

Abstract
While investigating microRNA targets, we have found that human genes divide into two roughly equal populations, based on the fraction of A plus T bases in their 3′ UTRs. Using the Gene Ontology database, we find significant functional differences between the two gene populations, with AT-rich genes implicated in transcription and translation processes, and GC-rich genes implicated in signal transduction and posttranslational protein modification. Better understanding of the background distribution of nucleotides in 3′ UTRs may allow improved prediction of microRNA-targeted genes in humans. We predict at least 1,200 KnownGene transcripts to be regulated by microRNAs. The large majority of these microRNA targets are in the AT-rich 3′ UTR population. However, notwithstanding this preference for AT-rich targets, microRNA targets are found preferentially to be regulatory genes themselves, including both transcription factors and posttranslational modifiers. These results suggest that some processes involving mRNA, of which microRNA regulation may be just one, require AT-richness of 3′ UTRs for functionality. A relationship, not simply one-to-one, between these 3′ UTR populations and large-scale genomic isochores is described.
Keywords: Gene Ontology, isochore, nucleotide content
MicroRNAs (miRNAs) are short (≈22 bp), single-stranded RNA molecules that bind specific mRNAs, their targets, and repress their translation (1, 2). Additionally, evidence suggests that miRNAs down-regulate message levels as well as protein levels (3–5). The large majority of both known and predicted target sites on mRNA molecules are within the 3′ UTRs (6). As a necessary condition for a target site of a particular miRNA, the mRNA (usually 3′ UTR) is believed to require six continuous nucleotides that form exact Watson–Crick base pairs to positions two through seven of the miRNA, where position one is the first base of the 3′ end of the miRNA (7, 8). Applying both experimental and comparative genomics techniques, a few groups have taken advantage of this hexamer binding condition to predict that a much larger number of human genes are regulated by miRNAs than at first believed, perhaps as many as several thousand (3, 6, 9–12). However, even with such a large number of regulated genes, six-nucleotide binding does not provide enough specificity for a miRNA to find its intended target. It does not seem likely that additional specificity is imparted by partial binding of the miRNA to more than seven positions of the target site in humans (6, 7), although such a mechanism may operate in Caenorhabditis elegans and Drosophila melanogaster (11).
We show that human miRNAs preferentially target a large, but nevertheless distinct, population of genes whose 3′ UTRs have a high proportion of A and T bases, not just near the miRNA binding site, but globally. Such genes tend also to be AT-rich in the third positions of their codons, where redundancy in the genetic code allows alternative choices of base. Because nearly half of all human genes are in this AT-rich population, the immediately implied gain in specificity is not large. However, our result is supportive of the conjecture that the additional specificity for miRNA binding lies in a global property of AT-rich target mRNAs (different from CG-rich mRNAs) not just adjacent to the target hexamer; an example would be three-dimensional conformational properties (13).
As additional evidence that a gene's AT-richness is not merely an artifact, but may be a fundamental aspect of its functioning, we find that some Gene Ontology classification (14) keywords correlate highly with AT-richness; we will show additional keyword differences, highly statistically significant, between genes that are miRNA targets and other AT-rich genes, meaning that miRNA targets are not just “typical” AT-rich genes, but a functionally distinctive subset thereof.
We have developed a variant of the method used by Lewis et al. (6), and similar to Krek et al. (9), but using a digraph background model, to predict miRNA targeted genes. For a list of predicted probabilities, by gene, of being a miRNA target, along with the set of miRNAs most likely to regulate the gene, see Table 4, which is published as supporting information on the PNAS web site.
Composition of 3′ UTRs
If we examine the nucleotide compositions of the ≈36,000 human KnownGene 3′ UTRs whose length is >100 bases (so that their composition is statistically determinable to within a reasonable error), an interesting pattern emerges. If we let A, C, G, and T represent the fraction of each base in a given 3′ UTR, the pattern is best seen by plotting A + T on one axis and C – G on the other, as is shown in Fig. 1 Top. (Animation, which is published as supporting information on the PNAS web site, shows all possible axes.) One sees two populations, only partially overlapping, distinguished primarily by their mean in A + T and secondarily by their dispersion in C – G. The ellipses in Fig. 1 are the 2-σ contours of a two-component Gaussian mixture model blindly fitted to the data (that is, with all parameters unguided by us). (See A. W. Moore's tutorial at www.autonlab.org/tutorials, and also ref. 15 for more on fitting Gaussian mixture models.) Such a model readily assigns, by the Bayes odds ratio method, a probability for each gene that it is in the AT-rich, versus AT-poor, population. For the fits shown in Fig. 1, and with x ≡ A + T, y ≡ C – G, the resulting assignment algorithm is
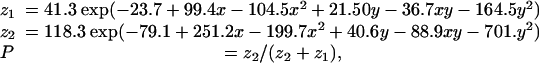 |
[1] |
yielding the value P as the probability of being in the AT-rich population. In the work described here, we carry forward this probability (“soft decision”) rather than make a hard assignment. The model places, statistically, ≈47% of genes in the AT-rich population, with a mean A + T of ≈0.63; 53% are in a CG-rich population with a mean C + G = 1 – (A + T) of ≈0.53. Additional fitted parameters are given in Supporting Text, which is published as supporting information on the PNAS web site.
Fig. 1.

Composition of human 3′ UTRs with A + T on the horizontal axis and C – G on the vertical axis. (Top) All human genes are plotted in red. The red ellipses are 2-σ contours of the maximum likelihood Gaussian mixture model with two components. (Middle) Invertebrates, including C. elegans and D. melanogaster, do not evidence more than a single population when plotted on the same axes. (Bottom) Same as Top (light red) with probable miRNA target genes now plotted in green (Lewis et al. “high signal-to-noise” set; ref. 6) and blue (probable targets as determined by the methods of this paper). miRNA targets lie in the right (AT-rich) component with ≈3:1 selectivity.
For the analyses, we have used the full set of KnownGene transcripts. Some of these transcripts refer to different splice forms of the same gene. Because mRNAs regulate at the message level, this is appropriate. However, we have also verified that very similar results are obtained if one uses unique genes from the RefSeq database.
The distribution of 3′ UTRs in A + T for organisms that are not warm-blooded vertebrates forms a single distinct population (e.g., C. elegans and D. melanogaster as shown in Fig. 1 Middle). The two-versus one-population phenomenon is related to the existence of isochores (16–19), which we discuss below.
Methods
Use of Word Counts in the Gene Ontology (GO) Database. We describe a recently developed method of identifying statistically significant functional differences between two large populations of genes using the GO database (14). We then apply the method to AT-versus CG-rich genes and probable miRNA targets versus all other genes.
One might think it straightforward to distinguish two large populations of genes by differences in how they are assigned to GO categories. Unfortunately, the “raw” GO data are very noisy for this purpose. Because the hierarchical GO categories are invented and populated with genes by a large community of individual investigators, they are very inhomogeneous, with breadth and depth varying widely according to the taste of the individual contributors. Also, it is not clear how one would assign a quantitative statistical significance to any differences found.
We found that it is useful to assign each gene the unweighted list of all biologically meaningful words (and word-like phrases) that occur in the descriptive titles of all of the gene's GO categories, rather than its GO category list. For example, we found that it is more meaningful (or at any rate less noisy) to take note of the term “nucleic acid” in one of a gene's GO categories, than it is to note in exactly which GO category that term occurs. As an additional set of keywords, we also include the HUGO gene name prefixes (e.g., “ZNF*,” designating zinc finger genes).
If δ(i, j) has the value 1 when a word j is associated with a gene i, and 0 otherwise, and if pi is the probability that gene i belongs to a population of interest, then we can form the probabilistic word counts for each word in that population and its complement
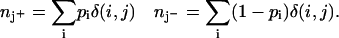 |
[2] |
Similar sums give the variance or expected error of these counts and the normalizing denominators:
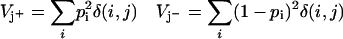 |
[3] |
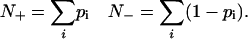 |
[4] |
Out of these sums we can form a t value (deviation in standard deviations) and a P value (two-tail probability) expressing the significance with which the word is associated (or negatively associated) with the set of interest
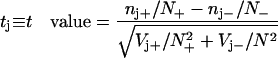 |
[5] |
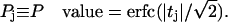 |
[6] |
The straightforward proof is given in Supporting Text.
Two criteria must be met before a difference between two gene populations can be considered as substantiated by this method. First, there must be a set of at least several words for which the P values, as calculated above, are highly significant (e.g., <10–4). This is a necessary Bonferroni constraint because the number of hypotheses (words) is large. Second, only slightly less objectively, there must be a thematic coherency among the highly significant words that makes sense biologically; this is necessary because one can readily imagine differences that, although statistically significant, are biologically uninteresting. For example, it would not be surprising to distinguish large populations of genes entered into the database by a single research group, simply by idiosyncrasies in their use of nonspecific words (e.g., “process,” “activity,” and “function”).
Digraph Probability Model. Using the assumption that functional regulatory binding sites are likely to be conserved, we look at conserved hexamers in 3′ UTRs from the multiple alignment of human, mouse, rat, dog, and chicken (20) similarly to Lewis et al. (6). The difficult part is determining the background rate of (noncausal) conserved hexamers. It seems unwise to use a background model that is the same for every gene, at the very least because we have identified two different populations of genes. Instead, to capture the background rate at which any given hexamer should occur, we use a digraph model that is specific to each gene; this will account not only for the bias implied by variable A + T content, but also for the known underrepresentation of CpG in the human genome and any other digraphic peculiarities of a given gene.
Ideally, one would model just the conserved regions in each gene. Unfortunately, the total conserved lengths in each gene are not enough to do this. Therefore, we make the assumption (or approximation) that conservation probability and digraph probability are independent, and we construct the digraph model of each gene from its entire (human) sequence.
Suppose a hexamer is abcdef. Then, to digraph order, we can write the probability relations
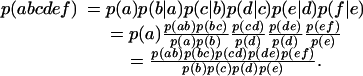 |
[7] |
Because Eq. 7 involves the product of many terms, it is convenient to work with log-probabilities, so in abbreviated notation we have
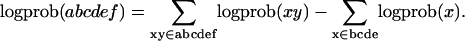 |
[8] |
The individual terms p(xy) or logprob(xy) are estimated by counting the number of times n that the digraph xy occurs in N opportunities. However, it is not a good idea to use an estimate like log(n/N) for the log-probability, because this is divergent for n = 0, and biased for small n.
Estimating Log-Probabilities with Small Number Counts. We proceed by writing a Bayesian estimate for the probability of a specific value of probability, call it ps, given the observed values n and N. (Note that, following usual statistical practice, commas are omitted in the following equations.) We include the possibility of having other information y associated with each gene, for example, whether it is in an AT-rich or CG-rich population. Bayes theorem and elementary manipulations give
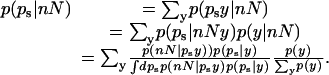 |
[9] |
We have replaced p(y|nN) by p(y) because the other information is assumed not to depend directly on nN. Using a binomial probability model for nN and a binomial conjugate prior for p(ps| y), we get
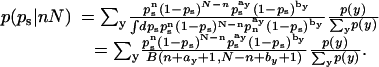 |
[10] |
Here B denotes the beta function. In the case that y varies over {AT-rich, CG-rich}, p(y) is given by the Gaussian mixture model previously discussed. Otherwise, one can simplify by assuming a single population y = 0 and deleting all references to p(y).
The constants ay and by parameterize the (conjugate) prior on ps. Although we had initial expectations that the use of good priors could have a beneficial effect, in practice one obtains as good or better results by taking a noninformative prior like ay = by = 1, or any small constant.
Note that (doing an integral) we have the expectation value
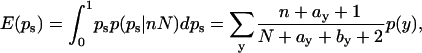 |
[11] |
and, for the case when log-probabilities are needed,
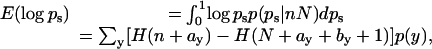 |
[12] |
where H(n) is the harmonic sum.
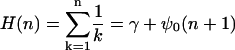 |
[13] |
Here, the second form is valid when n is not an integer, γ is the Euler–Mascheroni constant, and ψ0 is the digamma function. The harmonic sums play the role of logarithms, but now properly corrected for the possibility of small numbers of counts. Thus, Eq. 12 is asymptotically ≈log(n/N) as we might expect, but it remains regular as n and/or N go to zero. We recommend the use of Eqs. 11 and 12, as appropriate, whenever small-count data are being analyzed.
Identifying miRNA Target Genes. The digraph model and the observed number of conserved sites gives, for each gene, the expected number of conserved miRNA binding hexamers that should occur by chance and an error estimate (as described in Supporting Text). We can compare this to the number actually observed and thus assign a probability that any excess is causal, which we take to be the probability that the gene is an actual miRNA target. Our methodology for this is not conceptually different from Lewis et al. (6) and is detailed in Supporting Text. Of interest here, however, is a recently developed method that we have used to get model-free bounds for the total number of targeted genes.
Consider two histograms, “predicted” and “observed,” each giving the number of genes that contain i conserved miRNA binding sites. Each histogram has the same total number of genes. The idea is that “observed” is obtained from “predicted” by pushing some genes to the right in the histogram, that is, by adding (never subtracting) some causal conserved binding sites to the chance ones in that gene. Note that we are not using the correspondence gene-by-gene, because it is very noisy, but only the resulting histograms, which, because the number of genes is large, have good signal-to-noise.
Can we say anything about how many genes have been pushed to the right without knowing anything about the distribution of how far each gene was pushed? Yes. In fact, we can get both lower and upper bounds.
Let the numbers in bin i be mi for “predicted” and ni for “observed,” where i = 0, 1, 2,... Because the histograms have the same area (number of genes), the sum of the positive binwise differences must equal the sum of negative binwise differences. That is
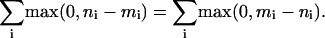 |
[14] |
The way to move the smallest number of genes is to take them strictly from bins with mi > ni and move them strictly to bins with ni > mi. If one does this starting from the right, then one can always achieve this by moving genes in the positive direction. A lower bound on the number of target genes is thus
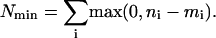 |
[15] |
One might at first think that the upper bound is just the number of extra counts in “observed,” spreading them out maximally with one new count per gene. This would give
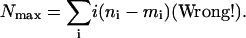 |
[16] |
The problem is that one can not always do this construction by moving genes strictly to the right. The actual bound is often substantially lower and thus more meaningful.
The bound is achieved by working from the right and building up the desired ni distribution, taking genes from the closest bin of mi that has any left to donate. That way, one never “wastes” a possible gene move by leaving a gene in place that could otherwise have been moved. (This is a little bit like Chinese checkers, but where one wants to avoid jumping one's marbles.) An explicit formula for the result is
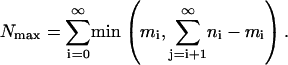 |
[17] |
In fact, it is easy to show that Eq. 16 is obtained if the first argument in the min is never used, that is, if one always has enough genes to move at each stage.
To give a sense of how much better Eq. 17 is than Eq. 16: For a typical histogram in this study, Eq. 17 yields an upper bound of 3,650 (genes), whereas Eq. 16 would yield a much less restrictive bound of 8,400. Eq. 15 gives a lower bound of 1,260. (In Results, we give values that include an additional allowance for statistical error, as described in Supporting Text.)
Results
GO Database Word Counts. Table 1 lists the 15 top words (or word-like phrases) that are positively associated with the AT-rich 3′ UTR population of genes, whereas Table 2 is the corresponding list that is positively associated with the CG-rich 3′ UTR population (that is, negatively associated with the AT-rich population). As shown by the listed t and P values, all of the associations are highly significant. However, note from the values of nj+ and nj– (the probabilistic word counts) that the word frequencies differ by at most ≈25% in the two populations. Virtually all biologically meaningful words occur, to a greater or lesser extent, in both populations. However, having large numbers of genes allows us to extract signal with high significance even from these modest differences.
Table 1. GO words most associated with AT-rich 3′ UTR genes.
| Word or phrase | t value | P value | nj+ | nj- |
|---|---|---|---|---|
| Nucleic acid | 8.75 | 0.000000 | 2,297 | 1,789 |
| Nucleus | 7.11 | 0.000000 | 1,722 | 1,365 |
| Transition metal | 6.80 | 0.000000 | 1,095 | 824 |
| Zinc | 6.65 | 0.000000 | 998 | 746 |
| Bound | 5.99 | 0.000000 | 2,398 | 2,042 |
| ZNF* | 5.87 | 0.000000 | 119 | 49 |
| RNA | 5.53 | 0.000000 | 613 | 448 |
| Organelle | 5.30 | 0.000000 | 2,489 | 2,169 |
| Cellular component | 4.63 | 0.000004 | 3,244 | 2,927 |
| Binding | 4.45 | 0.000009 | 4,405 | 4,054 |
| mRNA | 4.25 | 0.000022 | 102 | 53 |
| Metal | 4.11 | 0.000039 | 1,631 | 1,429 |
| Cycle | 4.07 | 0.000046 | 394 | 296 |
| DNA | 3.99 | 0.000067 | 1,324 | 1,149 |
| Nucleobase | 3.71 | 0.000205 | 1,468 | 1,297 |
Table 2. GO words most associated with CG-rich 3′ UTR genes.
| Word or phrase | t value | P value | nj+ | nj- |
|---|---|---|---|---|
| Receptor | -5.43 | 0.000000 | 852 | 1,085 |
| Signal transduction | -5.16 | 0.000000 | 968 | 1,204 |
| Signaling cascade | -5.13 | 0.000000 | 349 | 494 |
| Transducer | -4.88 | 0.000001 | 880 | 1,093 |
| Communication | -4.80 | 0.000002 | 1,172 | 1,413 |
| Signal | -4.56 | 0.000005 | 902 | 1,102 |
| Transmembrane | -4.37 | 0.000012 | 381 | 506 |
| Filament | -4.31 | 0.000016 | 86 | 150 |
| Cell | -3.83 | 0.000129 | 1,840 | 2,081 |
| Channel | -3.77 | 0.000159 | 151 | 222 |
| Immune | -3.62 | 0.000291 | 217 | 296 |
| Pore | -3.39 | 0.000708 | 162 | 227 |
| Defense | -3.30 | 0.000961 | 237 | 311 |
| Structural | -3.22 | 0.001281 | 241 | 314 |
| Development | -3.21 | 0.001300 | 518 | 625 |
It is striking that each of the two lists evidence a clear thematic coherency, and that the two lists are thematically very different. Genes with AT-rich 3′ UTRs are preferentially associated with transcription and translation events, especially nucleic acid and nucleic acid-binding processes (e.g., zinc finger motifs). These functions are evolutionary old. By contrast, the high GC population is associated with functions coupled to sensing and responding to the external environment. These include signaltransduction pathways and membrane transport. A unifying theme of the high-GC population is that its functions tend toward posttranslational protein modification and signaling interactions, as opposed to transcriptional regulation.
Although the evidence is only indirect, the strong association of AT-rich 3′ UTRs with genes that are implicated in RNA and mRNA processing supports the same conjecture as for miRNA target specificity. That is, some aspect of AT-richness in the 3′ UTR is necessary for at least some processes involving mRNA, of which regulation by miRNAs may be just one.
miRNA Target Genes. By the method of equations 15 and 17, we find among the ≈36,000 known genes a solid lower bound of 1,200 miRNA targets, and an upper bound of ≈5,000. However, this method does not identify which specific genes are likely to be targets. To accomplish this, and also to get a most probable total count of targets (between the two bounds), we use a Poisson odds-ratio method, as described in Supporting Text. However, this most probable value is model-dependent and rather less well determined. We get ≈1,400 ± 150, but we consider this value as likely subject to uncontrolled systematic errors. Lewis et al. (6) have identified a set of “high signal-to-noise” likely miRNA target genes. Although there is significant overlap, our set of most probable target genes is different in detail from this set. We believe that our use of a digraphic probability model, specific to each gene examined, ought to give superior predictions. However, a final verdict on this claim must await experimental evidence. (For our predictions by gene, see Table 5, which is published as supporting information on the PNAS web site.)
Fig. 1 Bottom is identical to Fig. 1 Top, with the Lewis et al. (6) likely targets now plotted in green. The association with the AT-rich population, in both A + T mean and C – G dispersion, is immediately apparent, and easy to substantiate statistically (P < 10–10). Genes that we predict to be miRNA targets with >50% probability are plotted in blue in Fig. 1. Using these probabilities, we can substantiate that ≈75% of miRNA target genes are in the AT-rich population, an ≈3:1 selectivity. However, there is no trend toward fewer targets in the CG-rich population as miRNA target probability goes to 1, indicating that the ≈25% minority of miRNA targets that are CG-rich are, in fact, genuine, although atypical.
We also find weak, but statistically significant, associations between the population of genes with AT-rich 3′ UTRs and those genes identified by the microarray analysis of Lim et al. (3) as being targets of two specific miRNAs, miR-1 (n = 82, P < 0.001) and miR-124 (n = 152, P <0.01).
We can perform the same GO keyword analysis as before on the population of (probabilistically known) miRNA targets. Knowing that miRNA targets lie strongly preferentially in the AT-rich population, we might expect such an analysis to yield an associated word list much like Table 1. The actual result, shown in Table 3, is unexpected and much more interesting. Comparing the two tables, it is striking that the multiple words that associated AT-rich genes with nucleic acid processes are completely absent from the miRNA preferential word list. Instead, the list is dominated by the word “regulation” and its closely related concepts. This finding provides statistically strong evidence that miRNA targets are themselves preferentially (although by no means exclusively) regulators.
Table 3. GO words most associated with probable miRNA target genes.
| Word or phrase | t value | P value | nj+ | nj- |
|---|---|---|---|---|
| Transcription regulator | 5.86 | 0.000000 | 134 | 1,114 |
| Transcription factor | 5.86 | 0.000000 | 129 | 1,068 |
| Regulation | 5.56 | 0.000000 | 315 | 3,215 |
| Regulation of transcription | 5.36 | 0.000000 | 205 | 1,970 |
| Development | 4.69 | 0.000003 | 140 | 1,326 |
| Protein modification | 4.65 | 0.000003 | 192 | 1,897 |
| Serine/threonine kinase | 4.42 | 0.000010 | 68 | 521 |
| Nucleus | 4.42 | 0.000010 | 319 | 3,477 |
| Phosphorylation | 4.30 | 0.000017 | 90 | 766 |
| Signal transduction | 4.09 | 0.000043 | 231 | 2,449 |
| Promoter | 4.07 | 0.000048 | 46 | 347 |
| Phosphate | 4.04 | 0.000052 | 133 | 1,286 |
| Signaling cascade | 4.02 | 0.000058 | 99 | 908 |
| Morphogenesis | 3.96 | 0.000075 | 66 | 567 |
| Kinase | 3.88 | 0.000106 | 133 | 1,311 |
| Phosphotransferase | 3.88 | 0.000106 | 105 | 977 |
| DNA | 3.82 | 0.000132 | 251 | 2,752 |
| Cell | 3.71 | 0.000155 | 30 | 205 |
| Intracellular | 3.72 | 0.000202 | 557 | 6,573 |
| Neurogenesis | 3.71 | 0.000205 | 30 | 205 |
What is also surprising, in view of the results of Tables 1 and 2, is that miRNA target preferences include both transcription factors and also posttranslational regulators, the latter evidenced in words such as “protein modification,” “phosphorylation,” “kinase,” “signaling cascade,” and so forth. The dominant theme of regulation is also seen in a set of words including and related to “development,” including “morphogenesis” and “neurogenesis.”
In other words, within the population of genes with AT-rich 3′ UTRs that miRNAs preferentially target, miRNAs tend to regulate other regulatory genes, even when the regulated processes are posttranslational and uncharacteristic of the AT-rich population generally. In particular, keywords like “signaling cascade” and “signal transduction” are among those strongly positively associated with miRNA targets, even though they are strongly negatively associated with AT-rich genes generally.
Because a smaller fraction (≈25%) of miRNA targets are genes with GC-rich, rather than AT-rich, 3′ UTRs, one may wonder whether those miRNA targets associated with posttranslational processes are associated with that fraction. The answer is no: keyword analysis of miRNA targets that are AT-rich (the majority), versus those that are CG-rich (the minority), show no significant differences. (By way of example, “protein modification” happens paradoxically to be the top word associated with AT-rich miRNA targets, whereas three of the five top words associated with CG-rich miRNA targets refer to transcription.)
Discussion
So-called isochores (16–19, 21) are long, megabase-scale regions of CG-richness that are found in the genomes of warm-blooded vertebrates, including human, and absent in lower organisms. Isochores span intron, exon, and intergene regions indiscriminately, as distinct from the comparatively tiny (≈1,000 base) scale of the individual 3′ UTRs discussed here. Although we do not provide a detailed discussion of the relationship between these very different scaled phenomena, we need here to remark on the obvious question as to whether our two populations of genes (characterized only by their 3′ UTRs) are located in CG-rich isochores, versus the complementary AT-rich isochores, in the genome. In other words, have we simply rediscovered a previously known phenomenon?
Interestingly, the answer is both yes and no. Analysis shows that, with a high degree of selectivity, AT-rich isochores contain only genes with AT-rich 3′ UTRs. However, CG-rich isochores contain an apparently random mixture of genes with CG- and AT-rich 3′ UTRs. Although this result sheds no new light, per se, on the (evolutionarily recent) origin of isochores, its relevance to our work is that it does add support to the idea that an AT-rich 3′ UTR is necessary for some functionally distinct subset of genes. Such genes would naturally resist the evolutionary trend that formed the CG-isochores (whatever it may have been; ref. 21), resulting in the mixture of genes seen in CG-rich isochores.
Given the observation that there are genes with AT-rich 3′ UTRs in both AT- and CG-rich isochores, it is also natural to ask whether one or the other set is dominantly responsible for the strong functional signal demonstrated in Table 1. The answer is that virtually all of the functional signal comes from those AT-rich 3′ UTR genes in CG-rich isochores. If AT-richness of the 3′ UTR is indeed functionally necessary for some genes, the most likely candidates for experimental verification should be sought in CG-rich isochores.
More speculatively, the evidence seems to indicate that, with respect to evolutionary pressure toward CG-richness, AT isochores were “never challenged,” as opposed to “challenged and resisted.” That is, AT isochores appear to include populations of AT-rich genes with functionalities that, had they been in a CG isochore, could have become CG-rich without difficulty (Table 2). Conversely, CG isochores include a functionally distinct population of AT-rich genes (Table 1) that seem to have strongly resisted such conversion.
Supplementary Material
Acknowledgments
We thank Arnold Levine, Gerald Joyce, Curt Callan, Richard Padgett, David Haussler, and Hagar Barak for reading various drafts and making numerous useful suggestions. John Kern provided important statistical insight. This work was supported in part by the Shelby White and Leon Levy Initiatives Fund.
Author contributions: H.R. and W.H.P. designed research, performed research, contributed new reagents/analytic tools, analyzed data, and wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: miRNA, microRNA; GO, Gene Ontology.
References
- 1.Bartel, D. P. (2004) Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 2.Ambros, V. (2004) Nature 431, 350–355. [DOI] [PubMed] [Google Scholar]
- 3.Lim, L. P., Lau, N. C., Garrett-Engele, P., Grimson, A., Schelter, J. M., Castle, J., Bartel, D. P., Linsley, P. S. & Johnson, J. M. (2005) Nature 433, 769–773. [DOI] [PubMed] [Google Scholar]
- 4.Liu, J., Valencia-Sanchez, M. A., Hannon, G. J. & Parker, R. (2005) Nat. Cell Biol. 7, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen, G. L. & Blau, H. M. (2005) Nat. Cell Biol. 7, 633–636. [DOI] [PubMed] [Google Scholar]
- 6.Lewis, B. P., Burge, C. B. & Bartel, D. P. (2005) Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- 7.Doench, J. G. & Sharp, P. A. (2004) Genes Dev. 18, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis, B. P., Shih, I. H., Jones-Rhoades, M. W., Bartel, D. P. & Burge, C. B. (2003) Cell 115, 787–798. [DOI] [PubMed] [Google Scholar]
- 9.Krek, A., Grun, D., Poy, M. N., Wolf, R., Rosenberg, L., Epstein, E. J., MacMenamin, P., da Piedade, I., Gunsalus, K. C., Stoffel, M. & Rajewsky, N. (2005) Nat. Genet. 37, 495–500. [DOI] [PubMed] [Google Scholar]
- 10.John, B., Enright, A. J., Aravin, A., Tuschl, T., Sander, C. & Marks, D. S. (2004) PLoS Biol. 2, 1862–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brennecke, J., Stark, A., Russell, R. B. & Cohen, S. M. (2005) PloS Biol. 3, 404–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grun, D., Wang, Y., Langenberger, D., Gunsalus, K. C. & Rajewsky, N. (2005) PLoS Comp. Biol. 1, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robins, H., Li, Y. & Padgett, R. W. (2005) Proc. Natl. Acad. Sci. USA 102, 4006–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris, M. A., Clark, J., Irel, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. (2004) Nucleic Acids Res. 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLachlan, G. & Peel, D. (2000) Finite Mixture Models (Wiley, New York).
- 16.Bernardi, G., Olofsson, B., Filipski, J., Zerial, M., Salinas, J., Cuny, G., Meunier-Rotival, M. & Rodier, F. (1985) Science 228, 953–958. [DOI] [PubMed] [Google Scholar]
- 17.Bernardi, G. (2000) Gene 241, 3–17. [DOI] [PubMed] [Google Scholar]
- 18.Cohen, N., Dagan, T., Stone, L. & Graur, D. (2005) Mol. Biol. Evol. 22, 1260–1272. [DOI] [PubMed] [Google Scholar]
- 19.Vinogradov, A. E. (2003) Nucleic Acids Res. 31, 5212–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karolchik, D., Baertsch, R., Diekhans, M., Furey, T. S., Hinrichs, A., Lu, Y. T., Roskin, K. M., Schwartz, M., Sugnet, C. W., Thomas, D. J., et al. (2003) Nucleic Acids Res. 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eyre-Walker, A. & Hurst, L. D. (2001) Nat. Rev. Genet. 2, 549–555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}