylation by HATs may be a universal and, probably, late post-translational modification that fully activates p53 as a transcription factor.
HATs are part of multiprotein HAT complexes that were first identified in Saccharomyces cerevisiae. In yeast, these HAT complexes promote transcription by bridging transcriptional activators with the TATA-binding protein (TBP). Besides several proteins that are responsible for the interaction with TBP, yeast HAT complexes contain yGcn5p, the HAT, and adaptor proteins, such as yAda2p and yAda3p, that interact with transcription factors (Grant et al., 1998; Sterner and Berger, 2000). PCAF and hGCN5 are both human homologs of yGcn5p, and hADA2 is homologous to yAda2p. The human homolog of yAda3p was identified by mass spectrometry after multiprotein HAT complexes were purified through interaction with FLAG-tagged PCAF or hGCN5 (Ogryzko et al., 1998). While the activities of HATs are quite well characterized, the roles of many of the other components of human HAT complexes, including hADA3, remain to be established.
Using a novel yeast p53 dissociator assay, we isolated hADA3 in a p53 dissociator screen of a HeLa cDNA expression library. Our characterization of hADA3 establishes that hADA3 and p53 preferentially interact after DNA damage has occurred and that proper hADA3 function is required for full p53 activity.
Results
The yeast p53 dissociator assay and p53 dissociator screens
The principles of the p53 dissociator assay have been described (Brachmann et al., 1996, 1998; Vidal et al., 1996). The reporter construct 1cUAS53::URA3 is activated by p53, thus enabling yeast cells to grow on medium lacking uracil (phenotype Ura+). Because the URA3 gene product is also involved in converting 5-fluoro-orotic acid (FOA) into a toxic substance, selection against p53 activity is possible as well (phenotype FOA-sensitive, FoaS). The FoaS phenotype provides the essential tool to perform cDNA expression library screens (‘dissociator screens’). Proteins that interfere with p53 activity lead to a reduction of URA3 expression and, thus, growth on FOA plates (phenotype FOA-resistant, FoaR). The p53 dissociator assay identifies proteins that inhibit p53, but proteins with very different functions in p53 biology are also isolated (see Figure 1).

Fig. 1. The p53 dissociator assay in S.cerevisiae and p53 dissociators. (A) SV40 large T antigen functions as a p53 dissociator. Independent transformants of RBy41 (expressing wild-type p53), containing either a plasmid for SV40 large T antigen (pTD1-1) or a control plasmid, were replica plated from SC-His-Leu to SC-His-Leu + 0.1% FOA at 30°C and evaluated after 3 days. (B) p53 dissociator screens using the improved diploid p53 reporter strain. The use of a diploid p53 reporter strain with two genomic copies each of the p53 expression cassette and the URA3 reporter gene (RBy99) eliminated 96% of the background of false-positives encountered with RBy41. For library screens, cDNA expression library plasmids were transformed into RBy99, transformants were placed on plates selecting for the plasmids (SC-His) and replica plated (‘RP’) after 3–5 days to plates containing FOA (SC-His + 0.1% FOA). Over the next 2–7 days, FoaR clones emerged which were then analyzed further as described in the text. (C) Mdm2 isolated as a p53 dissociator in a p53 dissociator screen. A p53 dissociator screen with a murine pre-B-cell cDNA expression library isolated four identical plasmids coding for Mdm2. RBy99 containing either the Mdm2 expression plasmid or control plasmid pPC86 was replica plated from SC-Trp to SC-Trp + 0.1% FOA at 30°C, and growth was evaluated after 3 days. (D) hADA3 and 53BP1 isolated as p53 dissociators in a p53 dissociator screen. RBy99 containing HeLa cDNA library clones for 53BP1 and hADA3 were replica plated from SC-His to SC-His + 0.075% FOA at 30°C, and growth was evaluated after 3 days. p2.5 was used as vector control.
The p53 dissociator assay was validated by transforming the haploid p53 reporter strain RBy41 with a plasmid expressing SV40 large T antigen (Clontech Laboratories), a viral antigen known to prevent DNA binding and transactivation by p53. SV40 large T antigen conferred an FoaR phenotype, while control strains were FoaS (Figure 1A). Our first p53 dissociator screen used a B-cell cDNA expression library. A total of 5500 of 5.5 × 106 transformants in RBy41 were FoaR. Of these, 3350 clones were analyzed further, but yielded no dissociator plasmids. The high percentage of 0.1% false-positives made screens in this form unfeasible. Further analysis established that 87% of false-positive FoaR clones were due to recessive mutations in the URA3 reporter gene, 9% due to recessive p53 mutations and 4% due to dominant-negative p53 mutations (Brachmann et al., 1996). This predicted that 96% of all false-positives would be eliminated with a diploid reporter strain with two genomic copies each of the p53 expression cassette and the URA3 reporter gene. The resulting new diploid p53 reporter strain RBy99 performed as expected: in three library screens, a total of 9.95 × 106 transformants resulted in 384 false-positives, representing 0.004% of all transformants (T.Kobayashi and R.K.Brachmann, unpublished data).
Figure 1B illustrates how p53 dissociator screens were performed. The cDNA expression library was transformed into RBy99, and transformants were placed on yeast plates selecting for the library plasmids. After 3–5 days, the lawn of transformants was replica plated (‘RP’) to selective plates containing FOA. FoaR clones emerged over the next 2–7 days. They were single colony purified and rechecked for the FoaR phenotype. After the plasmid dependency of the FoaR phenotype was determined, the plasmids were rescued and retransformed into RBy99 for phenotype confirmation prior to sequencing. A total of 1.5 × 105 transformants of a murine pre-B-cell cDNA expression library were screened and yielded 20 FoaR clones. Four clones passed all tests; their plasmids were identical and encoded murine Mdm2 (Figure 1C), a well characterized p53 inhibitor.
A p53 dissociator screen using a HeLa cell line-derived cDNA expression library
We next screened a HeLa cDNA expression library. A total of 1.73 × 106 transformants yielded 294 FoaR clones. For 204, the FoaR phenotype was confirmed after single colony purification, and 113 of them showed plasmid dependency of the FoaR phenotype. Sixty-one of the clones were FoaR upon retransformation of RBy99 after plasmid rescue. They represented seven candidate proteins not previously connected to p53, as well as 53BP1, a known p53 coactivator (Figure 1D; Iwabuchi et al., 1998).
hADA3 in the yeast p53 dissociator assay
One of the candidate proteins was hADA3 (eight of 61 library plasmids total, two independent clones; for phenotypes, see Figure 1D) that had just been identified as a component of multiprotein complexes containing either FLAG-tagged PCAF or hGCN5 (Ogryzko et al., 1998). hADA3 may have scored as a dissociator in the yeast p53 assay for various reasons, such as creation of a non-functional p53–hADA3 complex or disruption of endogenous yeast Ada complexes that are known to be required for p53 transcriptional acitivity in yeast (Candau et al., 1997). We decided to investigate the p53-related role of hADA3 in human cells, because its function was unknown, and it had been physically linked to PCAF, one of the HATs that can acetylate p53. An expressed sequence tag (EST) clone for full-length hADA3 (accession No. R17159; Ogryzko et al., 1998) was obtained, and expression plasmids for FLAG-tagged full-length hADA3 and the N- (amino acids 1–214) and C-terminal (amino acids 215–432) halves were constructed (Figure 2A).

Fig. 2. hADA3 interacts with components of HAT complexes. (A) Comparison of hADA3 with yAda3p and versions of hADA3 used in this study. Based on studies for yAda3p, the N-terminal half of hADA3 is predicted to interact with transcription factors, while the C-terminal half is predicted to interact with hADA2 and other members of HAT complexes. Besides full-length hADA3 and N-terminally truncated hADA3 from the HeLa library, the N- and C-terminal halves alone were also used in this study. (B) The C-terminal half of hADA3 interacts with hADA2 in yeast two-hybrid assays. PJ69-4A containing the indicated plasmid combinations were grown on SC-Leu-Trp plates and replica plated to SC-His at 30°C to evaluate activation of the reporter gene HIS3. Vector controls contained the GAL4 DNA-binding domain (GAL4-DBD) or GAL4 transactivation domain (GAL4-TAD) alone. Not all combinations of hADA2 and hADA3 could be evaluated, since full-length hADA3 and hADA3(aa1–214) fused to the GAL4-DBD activated the HIS3 reporter gene (data not shown). (C) The C-terminal half of hADA3 is in complexes with p300. FLAG-tagged full-length hADA3, the N-terminal half of hADA3 or the C-terminal half with NLS were co-expressed with p300 (CMVβ-p300) in 293 cells. After in vivo cross-linking with DTBP, anti-FLAG immunoprecipitation was followed by anti-p300 immunoblotting. ‘αp300 (before Co-IP)’ and ‘αFLAG (before Co-IP)’ show the amount of p300 and FLAG-tagged proteins in 4% of the cell lysate prior to co-immunoprecipitation.
Full-length hADA3 is 432 amino acids long, and both library clones lacked 5′ sequences [hADA3(aa119–432) and hADA3(aa168–432)]. hADA3 has 25% identity and 45% similarity with yAda3p; the C-terminal half shows higher conservation, while the N-terminal half of hADA3 is shorter and has significant gaps compared with yAda3p. Based on the alignment with and the biology of yAda3p, the N-terminal half of hADA3 is predicted to interact with transcription factors, and the C-terminal half with hADA2 and other components of HAT complexes (Figure 2A; Horiuchi et al., 1995; Candau and Berger, 1996; Ogryzko et al., 1998).
hADA3 interacts with components of HAT complexes
In yeast HAT complexes, the C-terminal half of yAda3p physically interacts with yAda2p, which in turn interacts with the yeast HAT yGcn5p (Horiuchi et al., 1995; Candau and Berger, 1996). We used yeast two-hybrid assays to establish the corresponding interaction between hADA3 and hADA2. Similarly to yeast, the C-terminal half of hADA3 was required for this interaction (Figure 2B).
The complexes of FLAG-tagged PCAF, hADA3 and other proteins surprisingly did not contain the HAT p300, even though PCAF had been described initially as a p300-interacting protein (Yang et al., 1996; Ogryzko et al., 1998). Since both PCAF and p300 can acetylate p53, we investigated whether hADA3 can be found in complexes with p300 as well. hADA3 co-immunoprecipitated p300, and the C-terminal half of hADA3 was sufficient for the complex formation with p300 (Figure 2C). Analogously to the experiments with hADA2, the N-terminal half of hADA3 was unable to co-immunoprecipitate p300, suggesting that the C-terminal half of hADA3 is crucial for linking it to HAT complexes.
hADA3 and p53 physically interact in human cells, and DNA damage dramatically enhances the interaction
We next determined whether hADA3 interacts with p53 in human cells. FLAG-tagged hADA3 co-immunoprecipitated endogenous wild-type p53. The N-terminal half of hADA3 was responsible for interaction with p53, while the C-terminal half immunoprecipitated little or no p53 (Figure 3A). We then investigated whether post-translational modifications of p53 modulate the interaction between hADA3 and p53 by subjecting the cells to γ-irradiation-induced DNA damage prior to co-immunoprecipitation. Following γ-irradiation, a dramatic increase in p53 co-immunoprecipitated with FLAG-tagged hADA3 was seen. In the same experiment, no change was found for p53 co-immunoprecipitated with SV40 large T antigen. As previously described, γ-irradiation resulted in increased levels of p53 protein phosphorylated at Ser15 (Figure 3B).

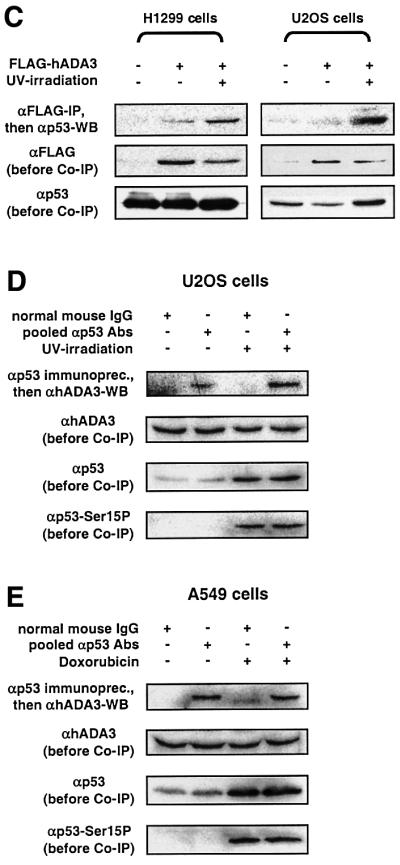
Fig. 3. The physical interaction of hADA3 and p53 in human cells requires the N-terminal half of hADA3 and is enhanced by DNA damage. (A) The N-terminus of hADA3 interacts with p53. FLAG-tagged full-length hADA3, the N-terminal half of hADA3 or the C-terminal half with NLS were expressed in 293 cells. Co-immunoprecipitation and immunoblotting of cell lysates were performed as in Figure 2C, except for the indicated antibodies. SV40 large T antigen and N-terminally truncated 53BP1 served as positive controls; the vector control was pFLAG-CMV2. (B) γ-irradiation markedly increases the amount of p53 co-immunoprecipitated with hADA3. Experiments were performed as in (A), except for the exposure to 80 Gy of γ-irradiation and lysis 1.5 h later. SV40 large T antigen was used as a control that did not show enhanced interaction with p53 after γ-irradiation. (C) UV-irradiation markedly increases the amount of p53 co-immunoprecipitated with hADA3. Experiments were performed as in (A) and (B), except for the exposure to 50 J/m2 UV-irradiation and lysis of cells 3 h later. p53-negative H1299 cells with transiently transfected p53 (pC53-SN3) do not show an increase of p53 protein after UV-irradiation due to high baseline p53 levels, while U2OS cells with endogenous wild-type p53 do. For H1299 cells, the enhanced p53–hADA3 interaction is seen despite unequal levels of FLAG-tagged hADA3 favoring the lane without UV-irradiation. (D and E) Endogenous hADA3 and p53 physically interact in human cells. To induce DNA damage, U2OS cells were exposed to 50 J/m2 UV-irradiation prior to lysis 3 h later (D) and A549 cells to 0.4 µg/ml doxorubicin for 12 h (E). After lysis, a mixture of five monoclonal anti-p53 antibodies, cross-linked to protein A– or protein G–agarose, was used to immunprecipitate p53, followed by detection of hADA3 by immunoblotting.
We further evaluated whether other types of DNA damage also lead to increased co-immunoprecipitation of p53 and hADA3. Using H1299 cells with transfected p53 and U2OS cells with endogenous p53, an enhanced p53–hADA3 interaction similar to that produced by γ-irradiation was seen in both cell lines after UV-irradiation (Figure 3C).
We also demonstrated the interaction between endogenous hADA3 and endogenous p53 in U2OS and A549 cells (Figure 3D and E). Both cell lines have wild-type p53 protein that is activated efficiently in U2OS cells by UV-irradiation (Figure 6A) and in A549 cells by doxorubicin, a chemotherapy agent that causes DNA double strand breaks (Figure 7A). U2OS cells after UV-irradiation showed an enhanced interaction between the endogenous proteins that was similar to experiments with FLAG-tagged hADA3 (compare Figure 3C and D). However, the interaction between hADA3 and p53 was not enhanced in A549 cells after doxorubicin treatment (Figure 3E). This result may be specific for A549 cells and doxorubicin. Alternatively, it may indicate that the interaction between p53 and hADA3 is transient and highly regulated by other factors. In the case of overexpressed FLAG-tagged hADA3, such cofactors may become limiting, thus leading to a more stable interaction between hADA3 and p53 that is then detected more readily in co-immunoprecipitation experiments.

Fig. 6. The N-terminal half of hADA3 prevents p53-mediated apoptosis. (A) U2OS cells with endogenous p53 were transfected with either vector control, SV40 large T antigen or hADA3(aa1–214) and membrane-bound GFP (pPCMVEGFPspectrin), and exposed to UV-irradiation (20 J/m2). GFP-positive cells were analyzed 24 h later for changes in the sub-G1 fraction of apoptotic cells. (C) SW480 cells were co-transfected with p53, pPCMVEGFPspectrin and vector control, SV40 large T antigen or hADA3(aa1–214). After 24 h, the sub-G1 fraction was determined as above. (B and D) The results were normalized to the percentage of apoptotic cells above background (expressed as 100%) that were induced by UV-irradiation in U2OS cells (B) or p53 transfection in SW480 cells (D) alone. Error bars represent the standard deviation for three independent experiments.
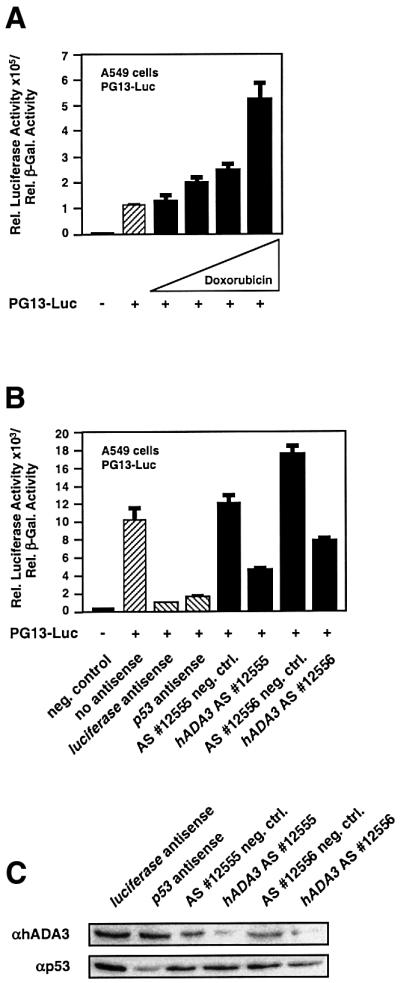
Fig. 7. Reduction of hADA3 protein results in decreased p53 transcriptional activity. (A) Doxorubicin increases p53 transcriptional activity in A549 cells. Reporter gene assays were performed as in Figure 5B, except that the transcriptional activity of endogenous p53 was evaluated in the presence of increasing amounts of doxorubicin (0.05, 0.1, 0.2 and 0.5 µg/ml). (B) Two antisense oligomers specific for hADA3 mRNA markedly reduce p53 transcriptional activity. Experiments were performed similarly to (A) with 0.5 µg/ml doxorubicin, except for the co-transfection of antisense oligomers. The results for the hADA3 oligomers were compared with specific control oligomers and oligomers for luciferase and p53 mRNA. p53 transcriptional activity in the absence of any antisense oligomers is also shown. (C) Two antisense oligomers specific for hADA3 mRNA reduce hADA3, but not p53 protein levels. Experiments were performed similarly to (B), except for anti-hADA3 and anti-p53 immunoblotting after cell lysis.
The enhanced interaction of hADA3 and p53 after DNA damage is governed by N-terminal phosphorylation of p53
We next evaluated whether mutations 22Q23S in the p53 transactivation domain that result in a transcriptionally inactive p53 protein (Lin et al., 1994) also affect the interaction with hADA3. Without DNA damage, approximately equal amounts of wild-type p53 and p53 22Q23S were immunoprecipitated with FLAG-tagged hADA3, suggesting a basal interaction of p53 with hADA3 and possibly other factors. However, contrary to wild-type p53, no increase of immunoprecipitated p53 22Q23S was seen after γ-irradiation (Figure 4A), indicating that the p53 N-terminus is important for the interaction with hADA3. The p53 N-terminus contains seven serine residues that can be phosphorylated in response to stress signals, such as DNA damage (Giaccia and Kastan, 1998; Ashcroft and Vousden, 1999; Chehab et al., 2000; Hirao et al., 2000; Oda et al., 2000; Shieh et al., 2000). To determine whether one or more of these phosphorylation sites are important for the enhanced p53–hADA3 interaction, we tested a p53 mutant that has serines 6, 9, 15, 20, 33, 37 and 46 replaced by alanines. This p53 mutant (p53-7A) did not show enhanced interaction with hADA3 after DNA damage, suggesting that phosphorylation events in the p53 N-terminus govern the interaction with hADA3 (Figure 4B).

Fig. 4. The enhanced interaction of hADA3 and p53 after DNA damage depends on N-terminal phosphorylation of p53. (A) γ-irradiation does not increase the amount of the transcriptionally inactive transactivation domain mutant p53 22Q23S co-immunoprecipitated with hADA3. Wild-type p53 and p53 22Q23S were transiently expressed in H1299 cells with hADA3 (or SV40 large T antigen as control), and co-immunoprecipitation was performed as in Figure 3A–C with and without 50 Gy of γ-irradiation. It is unclear why SV40 large T antigen co-immunoprecipitates p53 22Q23S as three bands. (B) One or several phosphorylation events in the p53 N-terminus govern the interaction with hADA3. Wild-type p53 and p53 with alanines instead of serines at positions 6, 9, 15, 20, 33, 37 and 46 (p53-7A) were evaluated with hADA3 as in (A) with and without 30 Gy of γ-irradiation.
Full-length hADA3 increases the transcriptional activity of p53
We then determined in reporter gene assays whether hADA3 influences p53 transcriptional activity in human cells (Brachmann et al., 1998). p53-negative H1299 cells were transiently transfected with p53 and increasing amounts of FLAG-hADA3. The results consistently showed a 2- to 4-fold increase in p53 transcriptional activity (Figure 5A and B). The protein levels of p53 were unaffected by co-expression of hADA3 (Figure 5A). The effect of hADA3 on reporter activation was p53 dependent, as hADA3 alone did not lead to luciferase expression. The stimulatory effect of hADA3 was only seen with overexpressed p53, suggesting that hADA3 is not limiting for endogenous p53 (data not shown).

Fig. 5. Full-length hADA3 increases and the N-terminal half of hADA3 inhibits p53 transcriptional activity. (A) p53 reporter gene assays with WWP-Luc. Increasing amounts of an expression plasmid for FLAG-tagged full-length hADA3 (50, 100 and 500 ng) were co-transfected into p53-negative H1299 cells with the p53 expression plasmid pC53-SN3 (100 ng), pIC400 expressing β-galactosidase (250 ng) and the reporter plasmid WWP-Luc containing luciferase under the control of the p21WAF1/CIP1/SDI1 promoter (500 ng). The cells were lysed after 24 h, and the luciferase activity, adjusted for β-galactosidase, was determined. Error bars represent the standard deviation for three independent experiments. p53 protein levels were assessed by anti-p53 immunoblotting after adjustment of the cell lysates from the shown reporter gene assays for β-galactosidase activity. (B) p53 reporter gene assays with PG13-Luc. The experiments were performed as described for (A), except that PG13-Luc containing a tandem array of 13 p53-binding sites and 100, 500 and 900 ng of FLAG-tagged hADA3 were used. (C) hADA3(aa1–214) inhibits p53 transcriptional activity. Increasing amounts of expression plasmids (100, 500 and 800 ng) for hADA3(aa1–214) with and without an NLS were co-transfected with 500 ng of PG13-Luc and 500 ng of pIC400 into 293 cells with endogenous wild-type p53. Luciferase activity was determined, and anti-FLAG immunoblotting for cell lysates with 800 ng of hADA3 expression plasmid was performed similarly to (A) for p53.
The N-terminal half of hADA3 inhibits p53 transcriptional activity
Our co-immunoprecipitation and yeast two-hybrid experiments (Figures 2 and 3) established that the C-terminal half of hADA3 is responsible for the interaction with components of HAT complexes (hADA2 and p300) and that its N-terminal half [hADA3(aa1–214)] interacts with p53. We reasoned that hADA3(aa1–214) might interfere with p53 transcriptional activity by creating a non-functional complex with p53. Consistent with this, the N-terminal half of hADA3 strongly inhibited p53 transcriptional activity in 293 cells with endogenous wild-type p53. This dominant-negative effect did not require the presence of an artificial nuclear localization signal (NLS; Figure 5C).
The N-terminal half of hADA3 prevents p53-mediated apoptosis
We investigated whether the reduction of p53 transcriptional activity in reporter gene assays by the N-terminal half of hADA3 correlated with suppression of p53-mediated apoptosis. U2OS cells with endogenous p53 were transfected with either vector control, SV40 large T antigen or hADA3(aa1–214) and membrane-bound green fluorescent protein (GFP) (Kalejta et al., 1997), and were exposed to UV-irradiation (20 J/m2). After 24 h, GFP-positive cells were evaluated by fluorescence-activated cell sorting (FACS) analysis for changes in the proportion of apoptotic cells, represented by the sub-G1 fraction. hADA3(aa 1–214) dramatically and consistently reduced the fraction of apoptotic cells to <30% of the control, i.e. UV-irradiation alone (Figure 6B; see Figure 6A for a representative FACS profile). This compared favorably with the effect seen with SV40 large T antigen. Very similar results were obtained in SW480 cells with mutant endogenous p53 in which apoptosis was triggered by overexpressing wild-type p53. In the presence of hADA3(aa1–214) or SV40 large T antigen, the fraction of apoptotic cells was reduced to <20% of the control, i.e. transfected p53 alone (Figure 6D; see Figure 6C for a representative FACS profile).
hADA3 is required for full transcriptional activity of p53
We then explored whether removal of hADA3 using antisense oligomers would also result in a significant reduction of p53 activity. We chose A549 cells since exposure of these cells to doxorubicin leads to a >5-fold increase in p53 transcriptional activity (Figure 7A) and since >90% of all cells take up antisense oligomers in a typical experiment (data not shown). Antisense oligomers 12555 and 12556 for hADA3 mRNA (Sequitur, MA) reduced p53 transcriptional activity to 35 and 43% of their specific negative controls, respectively (Figure 7B). The extent of p53 inhibition was similar to the reduction that was observed with antisense oligomers specific for luciferase and p53 mRNA (Sequitur, MA; Figure 7B). Both antisense oligomers specific for hADA3 mRNA reduced hADA3 protein to levels that corresponded well to the reduction in p53 activity, but did not affect p53 protein levels (Figure 7C).
Discussion
A novel yeast p53 dissociator assay that is based on the principles of the reverse two-hybrid system (Vidal et al., 1996) allowed us to identify hADA3 as an important player in p53 biology. The basic p53 dissociator assay was used previously to address other p53-related questions, such as the characteristics of dominant-negative p53 mutants and intragenic suppressor mutations for common p53 cancer mutations (Brachmann et al., 1996, 1998). We have optimized the p53 dissociator assay for highly efficient p53 dissociator screens to identify novel proteins important to p53 biology. This report establishes the feasibility of the reverse two-hybrid technology for such dissociator screens. The same strategies can be applied to many other protein–protein or protein–DNA interactions of interest.
In our collection of candidate proteins obtained from a yeast p53 dissociator screen, hADA3 was particularly interesting since it is homologous to yAda3p, a yeast protein that functions as an adaptor between transcription factors and HAT complexes. The N-terminus of yAda3p interacts with transcription factors, while its C-terminus contributes to the formation of yeast HAT complexes by interacting with yAda2p (Horiuchi et al., 1995; Candau and Berger, 1996). Human HAT complexes have a very similar composition to yeast HAT complexes, and, like its yeast counterpart, hADA3 is part of HAT complexes (Ogryzko et al., 1998). HAT complexes enhance p53 transcriptional activity, probably through two mechanisms: full transcriptional activation of p53 as a result of C-terminal acetylation by p300/CBP and PCAF (Gu and Roeder, 1997; Sakaguchi et al., 1998; Liu et al., 1999; Chao et al., 2000; Ito et al., 2001) and relaxation of chromatin at p53-responsive promoters through acetylation of histones (Giles et al., 1998; Struhl, 1998; Schiltz and Nakatani, 2000; Sterner and Berger, 2000).
hADA3 was first isolated in multiprotein complexes with FLAG-tagged PCAF or hGCN5, complexes that also contained hADA2 (Ogryzko et al., 1998). We show that, analogously to yAda3p, the C-terminal half of hADA3 interacts with hADA2. Even though PCAF and p300 have been demonstrated to interact, p300 surprisingly was not identified in the purified multiprotein complexes (Yang et al., 1996; Ogryzko et al., 1998). In this report, we demonstrate that hADA3 complexes with p300 and that the C-terminal half of hADA3 is responsible for this. Thus, hADA3 interacts with two HATs known to acetylate p53.
Using immunoprecipitation assays, we have clearly demonstrated an endogenous physical interaction between hADA3 and p53 in human cells. There appears to be a basal interaction between p53, hADA3 and probably other factors in the absence of DNA damage. This basal interaction between p53 and hADA3 is enhanced dramatically by γ- and UV-irradiation. Thus, the interaction with hADA3 appears to be part of a general response of p53 to DNA damage.
We found that, in the absence of DNA damage, the transcriptionally inactive double mutant p53 22Q23S was immunoprecipitated with hADA3 in amounts similar to wild-type p53. However, upon DNA damage, the basal interaction between the inactive p53 22Q23S and hADA3 was not augmented as was the interaction between wild-type p53 and hADA3. These data establish that the p53 N-terminus is instrumental in regulating the interaction with hADA3. The p53 mutant with alanines instead of serines at positions 6, 9, 15, 20, 33, 37 and 46 (p53-7A) behaved similarly to p53 22Q23S in co-immunoprecipitation experiments with hADA3. This further suggests that one or more specific phosphorylation event(s) in the p53 N-terminus are required for the enhanced interaction with hADA3.
Recent data in two murine knock-in models for the mutant p53 25Q26S, corresponding to the human mutant p53 22Q23S, showed that p53 25Q26S was transactivation deficient and unable to induce cell cycle arrest or apoptosis in response to DNA damage (Chao et al., 2000; Jimenez et al., 2000). While p53 25Q26S bound to p53 DNA-binding sites constitutively, its DNA-binding capacity did not increase with DNA damage, contrary to what was observed for wild-type p53 (Jimenez et al., 2000). Strikingly, and in contrast to wild-type p53, p53 25Q26S entirely lacked C-terminal acetylation at Lys317 and Lys379 (corresponding to human Lys320 and Lys382) after DNA damage, despite phosphorylation at Ser18 (human Ser15; Chao et al., 2000). These and our results establish an intriguing correlation between transcriptionally inactive p53, absence of p53 acetylation and lack of an enhanced p53–hADA3 interaction after DNA damage.
In our experiments, full-length hADA3 increased p53 transcriptional activity up to 4-fold without concomitant increases in p53 protein levels in transient transfection experiments overexpressing both hADA3 and p53. yAda3p has no known enzymatic activity, making it unlikely that hADA3 had a stimulatory effect on p53 through direct post-translational modifications of p53. A more likely scenario is that the endogenous level of hADA3 is not sufficient for the high level of overexpressed p53 protein. Thus, overexpression of hADA3 increases the pool of hADA3 protein, leading to enhanced p53 transcriptional activity.
The overexpression studies discussed above have established an important role for hADA3 in p53 transcriptional activity, and we have complemented these experiments with two loss-of-function analyses. In the first, we found that an N-terminally truncated form of hADA3 [hADA3(aa1–214)] interacts with p53, but not components of HAT complexes, perhaps leading to a non-functional complex with p53. We demonstrated that hADA3(aa1–214) had a strong dominant-negative effect on p53 transcriptional activity and interfered with p53-mediated apoptosis. In the second loss-of-function approach, we used two different antisense oligomers for hADA3 mRNA to substantiate further that p53 requires access to hADA3 for full transcriptional activity. The antisense oligomers reduced hADA3 protein levels and, as a consequence, p53 transcriptional activity. These data expand on previous experiments showing that yeast Ada3p, as well as yeast Ada2p and Gcn5p, are required for transcriptional activity of p53 in yeast (Candau et al., 1997).
p53 has been shown to interact directly with PCAF (Liu et al., 1999), p300 (Avantaggiati et al., 1997; Lill et al., 1997) and CBP (Gu et al., 1997; Scolnick et al., 1997), raising the question of how binding of p53 to hADA3 and various HATs is coordinated. Perhaps HATs are able to interact with p53 in vitro, but in living cells require the cooperation of additional factors to communicate efficiently with p53. In this case, hADA3 would promote a productive interaction between p53 and various HATs. This would be reminiscent of the differing abilities of HATs to acetylate free histones or nucleosomes depending on the absence or presence of a HAT complex (Struhl, 1998; Giles et al., 1998; Schiltz and Nakatani, 2000; Sterner and Berger, 2000). This scenario is also attractive as it provides a novel mechanism of specificity for HAT activities. By preferentially interacting with p53 protein that is stabilized through N-terminal phosphorylation after DNA damage, hADA3 may ensure that p53 is only acetylated by HATs when appropriate. Strictly speaking, our data are also consistent with a role for hADA3 in p53 transcriptional activity that is independent of HATs. This appears less likely though, based on the physical linkage of hADA3 to PCAF and p300 and the correlative data for the mutant p53 22Q23S.
In summary, our data suggest that hADA3 is an important cofactor for the transcription factor p53. It is very likely that hADA3 plays this role in the context of HAT complexes. The enhanced interaction of hADA3 and p53 after DNA damage depends on N-terminal phosphorylation event(s) of p53. The contribution of specific post-translational modifications of p53 to the interaction with hADA3 needs to be explored further, since it may represent a novel mechanism of specificity that regulates interactions between p53 and HAT complexes based on the post-translational state of p53. Likewise, it needs to be investigated whether and how absence of hADA3 function affects acetylation of p53. Since proper hADA3 function is required for full p53 activity, it raises the question of whether genetic alterations of hADA3 may account for decreased p53 activity in some human cancers with wild-type p53.
Materials and methods
cDNA libraries, plasmids and plasmid constructions
The screened cDNA expression libraries were derived from the following: B-cells (S.Malek and S.Desiderio, The Johns Hopkins University School of Medicine, Baltimore, MD, unpublished data), murine pre-B-cells (P.Stanhope and M.Schlissel, University of California, Berkeley, unpublished data) and the HeLa cell line [provided by M.Brasch, Life Technologies, Inc. (LTI)]. For the HeLa library, cDNAs were constructed using oligo(dT) NotI primers and 5′ SalI–MluI adaptors, as described in the Superscript Plasmid System, and ligated into SalI–NotI-digested p2.5 (Vidal et al., 1996). The library had 2.1 × 107 primary clones of which 1 × 107 were amplified. The average insert size was 1.3 kb, and 91% of vectors had inserts.
The following plasmids were used: pTD1-1 (Clontech Laboratories), pFLAG-CMV2 (Sigma), pPCMVEGFPspectrin (Kalejta et al., 1997), p53 22Q23S in pRC/CMV (Lin et al., 1994), pJL134 with SV40 large T antigen (J.Li, unpublished data), CMVβ-p300 (Eckner et al., 1994), pPC97 and pPC86 (Chevray and Nathans, 1992), pIC400 (Brachmann et al., 1998) and MG15-Luc, PG13-Luc, WWP-Luc and pC53-SN3 (Baker et al., 1990; el-Deiry et al., 1993).
Mdm2 was isolated from the murine pre-B-cell, and hADA3(aa119– 432), hADA3(aa168–432) and 53BP1(aa971–1972) were isolated from the HeLa cDNA expression library. EST clones R17159 and AA856985 (Washington University Genome Sequencing Center, St Louis) were the cDNA sources for hADA3 and hADA2, respectively. The first 598 bp of hADA3 were PCR amplified with primers RB117 (GAAGCTTAT GAGTGAGTTGAAAGA) and RB118 (AGCGCTGGGAGTAGTGC TTC) to introduce a 5′ HindIII site. The PCR product was combined with the remainder of the hADA3 cDNA, and the entire cDNA was sequence verified. A similar strategy was used for hADA2; the first 313 bp were amplified with primers RB119 (GAAGCTTATGGCCCTTTTAGAAGC) and RB120 (ATTCTTGCAAATCGCCTCAT). For two-hybrid assays, hADA3 and hADA2 were cloned into new versions of pPC97 and pPC86 (Chevray and Nathans, 1992) with markers TRP1 and LEU2, respectively. For experiments in mammalian cells, SV40 large T antigen, 53BP1(aa971–1972) and hADA3 were cloned into pFLAG-CMV2 (Sigma). Truncated versions of hADA3 were constructed by digestion and religation of full-length hADA3 expression plasmids. For the N-terminal half of hADA3, this resulted in the addition of the peptide PLSK in the case of the two-hybrid constructs and VASL for constructs in pFLAG-CMV2. Constructs with an NLS were derived from full-length hADA3 with an NLS in pFLAG-CMV2. To obtain a matching set of expression plasmids for p53 and p53-7A, the natural open reading frame (ORF) of p53 in pCMV-p53 (Clontech Laboratories) was replaced with designer p53 ORFs (T.Wang and R.K.Brachmann, unpublished data) encoding wild-type p53 or p53 with alanines at positions 6, 9, 15, 20, 33, 37 and 46.
Studies in S.cerevisiae
Techniques for yeast maintenance and the p53 dissociator assay have been described (Brachmann et al., 1996, 1998; Vidal et al., 1996). RBy99 resulted from the mating of two haploid strains, each containing the p53 expression cassette integrated at the LYS2 locus and the URA3 reporter construct. Two-hybrid studies were performed in strain PJ69-4A (James et al., 1996).
Cell culture, transfection and reporter gene assays
A549, H1299, 293 and SW480 cell lines were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM), and U2OS cells in McCoy 5A media, all with 10% fetal bovine serum. Lipofectamine (LTI) was used for all transfections, except for U2OS (SuperFect; Qiagen) and SW480 cells (Lipofectin; LTI). Reporter gene assays were performed as previously described (Brachmann et al., 1998). Immunoblotting was performed with the same, β-galactosidase adjusted, lysates used for reporter gene assays. Anti-p53 antibody PAb1801 (Oncogene Research Products, MA) was used at 2.5 µg/ml and anti-FLAG M5 (Sigma) at 5 µg/ml.
Co-immunoprecipitation experiments
Cells were seeded at 1 × 106 cells per 60 mm plate and transfected with 3 µg of total DNA. After 24 h, cells were either lysed, γ-irradiated with 30, 50 or 80 Gy and lysed 1.5 h later, or UV-irradiated with 50 J/m2 and lysed 3 h later. Cells were also exposed to doxorubicin (Sigma) at 0.4 µg/ml starting 1 h after transfection until lysis 24 h later. Cells were washed with phosphate-buffered saline (PBS), treated with 1 mM dimethyl 3,3′-dithiobispropionimidate (2HCl) (DTBP) for 20 min at room temperature (Xu and Reed, 1998) and lysed with 500 µl of ELB (50 mM HEPES, pH 7.2; 250 mM NaCl; 5 mM EDTA, pH 8.0; 0.5% NP-40) containing protease and phosphatase inhibitors. After gentle rocking for 5 min, samples were pipetted up and down 6–8 times for 10 min, centrifuged twice at 20 800 g for 10 min and exposed to FLAG-M2 beads (Sigma) for 1–2 h (0–4°C). Beads were washed three times with ELB and once with PBS, spun down for 2 min at 500 g, mixed with 20–30 µl of loading buffer and boiled for 4 min prior to standard immunoblotting analysis. The rabbit anti-phospho-p53 (Ser15) antibody (New England Biolabs, MA) was used at 1:1000, rabbit and goat anti-p53 antibodies FL-393(G) at 0.4 µg/ml and rabbit anti-p300 antibody (C20) at 0.4 µg/ml (Santa Cruz Biotechnology).
To demonstrate the interaction of endogenous p53 and hADA3, five anti-p53 antibodies, PAb421, PAb1620 (Oncogene Research Products, MA), PAb240, G59-12 and PAb122 (BD Pharmingen) were cross-linked to protein A– or protein G–agarose (Santa Cruz Biotechnology), with dimethyl pimelimidate (2HCl) (DMP) (Harlow and Lane, 1999), and a mixture of these was used to immunoprecipitate p53 from the lysate of five confluent 75 cm2 flasks of A549 or U2OS cells. Normal mouse IgG cross-linked to protein A– and protein G–agarose was the negative control. Co-immunoprecipitated hADA3 was detected using a rabbit polyclonal anti-hADA3 antibody at 1:500 (Brand et al., 1999).
Flow cytometry
Apoptosis assays were performed as described (Ozoren et al., 2000). A total of 1 × 106 U2OS cells were transfected with either pFLAG-CMV2, hADA3(aa1–214) or SV40 large T antigen and pPCMVEGFPspectrin, exposed to UV-irradiation (20 J/m2) after 12 h, and 24 h later fixed in 70% ethanol overnight and stained with 50 µg/ml propidium iodide. SW480 cells additionally were transfected with pC53-SN3 or pCMVneo and analyzed 24 h later. The results were normalized to the percentage of apoptotic cells above background (expressed as 100%) that were induced by UV-irradiation (U2OS) or p53 transfection (SW480) alone.
Antisense oligomer experiments
All antisense reagents and protocols used for A549 cells were from Sequitur, MA. The sequences of the hADA3 oligomers are: 12555, UCAUGGUCUCGACCCAGCUUCAGGA; 12156 (negative control for 12555), UGCCCAGCUGGUCUCUCGAUCAAGA; 12556, AAUUAG CUCCUCCUUGAUGCGGCUC; 12070 (negative control for 12556), CAGUUGACGUCUCUGUAACUUCCCG.
Acknowledgments
Acknowledgements
We thank A.Beavis, A.Bedi, M.Brasch, S.Desiderio, E.Harlow, P.James, M.Kastan, S.Korsmeyer, A.Levine, J.Li, D.Livingston, S.Malek, D.Nathans, J.Nathans, M.Schlissel, P.Stanhope and B.Vogelstein for providing reagents. Special thanks go to Y.Nakatani who generously provided the polyclonal antibody against hADA3. We thank C.B.Brachmann and D.Dean for comments on the manuscript. R.K.B. thanks J.D.Boeke for his unwavering support of the idea of the p53 dissociator assay during its earlier phases in his laboratory. This work was supported in part by grants from the James S.McDonnell Foundation, the Concern Foundation, the Edward Mallinckrodt, Jr Foundation and National Institutes of Health grant CA87468 (R.K.B.).
References
- Ashcroft M. and Vousden,K.H. (1999) Regulation of p53 stability. Oncogene, 18, 7637–7643. [DOI] [PubMed] [Google Scholar]
- Avantaggiati M.L., Ogryzko,V., Gardner,K., Giordano,A., Levine,A.S. and Kelly,K. (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell, 89, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Baker S.J., Markowitz,S., Fearon,E.R., Willson,J.K. and Vogelstein,B. (1990) Suppression of human colorectal carcinoma cell growth by wild-type p53. Science, 249, 912–915. [DOI] [PubMed] [Google Scholar]
- Brachmann R.K., Vidal,M. and Boeke,J.D. (1996) Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc. Natl Acad. Sci. USA, 93, 4091–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann R.K., Yu,K., Eby,Y., Pavletich,N.P. and Boeke,J.D. (1998) Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J., 17, 1847–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M., Yamamoto,K., Staub,A. and Tora,L. (1999) Identification of TATA-binding protein-free TAFII-containing complex subunits suggests a role in nucleosome acetylation and signal transduction. J. Biol. Chem., 274, 18285–18289. [DOI] [PubMed] [Google Scholar]
- Candau R. and Berger,S.L. (1996) Structural and functional analysis of yeast putative adaptors. Evidence for an adaptor complex in vivo. J. Biol. Chem., 271, 5237–5245. [DOI] [PubMed] [Google Scholar]
- Candau R., Scolnick,D.M., Darpino,P., Ying,C.Y., Halazonetis,T.D. and Berger,S.L. (1997) Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene, 15, 807–816. [DOI] [PubMed] [Google Scholar]
- Chao C., Saito,S., Kang,J., Anderson,C.W., Appella,E. and Xu,Y. (2000) p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J., 19, 4967–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehab N.H., Malikzay,A., Appel,M. and Halazonetis,T.D. (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev., 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- Chevray P.M. and Nathans,D. (1992) Protein interaction cloning in yeast: identification of mammalian proteins that react with the leucine zipper of Jun. Proc. Natl Acad. Sci. USA 89, 5789–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- el-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Giles R.H., Peters,D.J. and Breuning,M.H. (1998) Conjunction dysfunction: CBP/p300 in human disease. Trends Genet., 14, 178–183. [DOI] [PubMed] [Google Scholar]
- Grant P.A., Sterner,D.E., Duggan,L.J., Workman,J.L. and Berger,S.L. (1998) The SAGA unfolds: convergence of transcription regulators in chromatin-modifying complexes. Trends Cell Biol., 8, 193–197. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Gu W., Shi,X.L. and Roeder,R.G. (1997) Synergistic activation of transcription by CBP and p53. Nature, 387, 819–823. [DOI] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1999) Using Antibodies—A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Mdm2 promotes the rapid degradation of p53. Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong,Y.Y., Matsuoka,S., Wakeham,A., Ruland,J., Yoshida,H., Liu,D., Elledge,S.J. and Mak,T.W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase chk2. Science, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Horiuchi J., Silverman,N., Marcus,G.A. and Guarente,L. (1995) ADA3, a putative transcriptional adaptor, consists of two separable domains and interacts with ADA2 and GCN5 in a trimeric complex. Mol. Cell. Biol., 15, 1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Lai,C.H., Zhao,X., Saito,S., Hamilton,M.H., Appella,E. and Yao,T.P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J., 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K., Li,B., Massa,H.F., Trask,B.J., Date,T. and Fields,S. (1998) Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem., 273, 26061–26068. [DOI] [PubMed] [Google Scholar]
- James P., Halladay,J. and Craig,E.A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics, 144, 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez G.S., Nister,M., Stommel,J.M., Beeche,M., Barcarse,E.A., Zhang,X.Q., O’Gorman,S. and Wahl,G.M. (2000) A transactivation-deficient mouse model provides insights into trp53 regulation and function. Nature Genet., 26, 37–43. [DOI] [PubMed] [Google Scholar]
- Kalejta R.F., Shenk,T. and Beavis,A.J. (1997) Use of a membrane-localized green fluorescent protein allows simultaneous identification of transfected cells and cell cycle analysis by flow cytometry. Cytometry, 29, 286–291. [DOI] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat M.H., Jones,S.N. and Vousden,K.H. (1997) Regulation of p53 stability by Mdm2. Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lill N.L., Grossman,S.R., Ginsberg,D., DeCaprio,J. and Livingston,D.M. (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature, 387, 823–827. [DOI] [PubMed] [Google Scholar]
- Lin J., Chen,J., Elenbaas,B. and Levine,A.J. (1994) Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev., 8, 1235–1246. [DOI] [PubMed] [Google Scholar]
- Liu L., Scolnick,D.M., Trievel,R.C., Zhang,H.B., Marmorstein,R., Halazonetis,T.D. and Berger,S.L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol., 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K. et al. (2000) p53AIP1, a potential mediator of p53-dependent apoptosis and its regulation by ser-46-phosphorylated p53. Cell, 102, 849–862. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Kotani,T., Zhang,X., Schlitz,R.L., Howard,T., Yang,X.J., Howard,B.H., Qin,J. and Nakatani,Y. (1998) Histone-like TAFs within the PCAF histone acetylase complex. Cell, 94, 35–44. [DOI] [PubMed] [Google Scholar]
- Oren M. (1999) Regulation of the p53 tumor suppressor protein. J. Biol. Chem., 274, 36031–36034. [DOI] [PubMed] [Google Scholar]
- Ozoren N. et al. (2000) Homozygous deletion of the death receptor DR4 gene in a nasopharyngeal cancer cell line is associated with TRAIL resistance. Int. J. Oncol., 16, 917–925. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltz R.L. and Nakatani,Y. (2000) The PCAF acetylase complex as a potential tumor suppressor. Biochim. Biophys. Acta, 1470, 37–53. [DOI] [PubMed] [Google Scholar]
- Scolnick D.M., Chehab,N.H., Stavridi,E.S., Lien,M.C., Caruso,L., Moran,E., Berger,S.L. and Halazonetis,T.D. (1997) CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res., 57, 3693–3696. [PubMed] [Google Scholar]
- Shieh S.Y., Ahn,J., Tamai,K., Taya,Y. and Prives,C. (2000) The human homologs of checkpoint kinases chk1 and cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev., 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Sterner D.E. and Berger,S.L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Vidal M., Brachmann,R.K., Fattaey,A., Harlow,E. and Boeke,J.D. (1996) Reverse two-hybrid and one-hybrid systems to detect dissociation of protein–protein and DNA–protein interactions. Proc. Natl Acad. Sci. USA, 93, 10315–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q. and Reed ,J.C. (1998) Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol. Cell, 1, 337–346. [DOI] [PubMed] [Google Scholar]
- Yang X.J., Ogryzko,V.V., Nishikawa,J., Howard,B.H. and Nakatani,Y. (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]