

Abstract
Fet3p is a multicopper-containing glycoprotein localized to the yeast plasma membrane that catalyzes the oxidation of Fe(II) to Fe(III). This ferrous iron oxidation is coupled to the reduction of O2 to H2O and is termed the ferroxidase reaction. Fet3p-produced Fe(III) is transferred to the permease Ftr1p for import into the cytosol. The posttranslational insertion of four copper ions into Fet3p is essential for its activity, thus linking copper and iron homeostasis. The mammalian ferroxidases ceruloplasmin and hephaestin are homologs of Fet3p. Loss of the Fe(II) oxidation catalyzed by these proteins results in a spectrum of pathological states, including death. Here, we present the structure of the Fet3p extracellular ferroxidase domain and compare it with that of human ceruloplasmin and other multicopper oxidases that are devoid of ferroxidase activity. The Fet3p structure delineates features that underlie the unique reactivity of this and homologous multicopper oxidases that support the essential trafficking of iron in diverse eukaryotic organisms. The findings are correlated with biochemical and physiological data to cross-validate the elements of Fet3p that define it as both a ferroxidase and cuprous oxidase.
Keywords: ferroxidase, iron trafficking, metal homeostasis, multicopper oxidase
The link between copper nutrition and iron homeostasis in mammals was first described by Wintrobe and coworkers (1) in the 1950s. Almost all of the manifestations of decreased iron, e.g., ataxia, lowered hemocrit, and reduced mitochondrial function, were replicated in animals limited in copper. These symptoms were associated with loss of the activity of heme and iron, sulfur cluster-containing enzymes. There also existed biochemical markers specific to copper deficiency in these animals, including reduced activity of the serum enzyme ceruloplasmin (Cp) (1-3).
Cp was first characterized as a copper-containing enzyme by Holmberg and Laurell (4), who, based on its “sky blue” color, gave the protein its name (cæruleus). Cp is a member of a large family of proteins (>500 homologs found in all three super kingdoms) known as multicopper oxidases (MCOs). MCOs that contain ≈500 amino acid residues are composed of three Greek key β-barrel cupredoxin (plastocyanin-like) domains (5) that come together to form three spectroscopically distinct copper binding sites termed type 1 (T1), type 2 (T2), and type 3 (T3) (6, 7). With >1,100 residues, Cp differs from Fet3p through duplication of this trimeric cupredoxin assembly (8). Although it was evident why Cp activity would decline in copper deficiency, it was unclear why iron deficiency would correlate with this deficit. The molecular basis for this link was revealed through a demonstration of an activity toward Fe(II) by Cp not shared by other MCOs known at the time (9, 10). This ferroxidase reaction catalyzed by Cp is given in Eq. 1.
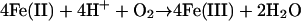 |
[1] |
Frieden and coworkers (9, 10) proposed that Fe(II) oxidation by Cp facilitates the transfer of iron to transferrin and that it was essential for iron trafficking from the intestinal epithelial cells to the liver and extrahepatic tissues. A role for Cp (and its paralog, hephaestin) in iron metabolism is now generally accepted (11), and the pathology observed in aceruloplasminemic patients is stark evidence for this role (12).
The molecular basis of the Fe(II) specificity exhibited by Cp but not by the other MCO proteins (e.g., several laccases and ascorbate oxidase) has not been fully elucidated, and how this ferroxidase activity is linked to iron homeostasis in mammals remains unclear. However, this copper-iron link was recognized to exist also in Saccharomyces cerevisiae and in this organism was demonstrated to correlate with the activity of the ferroxidase-dependent, high-affinity iron uptake complex in the yeast plasma membrane (13-19) (Fig. 1). This complex is composed of the iron permease Ftr1p and the MCO Fet3p, which cotranslocate to the plasma membrane from the secretory pathway. In yeast (and likely all other fungi and plants), environmental Fe(III) is mobilized by the reduction to Fe(II) by surface metalloreductases such as Fre1p, followed by oxidation by Fet3p. This Fet3p-bound Fe(III) is the substrate for permeation facilitated by Ftr1p (15). Uptake is strictly coupled to ferroxidation, as exogenous Fe(III) is not substrate for Ftr1p. The yeast system has thus provided a tractable model system to test the mechanisms that underlie the copper-iron connection in biology (19, 20). The 3D structure of Fet3p and supporting functional data presented here provide the framework for the delineation of this fundamental and apparently conserved metabolite trafficking pathway.
Fig. 1.

The link between copper and iron homeostasis in yeast. Cu(II) and Fe(III) are made substrates for permeation through reduction by the metalloreductase Fre1p. Cu(I) enters the cell via Ctr1p and is trafficked to the post-Golgi by the copper chaperone Atx1p. This Cu(I) is transferred to the P-type ATPase Ccc2p, which transports it into the lumen where it activates the apo form of Fet3p. In association with the high-affinity iron permease Ftr1p, Fet3p traffics to the plasma membrane. There, its ferroxidase activity supports the uptake of Fe via Ftr1p. Cells that lack copper also lack iron because of the absence of active Fet3p.
Materials and Methods
Protein Expression and Purification. Fet3p was overexpressed in yeast and purified as described (21). Briefly, a recombinant form of Fet3p is truncated at residue 555 and lacks its C-terminal transmembrane domain. This secreted Fet3p is isolated from the growth medium, purified by ion-exchange chromatography, and digested with endo-β-N-acetylglucosaminidase H (endo H) to remove high mannose carbohydrate.
Kinetic Analyses. Kinetic analyses of the ferroxidase reaction with ferrous ammonium sulfate (Sigma) as substrate were based on oxygen consumption by using an Oxygraph (Hansatech Instruments, Norfolk, U.K.). This method has been described in detail (22). All initial velocity, v, versus [S] data were analyzed by direct fitting to the Michaelis-Menten equation by using prism software (GraphPad, San Diego). Kinetic analyses of 59Fe uptake were performed as described (23) in yeast strain AJS04, a derivative of DEY1457 (24); the AJS04 genotype is MATα can1 his3 leu2 trp1 ura3 ade6 fet3::HIS aft1::AFT1upKAN. The AFT1up allele codes for a constitutively active form of the Aft1p transcription factor that drives expression of the FET3 locus.
Crystallization. Suitable crystals were obtained by using ≈80 mg/ml of endo H-treated Fet3p protein in 25 mM Mes (pH 6) and 400 μM FeCl3. Crystals grew in 7 days from hanging drops containing equal volumes (1-2 μl) of protein solution and reservoir solution containing 1.2-1.4 M sodium citrate, 0.1 M Hepes (pH 7.5), 3% ethanol, and 5% ethylene glycol. The crystals were merohedrally twinned, with twin fractions typically >0.4 as calculated by the online Crystal Twinning Server (25). Screening additives to the crystallization conditions, including known inhibitors of MCO activity fluoride and azide, produced two conditions with minimized crystal twinning, although these compounds were not observed in the final structure.
Data Collection, Structure Determination, and Refinement. Crystals were flash-cooled in liquid nitrogen before x-ray data collection at beamline 19BM at the Advanced Photon Source (Argonne, IL). Molecular replacement trials using laccase or ascorbate oxidase models from the Protein Data Bank (26) were unsuccessful, as were attempts at derivatization of the fragile crystals with heavy atoms. We thus pursued multiwavelength anomalous diffraction (MAD) phasing (27) with the intrinsic copper atoms. A three-wavelength copper MAD experiment was performed, and a 2.8-Å native data set was collected after translating the crystal in the beam after the MAD data collection. Diffraction data were processed with hkl-2000 (28), and data collection statistics are listed in Table 1. The native data were scaled by using scala, including the anisotropic B factor correction (29).
Table 1. Data collection.
| Data set | Resolution, Å | λ, Å | Completeness, %* | Rsym*† | I/σI* | Phasing power, iso/ano‡ | Rculliv iso/ano§ |
|---|---|---|---|---|---|---|---|
| Peak | 50—3.1 | 1.37837 | 98.2 (94.3) | 0.071 (0.418) | 12.9 (2.2) | 0.84/1.2 | 0.86/0.77 |
| Inflection point | 50—3.1 | 1.37944 | 98.3 (94.5) | 0.074 (0.459) | 12.2 (2.0) | 0.70/1.3 | 0.89/0.76 |
| High energy remote | 50—3.1 | 1.34766 | 97.2 (91.6) | 0.078 (0.481) | 10.8 (2.0) | —/0.92 | —/0.84 |
| Overall figure of merit to 3.1 Å: 0.44 | |||||||
| Native | 50—2.8 | 1.37837 | 99.5 (99.7) | 0.083 (0.491) | 13.4 (3.1) | — | — |
The numbers in parentheses correspond to the highest resolution bin
Rsym = Σ|Ihkl — 〈I〉|/Σ|Ihkl|, where Ihkl is the observed intensity and 〈I〉 is the average intensity of multiple symmetry-related observations of that reflection
Phasing power = 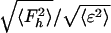 , where Fh is the heavy-atom structure factor amplitude and ε is the residual lack of closure error
, where Fh is the heavy-atom structure factor amplitude and ε is the residual lack of closure error
Rcullis = 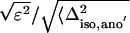 , where ε is the residual lack of closure error and Δiso,ano indicates the isomorphous (dispersive) or anomalous structure factor amplitude differences
, where ε is the residual lack of closure error and Δiso,ano indicates the isomorphous (dispersive) or anomalous structure factor amplitude differences
Copper positions were determined with shelxd (30). The Matthews parameter is ≈3.2 Å3/Da, with 62% solvent content (31). MAD phasing calculations to 3.1 Å yielded an overall figure of merit of 0.44 (32). Density modification in cns, including solvent flipping (33), was followed by calculation of a 6-fold real-space averaged electron density map by using rave (34). The initial Cα backbone trace was constructed from the similar regions of an ensemble of MCO structures [Protein Data Bank entries 1A65 (35), 1AOZ (36), 1GW0 (37), 1GYC (38), and 1KV7 (39)] superimposed by using dali (40). Regions unique to Fet3p were traced in 6-fold averaged 2 Fo - Fc and Fo - Fc σA-weighted (41) electron density maps calculated from iterative model building and refinement. refmac (42), including translation/libration/screw (TLS) refinement (43), was used for the later stages. “Tight” noncrystallographic symmetry restraint values were applied to each Fet3p monomer in the asymmetric unit, and each Fet3p chain represented a TLS group in refmac calculations. Manual model building was carried out in o (44) and completed with coot (45). Eleven N-linked glycosylation sites were modeled according to Fo - Fc σA-weighted electron density maps, and at some of the sites, mannose residues were observed. Refinement statistics are listed in Table 2. Figures were prepared with pymol (46).
Table 2. Refinement statistics.
| Resolution, Å | Rcryst* | Rfree† | F/σF | rms deviations bonds, Å/angles, ° | No. of protein atoms | No. of copper atoms | No. of carbohydrate atoms |
|---|---|---|---|---|---|---|---|
| 50—2.8 | 0.236 | 0.258 | >0 | 0.012/1.41 | 25,524 | 24 | 2,111 |
Rcryst = Σ||Fo| — |Fc\|/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively
Rfree = Σ||Fo,t| — |Fc,t||/Σ|Fo,t|, where Fo,t is from a test set (5% of reflections) not used in the structural refinement
Results
Structure Determination of Fet3p. The Fet3p structure was determined to 2.8-Å resolution by using MAD methods (Table 1). Producing crystals of a quality suitable for diffraction analysis from the highly glycosylated protein was a challenge. Sequence analysis predicts 13 N-linked glycosylation sites, and SDS/PAGE revealed that the Fet3p ectodomain migrated as a broad smear at ≈120 kDa. Attempts to crystallize this heterogeneous material were unsuccessful. When treated with endo H, the resulting Fet3p protein migrated in a much tighter band centered at ≈85 kDa (21). Fig. 5, which is published as supporting information on the PNAS web site, shows an SDS/PAGE gel of the Fet3 protein before and after endo H treatment. Crystals obtained from this treated material exhibited partial merohedral twinning; however, conditions were found that eliminated twinning in about half of the crystals in each drop. The untwinned crystals were used for the structure determination (see Fig. 6, which is published as supporting information on the PNAS web site, and Materials and Methods).
The Fet3p Fold. The Fet3p structure is shown in Fig. 2a, and the elements contributing to the formation of the copper sites are shown schematically in Fig. 2b. Within this tripartite cupredoxin fold, the T1 copper (Cu1) is liganded to side chains contributed solely by domain 3, whereas the trinuclear cluster composed of T2 (Cu4) and T3 (Cu2 and Cu3) copper atoms is found at the interface of domains 1 and 3. Interpretable electron density was observed for the Fet3p polypeptide except for the last five residues of the C terminus, which are part of the linker region connecting the Fet3p ectodomain to the transmembrane-spanning segment in the native form of the protein.
Fig. 2.

The structure and folding topology of Fet3p. (a) Ribbon diagram of Fet3p. The T1, T2, and T3 copper atoms are shown in blue, green, and yellow, respectively. The three plastocyanin-like domains are shown as blue, green, and gold. Included in red are the carbohydrates resolvable in the electron density map. (b) Schematic of Fet3p topology. The color scheme is as in a. The residues acting as ligands to the four copper atoms in Fet3p are mapped onto the Greek key connectivity of the cupredoxin fold that comprises each Fet3p domain.
Eleven of the 13 predicted N-linked sites are posttranslationally modified, and 5 of those 11 sites are fully digested by endo H, leaving one N-acetyl-d-glucosamine residue attached. Mannose branches are observed in the electron density for some sites, and they interact with the protein surface in some cases (see Fig. 7, which is published as supporting information on the PNAS web site). Three fully digested glycosylation sites are involved in contacts within or between asymmetric units, suggesting that endo H digestion was critical to producing this crystal form. Electrospray mass spectrometry analysis of this Fet3p preparation showed it to contain 15% (wt/wt) residual carbohydrate (21).
The Trinuclear Copper Cluster. Fig. 3a shows the trinuclear cluster (the T2 Cu and the binuclear T3 pair) superimposed on σA-weighted electron density. Although harvested Fet3p crystals are blue, indicating the oxidized state of the T1 copper atom, the crystals become colorless and reduced within 30-60 s in the x-ray beam. This conclusion is supported by the observation of a 5.1-Å Cu2-Cu3 distance in the binuclear T3 cluster and by the absence of electron density attributable to a bridging oxygen typically observed in oxidized forms of other MCO proteins. A similar finding was observed in the structure of the reduced trinuclear cluster in ascorbate oxidase (47). Analysis of the fluorescence spectrum from the Fet3p crystals reveals a shoulder at the midpoint (at ≈8,985 eV, see Fig. 8, which is published as supporting information on the PNAS web site) and a second shoulder at the Cu K absorption edge, a spectrum quite similar to that obtained in extended x-ray absorption fine structure studies on a reduced form of Fet3p that contained only the T3 binuclear pair (48). A preedge feature at 8,983-8,985 eV has been assigned to the 1 s → 4 pz plus ligand transition at Cu(I) (49).
Fig. 3.

Stereo views of the Fet3p trinuclear cluster. (a) The trinuclear cluster containing the T2 site (Cu4) and the binuclear T3 site (Cu2 and Cu3) superimposed on 2 Fo - Fc σA-weighted electron density contoured at 1.5 σ. The T3 site is reduced, as characterized by the loss of a bridging oxygen and a distance of 5.1 Å between Cu2 and Cu3. The color coding for the copper atoms is as in Fig. 2. (b) The amino acid residues D94 and D483 are observed in proximity to the two His residues that coordinate the T2 Cu, H81 and H416. Kinetic and spectral data indicate that both are required for dioxygen reduction at the Fet3p trinuclear cluster (59).
Substrate Targeting and Reactivity. Electron transfer from the reducing substrate occurs at the T1 Cu(II) center in MCOs, whereas the four-electron reduction of dioxygen to 2H2O occurs at the trinuclear cluster composed of the T2 and T3 copper atoms (7, 50). The Fet3p T1 Cu is coordinated by H413, C484, and H489 in a roughly trigonal planar geometry (51, 52). As in many of the laccases, the Fet3p T1 Cu lacks the fourth ligand that confers the more trigonal pyramidal coordination geometry found in hCp because of the ligation to the thioether sulfur of M1031 (8). In Fet3p, the side chain of L494 is oriented toward this fourth, apical T1 Cu coordination site preventing occupancy by a donor ligand.
The specificity for Fe(II) as substrate can be inferred from the array of potential iron ligands immediately adjacent to the T1 copper center, and this putative iron binding site is shown in Fig. 4a. Potential iron binding residues include E185, D283, and D409. A similar collection of acidic residues is observed adjacent to the ferroxidase site in hCp, and these have been described as providing a “holding site” for the Fe(III) product of the ferroxidase reaction. In soaking experiments, this site in Cp was demonstrated to bind transition metals (53). Kinetic and spectroscopic studies have shown that E185 in Fet3p contributes to Fe(II) binding, the intermolecular electron transfer from Fe(II) to the T1 Cu(II), and iron uptake via the Fet3p/Ftr1p complex (23, 54). Table 3 summarizes kinetic data on ferroxidation and iron uptake for the E185A, D283A, and D409A mutants that strongly support the inference that this trio of oxygen-containing side chains is an essential contributor to the iron binding [KM for Fe(II) in ferroxidation] and trafficking site on Fet3p (Vmax for 59Fe uptake).
Fig. 4.

Stereo views of the T1 Cu site viewed from the solvent showing the features that define Fet3p as a ferroxidase. (a) The Fet3p iron binding site. Hydrogen bonds (red dashes) are shown between D409 and H413 and between E185 and H489, components of the electronic matrix coupling pathway for electron transfer from the Fe(II) substrate to the T1 Cu(II). Kinetic and spectral data indicate that E185 and D409 also contribute to the binding of Fe(II) (Table 3) (23, 54). (b) Superposition of Fet3p (green and gold shading, as in Fig. 2) and Tv laccase (Protein Data Bank ID code 1GYC, gray shading) at the substrate binding site. The orientation is as in a. The residues present in Fet3p but absent in Tv laccase that confer Fet3p's specificity toward Fe(II) as substrate are shown.
Table 3. Kinetic constants for ferroxidation [Fe(II) turnover] and 59Fe-uptake.
| Fet3p species | Fe(II) oxidation (in vitro) KM, μM | 59Fe uptake (in vivo) V, % WT |
|---|---|---|
| WT | 5.4 ± 0.5 | 100 |
| E185A | 35.6 ± 2.8 | 0 |
| D409A | 18.8 ± 1.6 | 7 |
| D283A | 19.3 ± 1.5 | 3 |
| E185A/D409A | 4,000 ± 400 | 0 |
As in other MCOs, the two His ligands coordinate the T1 Cu via their Nδ 1 pyridine nitrogens. What is striking is that the Nε2 pyrrole NH groups of H413 and H489 appear to be hydrogen-bond donors to D409 and E185, respectively (Fig. 4a). This feature is similar to the apparent hydrogen bond between E272 and H1026, one of the ligands to the T1 Cu in domain 6 of hCp (8). No such hydrogen-bonding network is apparent at the T1 Cu sites in any of the laccases. The overlay of Fet3p and Trametes versicolor (Tv) laccase (55) structures shown in Fig. 4b illustrates this difference. In particular, the laccase structure lacks the carboxylate side chains found in Fet3p. Electron transfer data for both hCp and Fet3p indicate that the carboxyl-imidazole hydrogen bonds are part of the electronic coupling pathway that supports the outersphere, intermolecular electron transfer from Fe(II) to the T1 copper ion (51, 54). Electron transfer from organic reductants like hydroquinone in other MCOs, e.g., Tv laccase, is likely coupled by a direct H bond from the reductant to a coordinating histidine imidazole at the T1 Cu. The overlay shown in Fig. 4b demonstrates also how in Tv laccase substrate has direct access to these imidazoles and suggests that the carboxylate-coupled electron transfer pathway inferred for Fet3p is specific to the ferroxidase reaction and therefore an essential part of the function for which Cp, Fet3p, and their homologs have been adapted (51, 54).
Fet3p and hCp can also act as cuprous oxidases. Kinetic data indicate that this reaction occurs at the same site that supports ferrous iron turnover (22). This cuprous oxidase activity is essential for the resistance that yeast exhibit toward the toxicity coming from elevated copper levels in the environment (24). Fet3p does appear to provide possible Cu(I) ligands, most notably M281 and M345 (Fig. 4a). The bacterial cuprous oxidase CueO also uses methionine residues as Cu(I) ligands, although they are unlike those in Fet3p in that they come from a structurally distinct methionine-rich α-helix adjacent to the CueO active site (56, 57). Fet3p M345 is conserved in the sequences of 10 of the 11 archived fungal Fet proteins. The KM for Cu(I) but not for Fe(II) increases by ≈2-fold for Fet3p(M345A), consistent with the hypothesis that this residue contributes to cuprous ion oxidation (C.S.S. and D.J.K., unpublished work).
A feature of the MCO T3 site related to dioxygen turnover is the presence of an acidic side chain that appears to tether (by a hydrogen bond) a water molecule at the T2 Cu. This hydrogen-bonding network appears essential to the H+ transfer(s) that are coupled to dioxygen reduction (50, 52, 58, 59). This water molecule ligand to the T2 Cu was not observed in the Fet3p structure, possibly because of the relatively low-resolution limit of the x-ray diffraction data. In Fet3p, this acidic side chain comes from D94, and Fig. 3b reveals that it is placed at the trinuclear cluster 5.5 Å from the T2 Cu and 3.4 Å from the Nδ 1 (noncoordinating) atom of H81, one of the two T2 Cu histidine ligands. Furthermore, the structure shows that D458 has a comparable conformational relationship to the T2 Cu and its other ligand, H416, suggesting that both acidic side chains may contribute to oxygen binding and turnover at the T2, T3 cluster.
Discussion
Correlating Function with Structure. There are more site-directed mutants of Fet3p described than for all other MCO proteins combined. Residues targeted for mutagenesis through sequence homology and/or homology modeling fall into three groups. First are substitutions at ligands to one or more of the Fet3p copper atoms. These result in Fet3p proteins that are depleted of one or more of the copper atoms: Fet3p(T1Δ), Fet3p(T2Δ), and Fet3p(T1Δ/T2Δ) (48). In these Fet3p mutants, the remaining copper sites are spectroscopically like WT although the proteins themselves are inactive as oxidases. The inference that the residues mutated are copper ligands is confirmed by this structure.
The second group includes D94, a residue postulated to be at or near the T2 Cu through homology to the structure of ascorbate oxidase. Additionally, this residue was proposed to act as an acid catalyst of dioxygen turnover at the trinuclear cluster (52, 58). Indeed, kinetic and spectroscopic characterization of D94E and D94A mutants of Fet3p has provided support for this hypothesis (59), and the structure confirms this assumed conformational relationship between D94 and Cu4. The demonstration that D458 is related to D94 by a symmetry dyad suggests that these two residues are both potentially part of a hydrogen-bond, H+-transfer network that supports the coupled proton/electron transfers to O2 and its reduction intermediates (Fig. 3b).
The third group of mutants is the largest and includes residues suggested to be part of the structural motifs that provide the specificity toward Fe(II) as the substrate. Askwith and Kaplan (60) targeted E227, D228 and E330, but all of the alanine mutants at these positions retained oxidase and iron uptake activity (60). The Fet3p structure places these residues on the protein surface, distant from both the T1 and trinuclear sites. Two groups have investigated the activity of mutants constructed at E185 and D278 (and Y354) (23, 61) and to these we add D283 and D409. All of these were placed at or near the T1 Cu by homology modeling, and the structure reveals that the side chains of E185, D283, and D409 are oriented toward the T1 Cu, whereas the side chains of D278 and Y354 are not. However, all of these residues are part of the water-accessible (Connolly) surface of the protein, ensuring access from bulk solvent. A previous modeling study led to the proposal that D319 and D320 are part of a solvent-exposed, negative patch involved in Fe binding (62). In fact, these residues are part of an electrostatic “zipper” where the two carboxylate side chains are in salt bridges with the side chains of R463 and R427, respectively. Fet3p(D320A) is retained within the cell's endoplasmic reticulum, most likely because it fails to properly fold (C.S.S. and D.J.K., unpublished work).
The functions of E185, D409, and D283 can be one or more of the following: Fe(II) binding, electron transfer coupling, or Fe(III) trafficking to Ftr1p. Kinetic data indicate that E185 and D409 participate in all three processes (Table 3) (23, 54). Regarding electron transfer from bound Fe(II) to the T1 Cu, the structure allows for the estimation of the electronic matrix coupling (outersphere electron transfer) into this Cu(II) via the E185-H489 and D409-H413 pathways. The efficiency of this coupling was calculated by using the pathways program (63). The analysis suggested decay couplings for the two routes of 0.024 and 0.012, respectively. For comparison, the primary electron transfer pathway into Cp from Fe(II) is characterized by a decay coupling of 0.028 (54). Given the rate of outersphere electron transfer from Fe(II) to the T1 Cu(II) in these two proteins (>1,200 s-1), these pathways are quite efficient (51, 54). The prediction of two electron transfer pathways in Fet3p of comparable efficiency is a unique finding for MCOs that likely underscores a principal contribution to the ferrous iron specificity of this and the other fungal enzymes linked to iron trafficking.
In addition, the increased KM for Fe(II) in ferroxidation for the alanine mutants at positions E185 and D409 indicates that both residues participate in the binding of Fe(II) (Table 3) (23, 54). The 1,000-fold decrease in affinity for Fe(II) for the E185A/D409A double mutant is compelling evidence for this conclusion. However, the most striking phenotype of these mutations, along with the alanine substitution at D283, is the strongly reduced iron uptake in vivo (Table 3). This finding suggests that all of these residues function in the trafficking of Fe(III) to Ftr1p, and their surface exposure positions them to contribute to this pathway. Studies on Ftr1p have identified a DASE motif in an exocytoplasmic loop in this permease that is also likely to be part of this pathway (64). Together with the three acidic residues in Fet3p, D246 and E249 in Ftr1p are required for high-affinity iron permeation in the Fet3p/Ftr1p complex.
Distinguishing Ferroxidases from Laccases. The largest genus in the MCO family includes those proteins composed of three cupredoxin domains of ≈500 residues. In bacteria, fungi, and plants, this group includes the laccases and Fet3p homologs. What functionally distinguishes these species is the reactivity with Fe(II) exhibited by the ferroxidases. The Fet3p structure provides insight into the structural elements that distinguish the members of this MCO domain type. In Fig. 4b, Tv laccase is used for illustration where the protein fold surrounding the T1 Cu is superimposed onto the T1 Cu site in Fet3p (38, 55).
Although the folds of these two MCOs are similar, there are differences in the structural motifs that frame the T1 Cu site. First, a loop in domain 2 of Tv laccase including residues 162FPL164 provides one side of the binding site for an aromatic amine (or hydroxyl) substrate (55). In the laccase-substrate complex, the substrate heteroatom forms a hydrogen bond with the Nε2 pyrrole nitrogen of one of the His ligands to the T1 Cu(II), which likely represents the electronic matrix coupling pathway for the intermolecular electron transfer from reducing substrate to the T1 Cu(II). In Fet3p, this loop is longer, extending into the channel found in Tv laccase within which the large, hydrophobic substrate binds. In Fet3p, this loop is relatively hydrophilic and it is here that E185 resides (Fig. 4). As discussed, E185 is one of two residues in Fet3p involved in Fe(II) binding, intermolecular electron transfer, and Fe(III) trafficking to Ftr1p. The second feature distinguishing the laccase from the ferroxidase is a difference found in a loop of domain 3. In Tv laccase, this loop contains small and/or nonpolar residues, whereas in Fet3p it extends into the channel and contains D409, the other acidic residue critical to the three functions required of a ferroxidase. An additional difference is found in a second domain 2 loop that in Fet3p includes D283, the third residue linked to Fe(II) binding and Fe(III) trafficking. This loop is lacking in Tv laccase where the connectivity from β-strand to β-strand is relatively short and does not extend into the crevice in which the bulky aromatic substrate binds. A final difference in Fet3p is found in the strand containing M345. As mentioned, M345 is likely to be important to the cuprous oxidase activity exhibited by Fet3p. These differences are so clearly related to the specificity differences exhibited by the two proteins that one could easily and rationally propose to convert one into the other.
Supplementary Material
Acknowledgments
We thank the staff at beamline 19BM at the Advanced Photon Source, Ms. Susan Stoj for technical assistance, and Dr. Stephen Holloway for help with the figures. This work was supported by National Institutes of Health Grant DK53820 (to D.J.K.) and R. A. Welch Foundation Grant AQ-1399 (to P.J.H.). Support for the X-Ray Crystallography Core Laboratory at the University of Texas Health Science Center was provided by the Institutional Executive Research Council and the San Antonio Cancer Institute.
Author contributions: D.J.K. and P.J.H. designed research; A.B.T., C.S.S., and L.Z. performed research; A.B.T., C.S.S., L.Z., D.J.K., and P.J.H. analyzed data; and A.B.T., D.J.K., and P.J.H. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Cp, ceruloplasmin; MCO, multicopper oxidase; T1, type 1; T2, type 2; T3, type 3; endo H, endo-β-N-acetylglucosaminidase H; MAD, multiwavelength anomalous diffraction; Tv, Trametes versicolor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1ZPU).
References
- 1.Lahey, M. E., Gubler, M. S., Chase, G. E., Cartwright, G. E. & Wintrobe, M. M. (1952) Blood 7, 1053-1065. [PubMed] [Google Scholar]
- 2.Chase, M. S., Gubler, C. J., Cartwright, G. E. & Wintrobe, M. M. (1952) J. Biol. Chem. 199, 757-763. [PubMed] [Google Scholar]
- 3.Cartwright, G. E., Gubler, C. J. & Wintrobe, M. M. (1957) J. Biol. Chem. 224, 533-546. [PubMed] [Google Scholar]
- 4.Holmberg, C. G. & Laurell, C. B. (1948) Acta Chem. Scand. 2, 550-556. [DOI] [PubMed] [Google Scholar]
- 5.Murphy, M. E., Lindley, P. F. & Adman, E. T. (1997) Protein Sci. 6, 761-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoj, C. S. & Kosman, D. J. (2005) in Encyclopedia of Inorganic Chemistry, ed. King, R. B. (Wiley, New York), pp. 1134-1159.
- 7.Solomon, E. I., Sundaram, U. M. & Machonkin, T. E. (1996) Chem. Rev. 96, 2563-2605. [DOI] [PubMed] [Google Scholar]
- 8.Zaitseva, I., Zaitsev, V., Card, G., Moshkov, K., Bax, B., Ralph, A. & Lindley, P. (1996) J. Biol. Inorg. Chem. 1, 15-23. [Google Scholar]
- 9.Osaki, S., Johnson, D. A. & Frieden, E. (1966) J. Biol. Chem. 241, 2746-2751. [PubMed] [Google Scholar]
- 10.Freiden, E. & Hsieh, H. S. (1976) Adv. Exp. Med. Biol. 74, 505-529. [DOI] [PubMed] [Google Scholar]
- 11.Hellman, N. E. & Gitlin, J. D. (2002) Annu. Rev. Nutr. 22, 439-458. [DOI] [PubMed] [Google Scholar]
- 12.Xu, X., Pin, S., Gathinji, M., Fuchs, R. & Harris, Z. L. (2004) Ann. N.Y. Acad. Sci. 1012, 299-305. [DOI] [PubMed] [Google Scholar]
- 13.Stearman, R., Yuan, D. S., Yamaguchi-Iwai, Y., Klausner, R. D. & Dancis, A. (1996) Science 271, 1552-1557. [DOI] [PubMed] [Google Scholar]
- 14.Kosman, D. J. (2002) Adv. Protein Chem. 60, 221-269. [DOI] [PubMed] [Google Scholar]
- 15.Kosman, D. J. (2003) Mol. Microbiol. 47, 1185-1197. [DOI] [PubMed] [Google Scholar]
- 16.de Silva, D. M., Askwith, C. C., Eide, D. & Kaplan, J. (1995) J. Biol. Chem. 270, 1098-1101. [DOI] [PubMed] [Google Scholar]
- 17.Dancis, A., Yuan, D. S., Haile, D., Askwith, C., Eide, D., Moehle, C., Kaplan, J. & Klausner, R. D. (1994) Cell 76, 393-402. [DOI] [PubMed] [Google Scholar]
- 18.Askwith, C., Eide, D., Van Ho, A., Bernard, P. S., Li, L., Davis-Kaplan, S., Sipe, D. M. & Kaplan, J. (1994) Cell 76, 403-410. [DOI] [PubMed] [Google Scholar]
- 19.Askwith, C. & Kaplan, J. (1998) Trends Biochem. Sci. 23, 135-138. [DOI] [PubMed] [Google Scholar]
- 20.De Freitas, J., Wintz, H., Kim, J. H., Poynton, H., Fox, T. & Vulpe, C. (2003) Biometals 16, 185-197. [DOI] [PubMed] [Google Scholar]
- 21.Hassett, R. F., Yuan, D. S. & Kosman, D. J. (1998) J. Biol. Chem. 273, 23274-23282. [DOI] [PubMed] [Google Scholar]
- 22.Stoj, C. & Kosman, D. J. (2003) FEBS Lett. 554, 422-426. [DOI] [PubMed] [Google Scholar]
- 23.Wang, T.-P., Quintanar, L., Severance, S., Solomon, E. I. & Kosman, D. J. (2003) J. Biol. Inorg. Chem. 8, 611-620. [DOI] [PubMed] [Google Scholar]
- 24.Shi, X., Stoj, C., Romeo, A., Kosman, D. J. & Zhu, Z. (2003) J. Biol. Chem. 278, 50309-50315. [DOI] [PubMed] [Google Scholar]
- 25.Yeates, T. O. (1997) Methods Enzymol. 276, 344-358. [PubMed] [Google Scholar]
- 26.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrickson, W. A. (1991) Science 254, 51-58. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski, Z. & Minor, W. (2001) in International Tables for Crystallography, eds. Rossman, M. & Arnold, E. (Kluwer, Dordrecht, The Netherlands), Vol. F, pp. 226-235. [Google Scholar]
- 29.Evans, P. (1993) in Proceedings of CCP4 Study Weekend on Data Collection and Processing, eds. Sawyer, L., Isaacs, N. & Bailey, S. (SERC Daresbury Laboratory, Warrington, U.K.), pp. 114-122.
- 30.Uson, I. & Sheldrick, G. M. (1999) Curr. Opin. Struct. Biol. 9, 643-648. [DOI] [PubMed] [Google Scholar]
- 31.Matthews, B. W. (1968) J. Mol. Biol. 33, 491-497. [DOI] [PubMed] [Google Scholar]
- 32.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 33.Abrahams, J. P. & Leslie, A. G. (1996) Acta Crystallogr. D 52, 30-42. [DOI] [PubMed] [Google Scholar]
- 34.Kleywegt, G. J., Zou, J.-Y., Kjeldgaard, M. & Jones, T. A. (2001) in International Tables for Crystallography, eds. Rossman, M. & Arnold, E. (Kluwer, Dordrecht, The Netherlands), Vol. F, pp. 353-356, 366-368. [Google Scholar]
- 35.Ducros, V., Brzozowski, A. M., Wilson, K. S., Brown, S. H., Ostergaard, P., Schneider, P., Yaver, D. S., Pedersen, A. H. & Davies, G. J. (1998) Nat. Struct. Biol. 5, 310-316. [DOI] [PubMed] [Google Scholar]
- 36.Messerschmidt, A., Ladenstein, R., Huber, R., Bolognesi, M., Avigliano, L., Petruzzelli, R., Rossi, A. & Finazzi-Agró, A. (1992) J. Mol. Biol. 224, 179-205. [DOI] [PubMed] [Google Scholar]
- 37.Hakulinen, N., Kiiskinen, L. L., Kruus, K., Saloheimo, M., Paananen, A., Koivula, A. & Rouvinen, J. (2002) Nat. Struct. Biol. 9, 601-605. [DOI] [PubMed] [Google Scholar]
- 38.Piontek, K., Antorini, M. & Choinowski, T. (2002) J. Biol. Chem. 277, 37663-37669. [DOI] [PubMed] [Google Scholar]
- 39.Roberts, S. A., Weichsel, A., Grass, G., Thakali, K., Hazzard, J. T., Tollin, G., Rensing, C. & Montfort, W. R. (2002) Proc. Natl. Acad. Sci. USA 99, 2766-2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123-138. [DOI] [PubMed] [Google Scholar]
- 41.Read, R. J. (1986) Acta Crystallogr. A 42, 140-149. [Google Scholar]
- 42.Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D. 53, 240-255. [DOI] [PubMed] [Google Scholar]
- 43.Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001) Acta Crystallogr. D 57, 122-133. [DOI] [PubMed] [Google Scholar]
- 44.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 45.Emsley, P. & Cowtan, K. (2004) Acta Crystallogr. D 60, 2126-2132. [DOI] [PubMed] [Google Scholar]
- 46.DeLano, W. L. (2002) pymol (DeLano Scientific, San Carlos, CA).
- 47.Messerschimidt, A., Luecke, H. & Huber, R. (1993) J. Mol. Biol. 230, 997-1014. [DOI] [PubMed] [Google Scholar]
- 48.Blackburn, N. J., Ralle, M., Hassett, R. & Kosman, D. J. (2000) Biochemistry 39, 2316-2324. [DOI] [PubMed] [Google Scholar]
- 49.Kau, L.-S., Spira-Solomon, D. J., Penner-Hahn, J. E., Hodgson, K. O. & Solomon, E. I. (1987) J. Am. Chem. Soc. 109, 6433-6442. [Google Scholar]
- 50.Solomon, E. I., Chen, P., Metz, M., Lee, S. K. & Palmer, A. E. (2001) Angew. Chem. Int. Ed. Engl. 40, 4570-4590. [DOI] [PubMed] [Google Scholar]
- 51.Machonkin, T. E., Quintanar, L., Palmer, A. E., Hassett, R., Severance, S., Kosman, D. J. & Solomon, E. I. (2001) J. Am. Chem. Soc. 123, 5507-5517. [DOI] [PubMed] [Google Scholar]
- 52.Palmer, A. E., Quintanar, L., Severance, S., Wang, T. P., Kosman, D. J. & Solomon, E. I. (2002) Biochemistry 41, 6438-6448. [DOI] [PubMed] [Google Scholar]
- 53.Lindley, P. F., Card, G., Zaitseva, I., Zaitsev, V., Reinhammar, B., Selin-Lindgren, E. & Yoshida, K. (1997) J. Biol. Inorg. Chem. 2, 454-463. [Google Scholar]
- 54.Quintanar, L., Gebhard, M., Wang, T. P., Kosman, D. J. & Solomon, E. I. (2004) J. Am. Chem. Soc. 126, 6579-6589. [DOI] [PubMed] [Google Scholar]
- 55.Bertrand, T., Jolivalt, C., Briozzo, P., Caminade, E., Joly, N., Madzak, C. & Mougin, C. (2002) Biochemistry 41, 7325-7333. [DOI] [PubMed] [Google Scholar]
- 56.Singh, S. K., Grass, G., Rensing, C. & Montfort, W. R. (2004) J. Bacteriol. 186, 7815-7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts, S. A., Wildner, G. F., Grass, G., Weichsel, A., Ambrus, A., Rensing, C. & Montfort, W. R. (2003) J. Biol. Chem. 278, 31958-31963. [DOI] [PubMed] [Google Scholar]
- 58.Palmer, A. E., Lee, S. K. & Solomon, E. I. (2001) J. Am. Chem. Soc. 123, 6591-6599. [DOI] [PubMed] [Google Scholar]
- 59.Quintanar, L., Stoj, C., Wang, T. P., Kosman, D. J. & Solomon, E. I. (2005) Biochemistry 44, 6081-6091. [DOI] [PubMed] [Google Scholar]
- 60.Askwith, C. C. & Kaplan, J. (1998) J. Biol. Chem. 273, 22415-22419. [DOI] [PubMed] [Google Scholar]
- 61.Bonaccorsi di Patti, M. C., Felice, M. R., Camuti, A. P., Lania, A. & Musci, G. (2000) FEBS Lett. 472, 283-286. [DOI] [PubMed] [Google Scholar]
- 62.Bonaccorsi di Patti, M. C., Felice, M. R., De Domenico, I., Lania, A., Alaleona, F. & Musci, G. (2005) Yeast 22, 677-687. [DOI] [PubMed] [Google Scholar]
- 63.Betts, J. N., Beratan, D. N. & Onuchic, J. N. (1992) J. Am. Chem. Soc. 114, 4043-4046. [Google Scholar]
- 64.Severance, S., Chakraborty, S. & Kosman, D. J. (2004) Biochem. J. 380, 487-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}