

Abstract
Although the physiological role of tissue-specific translational control of gene expression in mammals has long been suspected on the basis of biochemical studies, direct evidence has been lacking. Here, we report on the targeted disruption of the gene encoding the heme-regulated eIF2α kinase (HRI) in mice. We establish that HRI, which is expressed predominantly in erythroid cells, regulates the synthesis of both α- and β-globins in red blood cell (RBC) precursors by inhibiting the general translation initiation factor eIF2. This inhibition occurs when the intracellular concentration of heme declines, thereby preventing the synthesis of globin peptides in excess of heme. In iron-deficient HRI–/– mice, globins devoid of heme aggregated within the RBC and its precursors, resulting in a hyperchromic, normocytic anemia with decreased RBC counts, compensatory erythroid hyperplasia and accelerated apoptosis in bone marrow and spleen. Thus, HRI is a physiological regulator of gene expression and cell survival in the erythroid lineage.
Keywords: apoptosis/erythropoiesis/heme-regulated eIF2α kinase/iron deficiency/translation
Introduction
Investigations into mechanisms that control gene expression in mammals have focused largely on transcription and, to a lesser degree, post-transcriptional events related to the fate of specific mRNAs and their cellular localization. Nevertheless, increasing biochemical evidence points to another level of regulation during somatic and germ cell differentiation, namely translational control. Translational regulation can be achieved either by modifying the concentrations or activities of general translational factors (Mathews et al., 2000) or in an mRNA-specific manner, dependent upon the interaction of trans-acting factors with cis-acting sequences present on the mRNA (Rouault and Harford, 2000).
Phosphorylation of the α-subunit of translational initiation factor 2 (eIF2α) has been recognized as a key mechanism of global inhibition of translational initiation. eIF2 is a general translational initiation factor composed of three subunits: α, β and γ. eIF2 forms two types of binary complexes: active eIF2·GTP and inactive eIF2·GDP. eIF2·GTP binds Met-tRNAi to form a ternary complex that is required for binding to the 40S subunit of ribosome and mRNA. Subsequently, upon joining of the 60S subunit, GTP from eIF2·GTP is hydrolyzed to GDP. In order to recycle to its active form and bind another Met-tRNAi, eIF2·GDP must be converted to eIF2·GTP through a guanylate exchange reaction catalyzed by eIF2B. eIF2 has a 400-fold greater affinity for GDP than for GTP. The exchange of tightly bound GDP for GTP requires eIF2B, which is in limiting concentrations and present at 15–25% of the amount of eIF2 (Trachsel, 1996).
The recycling of eIF2 is inhibited by phosphorylation of its α-subunit. Phosphorylation of the α-subunit of eIF2 at Ser51 is carried out by a family of eIF2α kinases. Phosphorylated eIF2 (αP)·GDP binds much more tightly to the regulatory sub-complex of eIF2B than eIF2·GDP and prevents the GDP–GTP exchange activity of eIF2B (Krishnamoorthy et al., 2001). Thus, once the amount of phosphorylated eIF2 exceeds the amount of eIF2B, protein synthesis is shut off.
Four eIF2α kinases have been identified in mammals: the double-stranded RNA-dependent eIF2α kinase (PKR), the mammalian ortholog of the yeast GCN2 protein kinase, the endoplasmic reticulum (ER) resident kinase (PERK) and heme-regulated eIF2α kinase (HRI). These eIF2α kinases share extensive homology in their kinase catalytic domains (Meurs et al., 1990; Chen et al., 1991; Ramirez et al., 1991; Berlanga et al., 1998; Shi et al., 1998; Harding et al., 1999). Although they all inhibit protein synthesis by phosphorylation of eIF2α, it is predicted that a differential physiological response may be produced as a consequence of their tissue distributions and the signals to which they respond. PKR is induced by interferon and regulated by dsRNA through two N-terminal dsRNA-binding domains (Kaufman, 2000). GCN2 is highly expressed in the liver and brain (Berlanga et al., 1999; Sood et al., 2000) and is activated under conditions of amino acid starvation through the C-terminal domain, which contains a His-tRNA synthase-like sequence (Hinnebusch, 1996). PERK is highly expressed in secretory tissues, particularly the pancreas, and is activated by ER stress. PERK contains a luminal domain that is similar to the sensor domain of the ER-stress kinase Irel (Ron, 2000). HRI is expressed predominantly in erythroid cells and is regulated by heme, the prosthetic group of hemoglobin, through the two heme-binding domains located in the N-terminus and the kinase insertion (Chen, 2000).
HRI has been extensively studied biochemically. It is well documented that protein synthesis in intact reticulocytes and their lysates is dependent upon the availability of heme. In heme deficiency, inhibition of protein synthesis correlates with the activation of HRI (Clemens, 1996; Chen, 2000). Expression of HRI in insect Sf9 cells causes global inhibition of protein synthesis. In addition, baculovirus-expressed HRI is a hemoprotein whose activity is regulated by micromolar concentrations of hemin, both in vitro and in vivo (Chefalo et al., 1994, 1998). HRI contains two distinct heme-binding sites. Heme bound to the N-terminal domain is stable and co-purifies with HRI to homogeneity. In contrast, heme binds to the kinase insertion domain reversibly and inhibits HRI kinase activity upon binding, thereby regulating HRI activity according to intracellular heme concentrations (Chefalo et al., 1998; Rafie-Kolpin et al., 2000). HRI protein, activity and mRNA are detected predominantly in red blood cell (RBC) precursors, and HRI mRNA levels increase during erythroid differentiation of mouse erythroleukemic (MEL) cells (Crosby et al., 1994). Small amounts of HRI mRNA are also found in non-erythroid tissues but no evidence of HRI protein expression has been reported (Mellor et al., 1994; Berlanga et al., 1998).
In order to elucidate the physiological role of HRI, in the context of a whole animal, we disrupted the HRI gene in mouse embryonic stem (ES) cells. HRI–/– mice appear to be normal, are fertile and present no gross abnormality of hematological parameters. However, in iron deficiency, the adaptive response of wild-type (wt) mice, characterized by RBC hypochromia and microcytosis, was dramatically altered. In HRI–/– mice, RBC size remained normal with hyperchromic rather than hypochromic anemia. Globins devoid of heme aggregated as inclusions within the RBC and its precursors, resulting in anemia with compensatory erythroid hyperplasia and accelerated apoptosis of erythroid precursors in bone marrow and spleen. Together, these results establish the physiological role of HRI in balancing the synthesis of α- and β-globins with the availability of heme in RBC precursors. Furthermore, this translational regulation of HRI in iron deficiency is necessary for the survival of erythroid precursors.
Results
Cloning of the murine HRI gene and generation of HRI–/– mice
We cloned a 19 kb mouse genomic DNA fragment that contains five exons of the mouse HRI gene (Figure 1A). These exons encode the second conserved kinase lobe of HRI with kinase domains VIa–XI and the entire C-terminus. We mapped the HRI gene to the very distal end of mouse chromosome 5, which corresponds to human chromosome 7p13q. So far, no known human or mouse disease is linked to these chromosomal loci.

Fig. 1. Targeted disruption of the HRI gene. (A) HRI wild-type locus (top), targeting construct (middle) and targeted homologous recombination at the HRI locus before and after Cre-mediated excision of the neomycin resistance gene (bottom). Exons were marked by the filled rectangles and labeled with Roman numerals. Abbreviations used for restriction enzymes: N, NdeI; B, BssHII; E, EcoRV; Bg, BglII. (B) Genotyping of the targeted disrupted mice by PCR. The primers P1 and P2 were used for amplification of the HRI+/+ DNA of 625 bp, whereas primers P1 and P3 were used for the amplification of HRI–/– DNA of ∼1000 bp. (C) HRI mRNA. HRI mRNA was determined by RT–PCR from RNA in E19.5 fetal livers of HRI +/+ and –/– embryos. (D) HRI protein and eIF2α kinase activity. The levels of HRI protein in the lysates of 1 × 106 reticulocytes of HRI +/+, +/– and –/– mice were examined by western blot analysis (top). The eIF2α kinase activity of HRI in reticulocyte lysates was determined using in vitro protein kinase assays followed by western blot analysis using antibody specific to the phosphorylated eIF2α (bottom).
The targeting construct was prepared by replacing a 5 kb DNA segment containing the three exons that encode kinase catalytic domains VIb–X with the neomycin phosphotransferase gene under the control of the phosphoglycerate kinase promoter (PGK-Neo). These three exons encode a region essential for the kinase activity of HRI. Thus, in the event that an HRI protein bearing this internal deletion were produced from the targeted HRI gene, it would be inactive. Correct targeting of the mouse HRI gene was confirmed by PCR analysis of tail DNA (Figure 1B).
HRI mRNA was examined in +/+ and –/– mice by RT–PCR (Figure 1C). RNA from +/+ fetal livers produced a diagnostic 1867 bp DNA fragment containing the entire coding sequence. RNA from –/– fetal livers produced a smaller DNA fragment of the expected size of 1463 bp with correct splicing of the mRNA over the three deleted exons of the targeted HRI gene.
Since HRI protein is expressed most abundantly in reticulocytes, we examined the expression of HRI protein in reticulocytes of +/+, +/– and –/– mice by western blot analysis using antibodies directed against the 138 N-terminal amino acids of mouse HRI, whose encoding exons were not removed by genomic targeting. HRI protein was detected in +/+ and heterozygote reticulocytes but not in –/– homozygotes (Figure 1D, top). These data suggest that the truncated HRI protein encoded by the targeted gene is unstable and demonstrate that HRI null mice are obtained.
HRI–/– mice are viable and fertile without gross morphological abnormalities. The distribution of progeny of HRI–/– mice follows closely the Mendelian rule, comprising 21% of total mice born from heterozygote crosses (Table I). In addition, the average litter size of homozygote crosses is very similar to that of the wt crosses. These results indicate that there is no significant death of HRI–/– embryos in the uterus.
Table I. Genotype of progeny of heterozygous crossesa.
| Genotype | Number | % |
|---|---|---|
| +/+ | 55 | 29.5 |
| +/– | 92 | 49.5 |
| –/– | 39 | 21.0 |
Average litter size 11.7.
aThe mice (total 186) were from 18 litters of pups. The genotype was determined by PCR of tail DNA as described in Figure 1B.
Mild macrocytosis and hyperchromia in otherwise normal HRI–/– mice in the absence of stress
Only minimal abnormalities were detected in the hematological parameters of HRI–/– mice in the absence of stress. There was a slight increase in mean RBC volume (MCV) accompanied by a moderate but significant increase in the mean RBC hemoglobin (MCH; see Figure 3A and B). As hemoglobin concentration per RBC is normally tightly regulated, no case of elevated MCH, referred to as hyperchromia, has ever been reported in humans or other mammals. There was also an increase in the number of Heinz body-containing RBCs in HRI–/– mice (see Figure 5B). We have also examined the phenotype of HRI–/– embryos and found no apparent defect in embryonic erythropoiesis under normal dietary conditions.

Fig. 3. Hematological analysis of HRI +/+ and –/– mice in iron deficiency. The RBC number, Hb (total hemoglobin) and the RBC indices, MCV and MCH were obtained from tail vein blood. Time courses of these changes from day 17 to 84 are shown. Four to six mice were used for each group. Wt + Fe, Wt mice in a normal diet; Wt – Fe, Wt mice in a low iron diet; Ko + Fe, knockout mice in a normal diet; Ko – Fe, knockout mice in a low iron diet. The differences in these red cell indices are statistically significant for a low iron diet (p <0.001 for all these four parameters).

Fig. 5. Precipitation of globins in RBCs of HRI–/– mice in iron deficiency. (A) Wright–Giemsa-stained peripheral blood smears. Peripheral blood smears were prepared from HRI +/+ and –/– mice maintained on an iron-deficient diet for 33 days. (B) Heinz body staining. Blood from mice at day 92 after receiving a low iron diet was collected and stained with crystal violet for the presence of Heinz bodies. (C) Precipitation of globins in the blood cells of HRI–/– mice. Precipitated proteins from the blood of Wt and HRI–/– mice at day 67 after receiving a low iron diet were collected by centrifugation and separated by 15% SDS–PAGE. Proteins were analyzed by Coomassie Blue staining (upper two panels) and by western blot analysis using anti-mouse hemoglobin antibody (lower two panels). P2, pellets of 2000 g centrifugation; P100, pellets of 100 000 g centrifugation.
Profound decrease in eIF2α phosphorylation in vivo in HRI–/– reticulocytes
Although HRI is the predominant eIF2α kinase in reticulocytes and nucleated erythroid precursors (Crosby et al., 1994; Lu et al., 2001), we wanted to determine whether other eIF2α kinases, such as PKR, might substitute for HRI in HRI–/– mice by examining the extent of eIF2α phosphorylation in intact reticulocytes. The level of eIF2α phosphorylation in intact reticulocytes was dramatically decreased in HRI–/– homozygotes compared with HRI+/+ mice (Figure 2A, middle), while the total eIF2α protein level was not altered significantly (Figure 2A, bottom). The low-level eIF2α phosphorylation observed in HRI–/– reticulocytes comes from other eIF2α kinases, such as PKR, which is known to be present in reticulocytes (Farrell et al., 1977). To assess HRI activity in vitro, eIF2α phosphorylation assays were performed with reticulocyte lysates in the presence of an excess of exogenous ATP, using endogenous eIF2 as a substrate. In vitro eIF2α phosphorylation occurred in reticulocyte lysates from +/+ mice and, to a lesser degree, in those of +/– heterozygotes. In contrast, little eIF2α phosphorylation was detected in the reticulocyte lysates of HRI–/– mice (Figure 1D, bottom). These data indicate that most of the eIF2α kinase activity normally found in reticulocytes under these experimental conditions is abrogated in HRI–/– mice and other eIF2α kinases are unable to compensate for this loss of HRI activity in reticulocytes.

Fig. 2. Characterization of the HRI–/– reticulocytes. (A) Protein synthesis and eIF2α phosphorylation in intact reticulocytes. Protein synthesis was carried out by labeling HRI +/+ or –/– reticulocytes (2 × 108 reticulocytes/ml) with [35S]methionine for 90 min. Samples (3 × 105 reticulocytes) were taken every 15 min to be analyzed for rate of protein synthesis (top, autoradiogram), eIF2α phosphorylation (middle, western) and total eIF2α (bottom, western). (B) Polysome profiles of HRI +/+ and –/– reticulocytes. Polysomal profiles were obtained with 5 × 107 reticulocytes. Numbers of ribosomes in the polysomes were labeled. (C) Northern blot analysis of β-globin mRNA. Total RNAs from HRI +/+ and –/– reticulocytes (0.5, 1 and 5 µg) were analyzed. (D) Loss of heme-dependent globin synthesis in HRI–/– reticulocytes. HRI+/+ and HRI–/– reticulocytes were incubated with [35S]methionine for 90 min in the presence of hemin (H) or cycloheximide (CHX). C, untreated control reticulocytes. Samples were taken every 30 min to be analyzed for globin synthesis.
Increased protein synthesis in HRI–/– reticulocytes and abolition of the regulation by heme
Since HRI is an inhibitor of protein synthesis, the rates of protein synthesis in intact reticulocytes of HRI +/+ and –/– mice were examined by measuring the incorporation of [35S]methionine into newly synthesized proteins. Compared with +/+ mice, there was a marked increase in protein synthesis seen globally for all proteins (Figure 2A, top). However, this effect was most pronounced for α- and β-globins since they are the most abundantly expressed and actively translated proteins in reticulocytes. Quantitation of [35S]methionine incorporation into the globin chains showed that there was a 7-fold increase in globin synthesis in HRI–/– reticulocytes as compared with HRI+/+ reticulocytes (Figure 2D). This increase in protein synthesis was accompanied by the increase in larger size polysomes in HRI–/– reticulocytes (Figure 2B). In addition, there was no difference in the β-globin mRNA levels between HRI +/+ and –/– reticulocytes (Figure 2C). Thus, these results demonstrate that HRI–/– reticulocytes have a higher rate of translation initiation, providing in vivo evidence for the role of eIF2α phosphorylation by HRI in the regulation of translational initiation.
We then examined the effect of heme on protein synthesis in HRI +/+ and –/– reticulocytes (Figure 2D). Incorporation of [35S]methionine into globins was increased to 208% of control by pre-treatment of +/+ reticulocytes with 40 µM hemin. In contrast, protein synthesis in HRI–/– reticulocytes was not affected by the addition of hemin. Protein synthesis of both wt and HRI–/– reticulocytes was inhibited equally well by cycloheximide (CHX), which inhibits translational elongation and is independent of HRI (Figure 2D).
Together, these results indicate that in the absence of HRI, the steady-state level of protein synthesis in reticulocytes is considerably increased in a heme-independent manner, with the main impact on the synthesis of α- and β-globin chains, the most abundant proteins translated in these cells.
Alteration of the hematological response of HRI–/– mice to diet-induced iron deficiency: anemia with erythroid hyperplasia and increased apoptosis of erythroid precursors
The profound biochemical changes in HRI–/– reticulocytes described above contrast with the minor alterations in the hematology of the mice in the absence of stress. Since we have hypothesized that HRI may act as a heme sensor that regulates translation in RBC precursors, one would expect to be able to exacerbate the phenotype of HRI–/– mice under conditions in which HRI is activated, particularly in heme deficiency. In order to induce heme deficiency, we put the mice on a low iron diet, since heme is synthesized by the insertion of iron into protoporphyrin IX via ferrochelatase during the last step of heme biosynthesis. In addition, iron deficiency is a very common condition found in humans and other mammals, and it may, in fact, be that HRI ultimately exists to protect RBCs from some of the major untoward consequences of iron deficiency. As shown in Table II, the total heme contents in the blood of +/+ and –/– mice were reduced by 27.4 and 46.2%, respectively, in iron deficiency, whereas the free protoporphyrin content was not significantly altered. In addition, there were also decreases in total hemoglobins (Hb; Figure 3D) in iron deficiency. Thus, heme deficiency is induced by iron deficiency. It is interesting that the decrease in heme content is more pronounced in HRI–/– blood. The role of HRI in iron homeostasis remains to be investigated.
Table II. Heme and protoporphyrin IX content in total blood (nmol/µl)a.
| Heme (± SD) | PPIX (± SD ) | |
|---|---|---|
| Wt + Fe | 26.82 ± 0.81 | 1.37 ± 0.06 |
| Wt – Fe | 19.73 ± 2.19 | 1.27 ± 0.08 |
| Ko + Fe | 22.74 ± 2.09 | 1.37 ± 0.10 |
| Ko – Fe | 12.24 ± 1.44 | 1.08 ± 0.13 |
an = 5 mice for each group.
The normal adaptive response to iron deficiency is well characterized in humans and mice. Both MCV and MCH decrease considerably, accompanied by a mild decrease in RBC counts, resulting in the classical microcytic and hypochromic anemia (Figure 3A and B; Wt – Fe). In HRI–/– mice, this physiological response to iron deficiency was dramatically altered (Figure 3; Ko – Fe). MCV was only slightly decreased compared with HRI–/– mice on a normal diet (Figure 3A), but was still similar to the Wt mice on a normal diet (Wt + Fe). Whereas MCH was slightly elevated compared with the Wt + Fe (Figure 3B), RBC counts decreased considerably (Figure 3C), resulting in a very unusual hyperchromic, normocytic anemia with a decrease in RBC counts. As seen above, an increase in MCH has never been reported in humans or mice. It is even more surprising to observe RBC hyperchromia in the setting of iron deficiency, when the normal response is characterized by a marked hypochromia.
These hematological changes were found in all animals examined continuously from 17 to 84 days of the low iron diet. No difference was observed in platelet counts. The hearts of HRI–/– mice were enlarged by ∼50% in iron deficiency compared with Wt + Fe, Wt – Fe or Ko + Fe mice. This cardiomegaly is most likely a secondary response to the anemia, in light of the fact that HRI is not expressed in the heart. A general pathological survey of tissues other than spleen and heart did not reveal significant morphological differences between –/– and +/+ mice placed on the low iron diet for 43–84 days.
Despite the hyperchromic RBCs, Ko – Fe mice were anemic due to a profound decrease in RBC numbers. To explore this further, we sought to determine whether the decreased RBC number was caused by decreased RBC production or increased RBC destruction. The spleens of Ko – Fe mice were on average 10-fold larger than those of Ko + Fe, Wt + Fe and Wt – Fe mice. Tissue sections of the bone marrow and the spleen of Ko – Fe mice showed marked expansion of erythroid precursors in bone marrow (data not shown) and the red pulp of spleen (Figure 4A).
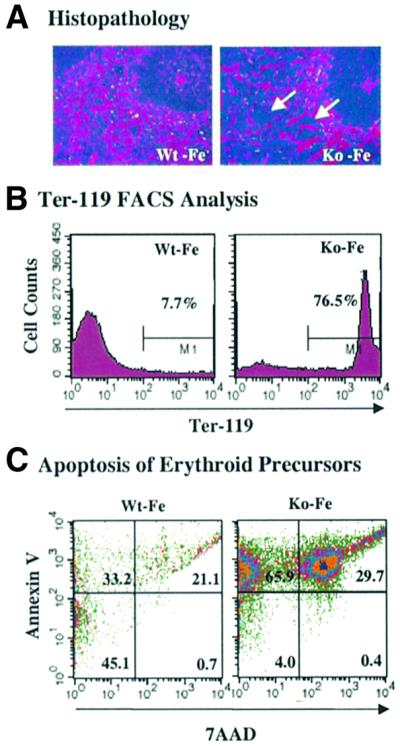
Fig. 4. Erythroid hyperplasia and increased apoptosis of HRI–/– erythroid precursors in iron deficiency. (A) Erythroid hyperplasia in the spleens of HRI–/– mice. The tissue sections of the spleens of Wt and HRI–/– mice both on normal and iron-deficient diets for 43 days were stained with hematoxylin and eosin. The arrows indicate the expanded red pulp. (B) Increased Ter-119+ cells in HRI–/– spleens. Single-cell suspensions from the spleens of HRI +/+ and –/– mice were gated with Ter-119 monoclonal antibody. (C) Increased apoptosis of erythroid progenitor cells in HRI–/– spleens. The Ter-119+ cells were further analyzed for their AnV and 7AAD stainings to gate the living cells (lower left quadrant), apoptotic cells (upper left quadrant) and dead cells (lower and upper right quadrant). The number in each quadrant represents the percentage of total cells.
The expansion of erythroid precursors was further established by fluorescence-activated cell sorter (FACS) analysis using Ter-119 monoclonal antibody, a marker for late erythroid cells from pro-erythroblast to mature erythrocytes (Kina et al., 2000). A 10-fold increase in Ter-119+ erythroid precursor cells was observed in the spleens of Ko – Fe mice (Figure 4B). There was a 2- to 3-fold increase in the colonies of BFU-E (burst forming unit-erythroid) and CFU-E (colony forming unit-erythroid), but no increase in CFU-GM (colony forming unit-granulocyte monocyte) was observed (data not shown). In addition, the reticulocyte count in the blood was significantly elevated in Ko – Fe mice (17.2%) compared with Ko + Fe, Wt + Fe and Wt – Fe mice (4.1%), providing additional evidence for erythroid hyperplasia of HRI–/– mice with iron deficiency.
We have examined the half-life of the RBCs by in vivo labeling with biotin (Hoffmann–Fezer et al., 1993; Browne et al., 1997). There was no apparent difference in the survival of RBCs from Wt and HRI–/– mice either on the normal or iron-deficient diet (data not show). The half-life of RBCs is ∼22 days, in good agreement with other published results (Levin, et al., 1999). All these features suggest that the considerable decrease in RBC count may be caused by abnormal destruction of red cell precursors.
We then examined the apoptotic death of Ter-119+ erythroid precursors in the spleen and bone marrow of HRI–/– mice in iron deficiency. The Ter119+ cells were further analyzed with annexin V (AnV) and 7-amino actinomycin D (7AAD). AnV binds to phosphatidylserine that has moved from the inner to the outer surface of the plasma membrane lipid bilayer, a very early event in apoptosis (Koopman et al., 1994). 7AAD was used to label the necrotic dead cells. Compared with Wt – Fe mice, there was a substantial increase in the percentage of apoptotic Ter-119+ cells from spleen (Figure 4C, upper left quadrant) and bone marrow (data not show) of Ko – Fe mice with a concomitant decrease in the percentage of live cells in spleen (Figure 4C, lower left quadrant). Thus, there is an increase in apoptosis of late erythroid precursors in HRI–/– mice with iron deficiency. No increase in apoptosis was observed in Ter-119– cells from HRI–/– mice (data not shown).
Collectively, these findings demonstrate that HRI–/– mice have dramatically altered the normal response to diet-induced iron deficiency by shifting from an adaptive decrease in RBC volume and intracellular hemoglobin content to an increased production of abnormally dense RBCs with decreasing red cell counts. The decrease in red cell number even in the presence of compensatory erythropoietic hyperplasia is the result of increased apoptosis of erythroid precursors.
Precipitation of globins in RBCs of iron-deficient HRI–/– mice
The molecular basis for the abnormal density of HRI–/– RBCs was investigated. Examination of Wright–Giemsa-stained blood smears under the light microscope showed the presence of multiple variably-sized eosinophilic inclusions within reticulocytes and, to a lesser extent, within fully mature RBCs in Ko – Fe mice (Figure 5A). These inclusions were not discernible in RBCs of +/+ mice. Upon staining of blood cells with crystal violet, Heinz bodies were seen in 80% of RBCs from Ko – Fe mice (Figure 5B). Heinz bodies were also seen in blood samples from Ko + Fe mice, albeit to a lesser extent (9.6%). In contrast, RBCs from Wt + Fe and Wt – Fe mice did not contain Heinz bodies. Heinz bodies have been extensively characterized in RBCs of human patients with unstable hemoglobin syndromes, thalassemias and various chemical, particularly oxidative, assaults. In all cases, Heinz bodies are composed of denatured proteins, primarily globins (Bull, 2001).
To characterize further the eosinophilic inclusion bodies found in the reticulocytes of HRI–/– mice, the blood cells were examined by transmission electron microscopy. The inclusion bodies were homogeneous in electron density and not bound by a membrane (Figure 6). Similar inclusions are present in RBCs and erythroblasts of thalassemic patients (Polliack and Rachmilewitz, 1973; Wickramasinghe and Bush, 1975; Wickramasinghe et al., 1980, 1984). In these cases, the inclusions have been shown to contain α- or β-globin in β- or α-thalassemia, respectively (Wickramasinghe et al., 1996).

Fig. 6. Electron microscopic examination of the inclusions in the reticulocytes of HRI–/– mice in iron deficiency. Blood from mice of all four groups at day 59 after receiving low iron diet was collected and processed for electron microscopy. The inclusion, indicated by an arrow, in a reticulocyte of HRI–/– mice is shown in (A) at a magnification of 30 000 ×. Inclusions were not observed in the blood samples of other groups. Higher magnification (150 000 ×) of the boxed area is shown in (B). The long arrow indicates the border of the inclusions and the shorter arrow indicates the membrane of a mitochondrion.
We have analyzed the components in the inclusion bodies biochemically. The inclusion bodies in blood samples were collected by centrifugation of blood lysates at 2000 g (P2), 14 000 g and 100 000 g (P100). No visible pellets were present at 14 000 g. The main protein in the pellet fractions of P2 and P100 was globin, as demonstrated by its molecular size and reactivity to anti-hemoglobin antibody (Figure 5C). Most importantly, there were more globins in the P2 and P100 fractions from Ko – Fe blood lysates than from Wt – Fe, Wt + Fe or Ko + Fe mice. These results demonstrate that there is a significant increase in the insoluble precipitated globins in HRI–/– red cells in iron deficiency. In addition, the majority of the insoluble globins were present in P2 fractions, indicating larger inclusion bodies, as were seen morphologically in Figures 5A and 6. These results provide further in vivo evidence that globins misfold and precipitate in the absence of proper binding to heme, as has been observed in vitro (Yip et al., 1972; Waks et al., 1973) and in the study of unstable hemoglobins caused by mutations that decrease heme incorporation (Dacie et al., 1967; Wajcman et al., 1992).
Increased sensitivity of HRI–/– RBCs to hemolytic reagents
States of iron deficiency have been and remain very common in humans. Thus, exposure to situations that render RBCs more fragile may often be concurrent with iron deficiency, such as in thalassemias, sickle cell disease, malaria and other infections, extreme climatic conditions, ingestion of natural toxins, heavy metals or drugs. Hence, we investigated whether HRI–/– mice were more sensitive to hemolytic agents than normal mice.
As shown in Figure 7A, the survival rate of iron-deficient HRI–/– mice upon phenylhydrazine-induced hemolytic erythroid stress was dramatically reduced. The standard regime employed three injections of phenylhydrazine on days 0, 1 and 3. After two injections, 90% of Ko – Fe mice died, in contrast to nearly 100% survival of Wt – Fe mice. Even in the absence of iron deficiency, HRI–/– mice were already more sensitive to phenylhydrazine with an IC50 of 50 mg/kg as compared with 55 mg/kg for wt mice (Figure 7B). Thus, although HRI–/– RBCs have a normal half-life on an iron-deficient diet, these RBCs are much more sensitive to additional stress, such as oxidative stress induced by the administration of phenylhydrazine. This finding indicates that HRI provides a protective role in maintaining the integrity of mature red cells, particularly during combined assaults of iron deficiency together with additional stress. Thus, even though HRI is no longer expressed in mature erythrocytes, its effect during erythroid differentiation persists into mature RBCs.

Fig. 7. Decreased survival of HRI–/– mice in phenylhydrazine-induced hemolysis. Both Wt and HRI–/– mice were injected with phenylhydrazine at the dosages indicated at days 0, 1 and 3. The mice were observed daily. (A) The time courses of survival of the iron-deficient Wt and HRI–/– mice at a dosage of 50 mg/kg. n = 12 for Wt mice; n = 11 for HRI–/– mice. (B) The percentage survival at different dosages at day 6 after initial phenylhydrazine injection. Six mice of each genotype were used for each dosage.
Collectively, our findings strongly suggest that the increased cell destruction seen in HRI–/– mice with iron deficiency is caused by the excessive accumulation and precipitation of heme-free globin chains. The lack of HRI relieves the normal feedback regulation of globin translation in heme deficiency. Furthermore, in the absence of HRI, RBCs become much more sensitive to hemolytic agents, particularly in iron deficiency.
Discussion
HRI was first identified as an inhibitor of protein synthesis in reticulocytes in 1960 (reviewed in Chen, 1993). Although HRI has been extensively studied biochemically, its physiological function in the whole animal was unknown. To date, no disease in human or mouse is known to be associated with mutations in the HRI gene, yet orthologs of HRI appear widely conserved from fish to humans, suggesting an important role for HRI.
Here, we have uncovered a clear physiological function of HRI by analyzing the phenotype of mice with a targeted disruption of the HRI gene. We demonstrate that HRI is essential to shut off α- and β-globin synthesis in erythroid cells in iron deficiency. As expected from a protein expressed predominantly in erythroid cells, only RBCs and their precursors were directly affected by the lack of HRI. Our findings are summarized in the model shown in Figure 8A. One molecule of heme is incorporated into each of the α- and β-globin chains in the formation of a stable α2β2 hemoglobin tetramer. HRI serves as a feedback inhibitor of globin synthesis by sensing heme. Heme binds to the kinase insertion domain of HRI and prevents its activation by autophosphorylation. Once HRI is activated, it phosphorylates eIF2α and inhibits the recycling of eIF2 for another round of protein synthesis. Consistent with previous biochemical studies, a 7-fold increase in protein synthesis, which also became insensitive to heme, was observed in the reticulocytes of HRI–/– mice with a corresponding decrease in eIF2α phosphorylation (Figure 2).

Fig. 8. Models of the role of HRI during erythroid differentiation. (A) Regulation of α- and β-globin synthesis by HRI and heme. (B) Altered hematological response of HRI–/– mice to iron deficiency.
As illustrated in Figure 8B, the mild alterations of RBCs observed in HRI–/– mice in the absence of stress became profound under conditions of diet-induced iron deficiency, which decreases the intracellular concentration of heme. The normal hematological response of wt mice to iron deficiency, characterized by a microcytic and hypochromic anemia, switched to a hyperchromic anemia with increased destruction of the late red cell precursors and compensatory erythroid hyperplasia. Destruction of mature RBCs was exacerbated in the presence of hemolytic agents. Together, these data are consistent with the above model in which HRI normally ensures that no globin chains are translated in excess of what can be assembled into hemoglobin tetramers for the amount of heme available. Therefore, the critical role of HRI becomes apparent only when heme concentrations in RBC precursors decline, as is commonly found in iron deficiency.
The lesser of two evils: microcytic anemia rather than hemolytic anemia
Iron deficiency is very common with an incidence of ∼2 billion cases worldwide. It is most often a consequence of a low iron diet or blood loss. When available iron and, as a consequence, heme declines below a certain threshold, the occurrence of anemia by decreased hemoglobin tetramer production is unavoidable. However, our data indicate that, in the absence of HRI, the consequences of iron deficiency are much more deleterious than with HRI. These findings are consistent with the hypothesis that the least detrimental adaptive response is to decrease globin production, resulting in mild well-tolerated microcytic and hypochromic anemia, rather than allowing globin translation to continue. If this occurs, as in the HRI–/– mice, free globins precipitate and add a major cell destruction component to the pathophysiology of the anemia.
Microcytosis and hypochromia have been one of the first biological signs identified in human medicine and remain one of most frequent anomalies found in patients. Yet, no molecular mechanism had been identified for this phenomenon. Our data indicate that HRI is responsible for this physiological adaptation of RBCs to iron deficiency. In addition, the occurrence of situations that render RBCs more fragile often co-exists, as in thalassemias, sickle cell disease, unstable hemoglobins, malaria and other infections, extreme climatic conditions, ingestion of natural toxins, heavy metals or drugs. In the absence of HRI, the combination of iron deficiency with one of these situations may be rapidly fatal.
Control of the expression of globin genes by a general translational factor eIF2
Control of gene expression of single genes most often involves a form of regulation that targets a specific trans-acting factor acting on a gene-specific cis-acting element in isolation or as part of a complex of factors. It might, therefore, be surprising that the control of the expression of α- and β-globins by heme ultimately acts on the general translational factor eIF2. In this case, the specificity is achieved by the restrictive expression of the sensor/regulator HRI for the erythroid lineage, coupled to the fact that globin mRNAs are the main template for protein synthesis in reticulocytes. Why does the negative control exerted by heme deficiency inhibit translation rather than transcription? Since heme plays a role in the co-ordinated expression of many erythroid-specific genes, which include the globin genes and the HRI gene itself (Sassa and Nagai, 1996), by activating their transcription, it can be hypothesized that a negative control exerted by heme deficiency is more readily achieved at another level of the gene expression machinery. Since at least 25% of globin protein synthesis occurs in reticulocytes after nuclei have been extruded, translational regulation is the main level of control remaining (Bull, 2001).
Although the hyperplasia of hematopoietic organs may be a compensatory reaction to the accelerated RBC destruction in HRI–/– mice in iron deficiency, it may also be due to direct proliferative or anti-apoptotic effects triggered by the loss of HRI in RBC precursors. Overexpression of dominant-negative mutants of HRI in MEL cells has been shown to increase the proliferative capacity of these cells upon induction of terminal differentiation by dimethylsulfoxide (DMSO; Crosby et al., 2000).
eIF2α kinases as specific sensors and effectors that protect against environmental stress
In addition to HRI, three other eIF2α kinases (PKR, GCN2 and PERK) have been found in mammalian cells. The observation that the pathological consequences of the disruption of the mouse HRI gene are revealed under conditions of diet-induced iron deficiency is consistent with the fact that all eIF2α kinases are activated under various stress conditions. PKR–/– mice are also viable without significant phenotypic change until challenged by viral infection (Yang et al., 1995; Abraham et al., 1999; Stojdl et al., 2000). In yeast, GCN2 is non-essential under optimal growth conditions (Hinnebusch, 1996). The phenotypic changes of HRI–/– mice in erythroid cells in iron deficiency reported here are very similar to the uncontrolled translation and apoptotic cell death in pancreatic cells of PERK–/– mice reported recently (Harding et al., 2001). Thus, the physiological role of members of this class of kinases may be to act as homeostatic guardians against major environmental stress by ultimately regulating protein synthesis in response to specific exogenous signals and ensuring cell survival.
Our findings provide important insights into the function of HRI in co-ordinating the synthesis of globins in RBC precursors to the concentration of heme in vivo. They also warrant the search for possible mutations in the HRI gene in cases of hematological syndromes of unknown origin, in which normocytic and hyperchromic anemia are observed with Heinz bodies and inclusions in RBC and late erythroid precursors. Beyond the example of HRI, our study provides a demonstration that translational control of gene expression does play an important physiological role in somatic cell differentiation in vivo.
Materials and methods
Targeted disruption of the murine HRI gene
A λ-FixII phage genomic DNA library of mouse strain 129/SV (Stratagene) was screened for the HRI gene using the entire coding sequence of rabbit HRI cDNA (Chen et al., 1991) as a probe. Clone 19, which contains five exons, was used for targeting construct preparation. The 5 kb fragment containing three exons of HRI (encoding kinase domains VIb–X) was excised with NdeI and replaced with floxed PGK-Neo (Figure 1A). HSV-thymidine kinase (TK) was ligated to the EcoRV site. The HRI targeting construct was linearized with SalI and electroporated into ES cells. Of two homologous-recombined ES clones, one with a normal karyotype was injected into blastocysts to produce chimeric mice and subsequently the heterozygous HRI+/– mice. Heterozygotes were crossed with the GATA-1–Cre mice to remove the PGK-Neo gene in the germline (Mao et al., 1999). The PGK-Neo minus HRI+/– heterozygotes were interbred to generate HRI–/– homozygotes.
Genotyping was performed by PCR of tail DNA. PCR reaction 1 was carried out with primers 1 (5′-AGCTCCACCCTGACGATCTA-3′) and 2 (5′-ATGTGCAGGGCTGAAGAGAT-3′) and PCR reaction 2 with primers 1 and 3 (5′-CATGCTGGGGGTCAAATAGT-3′), as illustrated in Figure 1A. The conditions for PCR were denaturation at 95°C for 2 min, followed by 30 cycles of amplification (denaturing at 95°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 2 min) and a subsequent extension at 72°C for 10 min, using Taq polymerase, 5 pmol of each primer, 2.5 mM MgCl2 and 400 µM dNTPs.
Cloning of the mouse HRI cDNA and production of anti-HRI polyclonal antibodies
For the characterization of the phenotype of HRI knockout (Ko) mice, mouse HRI cDNA was cloned from a λ-gt11 cDNA library of DMSO-induced MEL cells. The full-length mouse HRI cDNA (AY033898) encodes 619 amino acids and exhibits high homology (82%) to rabbit HRI. The mouse 138 N-terminal amino acids and the kinase insertion sequence (amino acids 241–405) were expressed in Escherichia coli BL-21 cells, purified and then used to produce polyclonal antibodies (HTI Bio-Products).
Production of mouse reticulocytes, protein synthesis, protein kinase assays and western blot analysis
Mice were injected with phenylhydrazine at 40 mg/kg on days 0, 1 and 3. Blood samples were collected by heart puncture on day 7 when the reticulocyte counts were 85–95%. Reticulocytes were washed twice with ice-cold phosphate-buffered saline (PBS) supplemented with 5 mM glucose. For protein synthesis assays, reticulocytes were resuspended in Dulbecco’s modified Eagle’s medium (DMEM; 2 × 108 cells/ml) with 1/10 concentration of methionine plus 2% dialyzed fetal bovine serum (FBS) and pre-incubated for 30 min at 37°C for recovery. The reticulocytes were treated with hemin (40 µM) or CHX (2 µM) for 30 min as indicated and then labeled with [35S]methionine (5 Ci, 3000 Ci/mmol). At the times indicated in Figure 2, 200 µl of cell suspension were lysed in SDS sample buffer containing EDTA (1 mM) and NaF (50 mM). Incorporations of the [35S]methionine into the globin chains and other proteins were analyzed by 15% SDS–PAGE, followed by transfer to nitrocellulose membranes and autoradiography. Globin synthesis in reticulocytes was quantitated by scintillation counting of the nitrocellulose strips containing globin chains. Phosphorylation of HRI and eIF2α in isolated reticulocytes was analyzed by western blot analysis following 7.5 and 12% SDS–PAGE, respectively. In vitro protein kinase assays using the endogeneous eIF2 as a substrate were performed as described previously (Chen et al., 1989).
Northern blot and polysome analyses
Total RNAs from HRI +/+ and –/– reticulocytes (8 × 107 cells) were isolated using TriPure reagents (Boehringer Mannheim). For northern blot analysis of β-globin mRNA, RNAs were separated by 1.2% agarose gel electrophoresis, probed with 32P-labeled mouse β-globin cDNA and detected by autoradiography.
Polysome profiling of reticulocytes was performed as described in Martin and Berry (2001) using 15–50% sucrose gradients and centrifugation at 45 000 r.p.m. for 100 min at 4°C in a Beckman SW 50.1 rotor. Reticulocytes, prepared as described above, were washed once with PBS containing CHX (50 µg/ml) prior to lysis of the cells
Diet-induced iron deficiency and hematological and pathological analyses
A state of iron deficiency was induced by placing newborn mice (after weaning) on a low iron diet containing 5 p.p.m. Fe. Some of the littermates were fed a normal diet containing 196 p.p.m. Fe as controls. Hematological analysis of the peripheral blood collected from the tail vein was performed bi-weekly using a Hemavet® 800 instrument (CDC Technologies Inc.). Heme and protoporphyrin contents in total blood of mice on the low iron diet or normal diet for 2 months were determined as described (Sassa, 1976).
Reticulocyte counts were analyzed by FACS after staining with thiazole orange (Aldrich Chem. Co.) as described (Lee et al., 1986). Heinz bodies in the blood samples were determined by staining live cells with crystal violet (Beutler, 1983). Tissues were fixed in formalin, and processed for paraffin embedding and sectioning using standard procedures. Electron microscopy was performed on Karnofsky’s fixed EDTA-anticoagulated blood using standard methods.
FACS analysis of Ter-119+ cells and apoptosis
A single-cell suspension of spleen from mice on the low iron diet for 2 months was prepared by homogenizing the tissue with a Dounce homogenizer in PBS plus 2% FBS. A single-cell suspension of bone marrow was obtained by flushing both femurs with a 21-gauge needle. The mature erythrocytes were removed by hypotonic lysis (5 mM NH4Cl in 0.1 × PBS). Cells were then washed twice with PBS plus 2% FBS and labeled with phycoerythrin-conjugated Ter-119 antibody (1 µg/106 cells; PharMingen) for 60 min in ice. After washing twice in PBS plus 2% FBS, 50 000 cells were gated by Ter-119 signal and the data were collected on a FACSCalibur and analyzed using CellQuest software (Becton Dickinson).
For apoptosis analysis, cells were labeled with fluorescein isothiocyanate-conjugated AnV (5 µl/106 cells; PharMingen) after labeling with Ter-119 antibody. Cells were subsequently stained with 7AAD (20 µg/ml; Sigma) in ice for 15 min to label the necrotic dead cells. Labeled cells were then gated first with Ter-119 signal and were further sorted by AnV and 7AAD signals.
Identification of globins in inclusion bodies
RBCs (8.8 × 107 cells) from both Wt and HRI–/– mice were washed twice with PBS supplementing 5 mM glucose and were then lyzed with 400 µl of 0.1 × PBS on ice for 20 min. The whole lysates were first centrifuged at 2000 g for 30 min at 4°C. The pellets (designated P2) were washed with 3 ml of 0.1 × PBS and dissolved in 100 µl of 1 × SDS sample buffer. Supernatant (300 µl) was further centrifuged at 100 000 g for 3 h at 4°C. The pellets were designated P100 and dissolved in 50 µl of 0.1 × PBS. Proteins in P2 (10 µl) and P100 (5 µl) fractions were separated by 15% SDS–PAGE and stained with Coomassie Blue. For western blot analysis of globins, 1 µl of the P2 and 0.5 µl of the P100 samples were used. The globin chains were detected with a rabbit anti-mouse hemoglobin antibody (Capple).
Acknowledgments
Acknowledgements
This work was supported by NIH grant DK-16272 to J.-J.C.
References
- Abraham. N.S.D. et al. (1999) Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem., 274, 5953–5962. [DOI] [PubMed] [Google Scholar]
- Berlanga J.J., Herrero,S. and de Haro,C. (1998) Characterization of the hemin-sensitive eukaryotic initiation factor 2α kinase from mouse nonerythroid cells. J. Biol. Chem., 273, 32340–32346. [DOI] [PubMed] [Google Scholar]
- Berlanga J.J., Santoyo,J. and DeHaro,C. (1999) Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2α kinase. Eur. J. Biochem., 265, 754–762. [DOI] [PubMed] [Google Scholar]
- Beutler E. (1983) Heinz body staining. In Williams,W.J., Beutler,E., Erslev,A.J. and Lichtman,M.A. (eds), Hematology. McGraw-Hill Book Co., USA, p. 1603.
- Browne P.V., Shalev,O., Kuypers,F.A., Brugnara,C., Solovey,A., Mohandas,N., Schrier,S.L. and Hebbel,R.P. (1997) Removal of erythrocyte membrane iron in vivo ameliorates the pathobiology of murine thalassemia. J. Clin. Invest., 100, 1459–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull B.S. (2001) Morphology of the Erythron. In Beutler,E., Lichtman,M.A., Coller,B.S., Kipps,T.J. and Seligsohn,U. (eds), Williams Hematology. McGraw-Hill Book Co., USA, pp. 271–288.
- Chefalo P.J., Yang,J.M., Ramaiah,K.V.A., Gehrke,L. and Chen,J.-J. (1994) Inhibition of protein synthesis in insect cells by baculovirus-expressed heme-regulated eIF-2α kinase. J. Biol. Chem., 269, 25788–25794. [PubMed] [Google Scholar]
- Chefalo P., Oh,J., Rafie-Kolpin,M. and Chen,J.-J. (1998) Heme-regulated eIF-2α kinase purifies as a hemoprotein. Eur. J. Biochem., 258, 820–830. [DOI] [PubMed] [Google Scholar]
- Chen J.-J. (1993) Translational regulation in reticulocytes: the role of heme-regulated eIF-2α kinase. In Ilan,J. (ed.), Translational Control of Gene Expression 2. Plenum Press, New York, NY, pp. 349–372.
- Chen J.-J. (2000) Heme-regulated eIF-2α kinase. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 529–546.
- Chen J.-J., Yang,J.M., Petryshyn,R., Kosower,N. and London,I.M. (1989) Disulfide bond formation in the regulation of eIF-2α kinase by heme. J. Biol. Chem., 264, 9559–9564. [PubMed] [Google Scholar]
- Chen J.-J., Throop,M.S., Gehrke,L., Kuo,I., Pal,J.K., Brodsky,M. and London,I.M. (1991) Cloning of the cDNA of the heme-regulated eukaryotic initiation factor 2α (eIF-2α) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double-stranded-RNA-dependent eIF-2α kinase. Proc. Natl Acad. Sci. USA, 88, 7729–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens M.J. (1996) Protein kinases that phosphorylate eIF-2 and eIF-2B and their role in eukaryotic cell translational control. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 139–172.
- Crosby J.S., Lee,K., London,I.M. and Chen,J.-J. (1994) Erythroid expression of the heme-regulated eIF-2α kinase. Mol. Cell. Biol., 14, 3906–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby J.S., Chefalo,P.J., Yeh,I., Ying,S., London,I.M., Leboulch,P. and Chen,J.-J. (2000) Regulation of hemoglobin synthesis and proliferation of differentiating erythroid cells by heme-regulated eIF-2α kinase. Blood, 96, 3241–3247. [PubMed] [Google Scholar]
- Dacie J.V., Shinton,N.K., Gaffney,P.J. and Lehmann,H. (1967) Haemoglobin hammersmith [β-42 (CDI) Phe replaced by Ser]. Nature, 216, 663–665. [DOI] [PubMed] [Google Scholar]
- Farrell P., Balkow,K., Hunt,T., Jackson,R.J. and Trachsel,H. (1977) Phosphorylation of initiation factor eIF-2 and the control of reticulocyte protein synthesis. Cell, 11, 187–200. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zhang,Y. and Ron,D. (1999) Protein translation and folding are coupled by an endoplasmic reticulum-resident kinase. Nature, 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zeng,H., Zhang,Y., Jungries,R., Chung,P., Plesken,H., Sabatini,D.D. and Ron,D. (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk–/– mice reveals a role for translational control in secretory cell survival. Mol. Cell, 7, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. (1996) Translational control of GCN4: gene-specific regulation by phosphorylation of eIF2. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 199–244.
- Hoffmann-Fezer G., Mysliwietz,J., Mortlbauer,W., Zeitler,H.J., Eberle, E., Honle,U. and Thierfelder,S. (1993) Biotin labeling as an alternative nonradioactive approach to determination of red cell survival. Ann. Hematol., 67, 81–87. [DOI] [PubMed] [Google Scholar]
- Kaufman R.J. (2000) Double-stranded RNA-activated protein kinase PKR. In Sonenberg,N., Hershey,J.W.B. and Matthews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 503–528.
- Kina T., Ikuta,K., Takayama,E., Wada,K., Majumdar,A.S., Weissman,I.L. and Katsura,Y. (2000) The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br. J. Haematol., 109, 280–287. [DOI] [PubMed] [Google Scholar]
- Koopman G., Reutelingsperger,C.P., Kuijten,G.A., Keehnen,R.M., Pals,S.T. and van Oers,M.H. (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood, 84, 1415–1420. [PubMed] [Google Scholar]
- Krishnamoorthy T., Pavitt,G.D., Zhang,F., Dever,T.E. and Hinnebusch,A.G. (2001) Tight binding of the phosphorylated α-subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol., 21, 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L.G., Chen,C.H. and Chiu,L.A. (1986) Thiazole orange: a new dye for reticulocyte analysis. Cytometry, 7, 508–517. [DOI] [PubMed] [Google Scholar]
- Levin J., Peng,J.P., Baker,G.R., Villeval,J.L., Lecine,P., Burstein,S.A. and Shivdasani,R.A. (1999) Pathophysiology of thrombocytopenia and anemia in mice lacking transcription factor NF-E2. Blood, 94, 3037–3047. [PubMed] [Google Scholar]
- Lu L., Han,A. and Chen,J.-J. (2001) Translation initiation control by heme-regulated eIF2α kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol., 21, 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Fujiwara,Y. and Orkin,S.H. (1999) Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc. Natl Acad. Sci. USA, 96, 5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G.W. III, and Berry,M.J. (2001) Selenocysteine codons decrease polysome association on endogenous selenoprotein mRNAs. Genes Cells, 6, 121–129. [DOI] [PubMed] [Google Scholar]
- Mathews M.B., Sonenberg,N. and Hershey,J.W.B. (2000) Origins and Principles of Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Mellor H., Flowers,K.M., Kimball,S.R. and Jefferson,L.S. (1994) Cloning and characterization of cDNA encoding rat hemin-sensitive initiation factor-2α (eIF-2α) kinase. J. Biol. Chem., 269, 10201–10204. [PubMed] [Google Scholar]
- Meurs E., Chong,K., Galabru,J., Thomas,N.S.B., Kerr,I.M., Williams,B.R.G. and Hovanessian,A.G. (1990) Molecular cloning and characterization of human double-stranded RNA activated protein kinase induced by interferon. Cell, 62, 379–390. [DOI] [PubMed] [Google Scholar]
- Polliack A. and Rachmilewitz,E.A. (1973) Ultrastructural studies in β-thalassaemia major. Br. J. Haematol., 24, 319–326. [DOI] [PubMed] [Google Scholar]
- Rafie-Kolpin M., Chefalo,P.J., Hussain,Z., Hahn,J., Uma,S., Matts,R.L. and Chen,J.-J. (2000) Two heme-binding domains of heme-regulated eIF-2α kinase: N-terminus and kinase insertion. J. Biol. Chem., 275, 5171–5178. [DOI] [PubMed] [Google Scholar]
- Ramirez M., Wek,R.C. and Hinnebusch,A.G. (1991) Ribosome association of GCN2 protein kinase, a translational activation of the GCN4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol., 11, 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. and Harding,H.P. (2000) PERK and translational control by stress in the endoplasmic reticulum. In Sonenberg,N., Hershey,J.W.B and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 547–560.
- Rouault R.A. and Harford,J.B. (2000) Translational control of ferritin synthesis. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 655–670.
- Sassa S. (1976) Sequential induction of heme pathway enzymes during erythroid differentiation of mouse Friend leukemia virus-infected cells. J. Exp. Med., 143, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa S. and Nagai,T. (1996) The role of heme in gene expression. Int. J. Hematol., 63, 167–178. [DOI] [PubMed] [Google Scholar]
- Shi Y., Vattem,K.M., Sood,R., An,J., Liang,J., Stramm,L. and Wek,R.C. (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol., 18, 7499–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood R., Porter,A.C., Olsen,D., Cavener,D.R. and Wek,R.C. (2000) A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2α. Genetics, 154, 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl D., Abraham,N., Knowles,S., Marius,R., Brasey,A., Lichty,B.D., Brown,E.G., Sonenberg,N. and Bell,J.C. (2000) The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol., 74, 9580–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachsel H. (1996) Binding of initiator methionyl-tRNA to ribosomes. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 113–138.
- Wajcman H., Kister,J., Vasseur,C., Blouquit,Y., Trastour,J.C., Cottenceau,D. and Galacteros,F. (1992) Structure of the EF corner favors deamidation of asparaginyl residues in hemoglobin: the example of Hb La Roche-sur-Yon [β-81 (EF5) Leu-His]. Biochim. Biophys. Acta, 1138, 127–132. [DOI] [PubMed] [Google Scholar]
- Waks M., Yip,Y.K. and Beychok,S. (1973) Influence of prosthetic groups on protein folding and subunit assembly: recombination of separated human α- and β-globin chains with heme and alloplex interactions of globin chains with heme-containing subunits. J. Biol. Chem., 248, 6462–6470. [PubMed] [Google Scholar]
- Wickramasinghe S. and Bush,V. (1975) Observations on the ultrastructure of erythropoietic cells and reticulum cells in the bone marrow of patients with homozygous β-thalassemia. Br. J. Haematol., 30, 395–399. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S.N., Hughes,M., Hollan,S.R., Horanyi,M. and Szelenyi,J. (1980) Electron microscope and high resolution autoradiographic studies of the erythroblasts in haemoglobin H disease. Br. J. Haematol., 45, 401–404. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S., Hughes,M., Fucharoen,S. and Wasi,P. (1984) The fate of excess β-globin chains within erythropoietic cells in α-thalassaemia 2 trait, α-thalassaemia 1 trait, haemoglobin H disease and haemoglobin Q-H disease: an electron microscope study. Br. J. Haematol., 56, 473–482. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe S.N., Lee,M.J., Furukawa,T., Eguchi,M. and Reid,C.D.L. (1996) Composition of the intra-erythroblastic precipitates in thalassaemia and congenital dyserythropoietic anaemia (CDA): identification of a new type of CDA with intra-erythroblastic precipitates not reacting with monoclonal antibodies to α- and β-globin chains. Br. J. Haematol., 93, 576–585. [DOI] [PubMed] [Google Scholar]
- Yang Y.-L., Reis,L.F.L., Pavlovic,J., Aguzzi,A., Schafer,R., Kumar,A., Williams,B.R.G., Aguet,M. and Weissman,C. (1995) Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J., 14, 6095–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip Y.K., Waks,M. and Beychok,S. (1972) Influence of prosthetic groups on protein folding and subunit assembly. I. Conformational differences between separated human α- and β-globins. J. Biol. Chem., 247, 7237–7244. [PubMed] [Google Scholar]