

Abstract
Protein targeting by the signal recognition particle (SRP) pathway requires the interaction of two homologous GTPases that reciprocally regulate each other’s GTPase activity, the SRP signal peptide- binding subunit (SRP54) and the SRP receptor α-subunit (SRα). The GTPase domain of both proteins abuts a unique ‘N domain’ that appears to facilitate external ligand binding. To examine the relationship between the unusual regulation and unique architecture of the SRP pathway GTPases, we mutated an invariant glycine in Escherichia coli SRP54 and SRα orthologs (‘Ffh’ and ‘FtsY’, respectively) that resides at the N–GTPase domain interface. A G257A mutation in Ffh produced a lethal phenotype. The mutation did not significantly affect Ffh function, but severely reduced interaction with FtsY. Likewise, mutation of FtsY Gly455 produced growth defects and inhibited interaction with Ffh. The data suggest that Ffh and FtsY interact only in a ‘primed’ conformation which requires interdomain communication. Based on these results, we propose that the distinctive features of the SRP pathway GTPases evolved to ensure that SRP and the SR engage external ligands before interacting with each other.
Keywords: GTPase/protein targeting/SRP/SRP receptor
Introduction
Proteins that bind and hydrolyze GTP (GTPases) regulate a wide array of cellular processes including signal transduction, translation, cytoskeletal organization and protein trafficking (Bourne et al., 1991). Although some of these processes are controlled by a single GTPase, many others are regulated by multiple GTPases or GTPase cascades (Chant and Stowers, 1995). Virtually all GTPases studied to date act as ‘molecular switches’ that cycle between GTP-bound and GDP-bound conformations. Interconversion between the two states allows GTPases to interact in temporal succession with different macromolecules and thereby ensure the unidirectionality of a biological process (Bourne et al., 1990). The alternation between the ‘active’ GTP-bound and ‘inactive’ GDP-bound conformations generally is catalyzed (or inhibited) by a separate set of GTPase-activating proteins (GAPs), guanine nucleotide exchange factors (GEFs) and guanine nucleotide dissociation inhibitors (GDIs).
A striking exception to the molecular switch paradigm is provided by the GTPases that regulate the release of nascent polypeptides from the signal recognition particle (SRP). SRP is a multisubunit ribonucleoprotein complex found in all organisms (reviewed in Keenan et al., 2001). The most highly conserved subunit is an ∼54 kDa GTPase (SRP54). In mammalian cells, SRP54 recognizes both signal sequences of secreted proteins and transmembrane segments of integral membrane proteins that lack a discrete signal peptide as they emerge during translation. Subsequently, SRP targets ribosome–nascent chain complexes to the endoplasmic reticulum (ER). Upon arrival at the ER, SRP54 binds to a homologous GTPase, the α-subunit of the heterodimeric SRP receptor (SRα). This interaction activates a ‘concerted switch’ in which both proteins bind GTP simultaneously and the nascent chain is transferred to a protein-conducting channel (‘translocon’) (Rapiejko and Gilmore, 1997). Curiously, neither SRP54 nor SRα contains a stably bound guanine nucleotide prior to their interaction. At the end of the targeting cycle, SRP54 and SRα act as GAPs that cross-stimulate each other’s GTPase activity (Powers and Walter, 1995; Rapiejko and Gilmore, 1997), and SRP is released from the membrane (Connolly et al., 1991). Since both proteins have a very low affinity for GDP (Miller et al., 1993; Moser et al., 1997), they probably return to a nucleotide-free state rapidly. Many bacteria contain a simplified SRP consisting of only an SRP54 ortholog (‘Ffh’) and a RNA (‘4.5S RNA’) that is much smaller than eukaryotic SRP RNA, and an SR consisting of only an SRα homolog (‘FtsY’). The genes that encode all of these molecules are essential for viability in Escherichia coli (Brown and Fournier, 1984; Phillips and Silhavy, 1992; Luirink et al., 1994). Despite the absence of most of the SRP subunits and the conversion of the SR from an integral to a peripheral membrane protein (Luirink et al., 1994), the bacterial SRP pathway functions similarly to its eukaryotic counterpart. In E.coli, SRP targets integral membrane proteins to the cytoplasmic membrane co-translationally (de Gier et al., 1996; Seluanov and Bibi, 1997; Ulbrandt et al., 1997; Neumann-Haefelin et al., 2000). Moreover, the properties of the SRP–SR interaction are conserved (Miller et al., 1994).
The unusual mechanism by which SRP54 and SRα regulate the SRP pathway correlates with a distinctive architecture that is not found in any other GTPases. The two proteins share ∼30 kDa of homology, including closely related GTPase domains and a unique N-terminal extension (‘N domain’). Although the core of the GTPase domains is structurally similar to small GTPases such as Ras (Freymann et al., 1997; Montoya et al., 1997), they also contain unique features that imply evolution from a common ancestor. One such feature is an insertion (‘I box’) which may act as an internal GEF (Moser et al., 1997). The N domain is a four-helix bundle that forms extensive contacts with the GTPase domain (Figure 1A), and the two domains can be isolated as a proteolytically stable fragment from both mammalian SRP54 and E.coli Ffh (Römisch et al., 1990; Zopf et al., 1990; Zheng and Gierasch, 1997). Both SRP54 and SRα GTPase domains contain a conserved motif (‘DAR/KGG’) overlapping the start of a short α-helix (α6) that sits at the interface between the N and GTPase domains (Figure 1A). This motif is in close proximity to the the guanine ring-binding site and faces a conserved loop (‘ALLEADV’) in the N domain. In addition to the N and GTPase domains, SRP54 has a conserved C-terminal domain (‘M domain’) that mediates interactions with both SRP RNA and signal peptides, and SRα has a highly variable N-terminal segment. In E.coli, the N-terminus of FtsY (‘A’ domain) is highly enriched in acidic amino acids. The orientation of the SRP54 M domain and the SRα N-terminal segment with respect to the NG domain is unknown.

Fig. 1. Mutation of an invariant glycine distorts the N–GTPase domain interface of Ffh and FtsY. (A) A ribbon diagram of the N and GTPase domains of Thermus aquaticus Ffh and E.coli FtsY (Freymann et al., 1997; Montoya et al., 1997) reveals the similarity of the overall fold and the close association of the loop following N domain α-helix 2 (α2) and GTPase domain α-helix 6 (α6). Conserved sequence motifs are found on both sides of the N–GTPase domain interface. The N domain motif (‘ALLEADV’) begins at the C-terminus of α2 and ends in the adjacent loop. The GTPase domain motif (‘DAR/KGG’) begins in the loop following the guanine ring-binding site (*) and ends in α6. The second glycine is invariant in all SRP54/Ffh and SRα/FtsY orthologs sequenced to date. Key amino acid side chains in the N domain and an alanine or valine in place of the invariant glycine in the DAR/KGG motif in Ffh and FtsY, respectively, are depicted as ball-and-stick representations. (B) A close-up view of the N–GTPase domain interface of Ffh and FtsY is shown. The van der Waals radii of the amino acid side chains highlighted in (A) are depicted as spheres. In Ffh, the G257A mutation impinges on the N domain polypeptide backbone between residues 42 and 43. The amino acid numbering is based on the sequence of E.coli orthologs.
Recent results have begun to shed light on the function of the SRP54 and SRα N domains. Mutation of the conserved hydrophobic amino acids in the ALLEADV motif of mammalian SRP54 moderately reduces signal peptide binding affinity (Newitt and Bernstein, 1997). Since the signal peptide-binding pocket resides in the C-terminus of the protein, this observation suggests that the N domain stimulates substrate binding, possibly by acting as a ‘lid’ that keeps signal peptides tightly bound prior to docking on the ER or bacterial cytoplasmic membrane. Likewise, the N domain of E.coli FtsY appears to play a role in the binding of external ligands. Using a membrane floatation assay, an FtsY fragment consisting of the A and N domains (but not fragments consisting of either domain alone) was shown to mediate interactions between FtsY and phosphatidylethanolamine and membrane proteins (Millman and Andrews, 1999; Millman et al., 2001). Interaction between liposomes and the AN fragment also causes a conformational change in which a protease-sensitive site between the A and N domains is exposed. In addition, a separate study provided evidence for a lipid-binding site in the FtsY NG fragment and suggested that conformational changes in the protein induced by lipid binding stimulate its GTPase activity (de Leeuw et al., 2000).
To begin to investigate the relationship between the non-canonical GTP cycles of SRP54 and SRα and their unique structure, we examined the function of the highly conserved N–GTPase domain interface. Indeed the proximity of this interface to the GTP-binding site as well as provocative evidence from structural studies indicating that the interface is dynamic (Freymann et al., 1999) suggest that intradomain communication might play a role in controlling nucleotide occupancy and therefore influence the SRP–SR interaction. To test this hypothesis, we perturbed the interface by mutagenizing residues in the DAR/KGG motif of E.coli Ffh and FtsY. We found that mutation of the second glycine, which is invariant in all known SRP54 and SRα orthologs, produced strong phenotypes. Consistent with our hypothesis, the mutations in both proteins inhibited the SRP–SR interaction. The data strongly suggest the existence of a previously unrecognized step in the targeting pathway between signal peptide binding and membrane docking that depends on communication between the N and GTPase domains of SRP54 and SRα. Based on these and previous results, we propose that the N domain evolved to link the binding of external factors to the GTPase cycles of both proteins.
Results
A mutation that slightly distorts the Ffh N–GTPase domain interface produces a strong phenotype
An alignment of all 60 SRP54/Ffh orthologs in the SRP database (http://psyche.uthct.edu/dbs/SRPDB/SRPDB.html) reveals that the loop and short α-helix (α6) in the GTPase domain that is situated between the guanine ring-binding site (Figure 1A, ‘*’) and the N domain contains one of the most highly conserved sequences (‘DAR/KGG’) in the entire protein. Although the aspartic acid of this motif is conserved only in prokaryotic orthologs, the basic residue and the first glycine are nearly invariant in all versions of SRP54/Ffh. Most strikingly, the second glycine is completely universal. This residue is buried in the hydrophobic interface between the N and GTPase domains and points toward the polypeptide backbone of the N domain between the highly conserved aspartic acid and valine residues of the ALLEADV motif. Because the N and G domains are packed together so tightly at this position, it is likely that only a hydrogen atom can be accommodated. The addition of even a methyl group is predicted to displace the adjacent region of the N domain slightly (Figure 1B).
Genetic analysis confirmed that the invariant glycine is situated at a particularly critical location in the protein. To determine the significance of the conserved DAR/KGG motif, we individually mutated amino acids 253–257 in E.coli Ffh to a closely related residue and assessed the biological consequences of the mutations in a complementation assay. The desired mutations were introduced into pHDB6, a plasmid in which the expression of wild-type ffh is under the control of a lac promoter, and pHDB1, a plasmid in which ffh under the control of its native promoter is amplified and constitutively overexpressed. WAM121 (PBAD-ffh ffh::kan) transformed with pHDB6 or a mutant version of the plasmid were streaked on LB agar containing either 0.2% arabinose or 200 µM isopropyl-β-d-thiogalactopyranoside (IPTG) to drive expression of the chromosomal copy or plasmid-borne copy of ffh, respectively. In the presence of 200 µM IPTG, WAM121 transformed with pHDB6 produce approximately the same amount of Ffh as the parent ffh+ strain MC4100 (data not shown). As expected, cells transformed with pHDB6 grew equally well on plates containing arabinose and IPTG (Figure 2). In contrast, cells transformed with a plasmid expressing the ffh G257A allele [pHDB6(G257A)] were inviable in the presence of IPTG on either LB or minimal agar at all temperatures (Figure 2 and data not shown). Transforming the cells with a pHDB1 derivative that drives overexpression of the ffh G257A allele did not overcome the lethal phenotype (Table I). Surprisingly, cells that expressed the ffh D253N, A254L, R255N or G256A alleles showed no growth defects. These results suggest that even a mild perturbation of the N–GTPase interface can be highly deleterious.
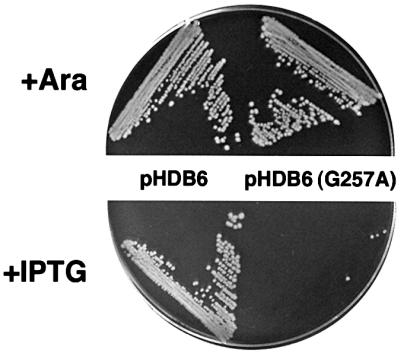
Fig. 2. E.coli that express the ffh G257A allele are inviable. WAM121 (PBAD-ffh ffh::kan) transformed with pHDB6 or pHDB6 (G257A) was streaked on LB agar containing 0.2% arabinose or 200 µM IPTG and incubated at 37°C for 18 h.
Table I. Growth of WAM121 containing plasmid-borne ffh alleles.
| Allele | Vector |
|
|---|---|---|
| pHDB6 | pHDB1 | |
| Wild type | + | + |
| G110S | – | + |
| P142L | – | + |
| A192D | – | + |
| L195P | – | + |
| D253N | + | n.d. |
| A254L | + | n.d. |
| R255N | + | n.d. |
| G256A | + | n.d. |
| G257A | – | – |
Cells transformed with pHDB6 and pHDB1 derivatives were streaked on LB agar containing 200 µM IPTG and plain LB agar, respectively. +, wild-type colony size; –, little or no growth; n.d., not done.
The ffh G257A mutation does not significantly impair protein stability, substrate binding or GTPase activity
We performed a series of biochemical assays in an effort to explain the basis for the strong phenotype produced by the ffh G257A mutation. Two lines of evidence suggest that the mutation affects the function of the protein rather than its overall structure. First, we found that the mutant protein is stable in vivo. WAM121 transformed with pHDB6, pHDB6(G257A) or the cloning vector pJN3 were grown in minimal medium without arabinose to repress expression of the chromosomal ffh gene and deplete endogenous Ffh. Expression of plasmid-borne ffh was then induced by adding IPTG, and the relative half-life of wild-type and mutant Ffh was determined by immunoprecipitating the protein from cells that were pulse-labeled and incubated for a chase period of up to 60 min. A nearly identical amount of radiolabeled Ffh remained in cells containing pHDB6 or pHDB6(G257A) at each time point (Figure 3A, lanes 2–11). All of the protein was derived from the plasmid-encoded genes since no Ffh was detected in cells transformed with pJN3 (Figure 3A, lane 1). Furthermore, western blot analysis showed that cells producing wild-type and mutant Ffh contained the same steady-state level of protein after growth in LB (data not shown). These data strongly suggest that the mutant protein folds relatively normally. In a second set of experiments, we incubated purified wild-type or mutant Ffh with V8 protease at either 25 or 37°C and removed aliquots at various times. Consistent with previous results (Zheng and Gierasch, 1997), the M domain of wild-type Ffh was degraded rapidly at 25°C, but the NG fragment was protease resistant (Figure 3B, lanes 1–5). Proteolysis of full-length Ffh was accelerated at 37°C, but a stable NG fragment (as well as a slightly smaller product) was still observed at the higher temperature). Interestingly, digestion of the mutant protein also yielded a stable NG fragment at both temperatures (Figure 3B, lanes 6–10). Since the NG fragment is readily destabilized by signal peptides (Zheng and Gierasch, 1997) or SDS (data not shown), the data imply that the Ffh G257A mutation does not grossly alter the tertiary structure of the N and GTPase domains.

Fig. 3. The G257A mutation does not destabilize Ffh. (A) WAM121 transformed with pJN3, pHDB6 or pHDB6(G257A) were depleted of endogenous Ffh and expression of the plasmid-borne ffh allele was induced by the addition of IPTG. Pulse-labeled cells were incubated for the indicated chase period, and Ffh was immunoprecipitated from cell extracts. The percentage of radiolabeled Ffh remaining at each time point is shown. Lane 1, cells containing pJN3; lanes 2–6, cells containing pHDB6; lanes 7–11, cells containing pHDB6(G257A). (B) Purified wild-type (WT) or mutant (G257A) Ffh was subjected to partial proteolysis with V8 protease at either 25 or 37°C. Aliquots were removed from the reactions at the indicated times and analyzed by SDS–PAGE on 16% gels. Lanes 1–5, wild-type Ffh; lanes 6–10, Ffh G257A.
We next assessed the ability of the mutant protein to recognize targeting signals. Previous work has shown that like native mammalian SRP, a chimeric SRP containing Ffh in place of SRP54 binds to signal peptides in cell-free translation reactions and specifically inhibits the synthesis of full-length pre-secretory proteins (Bernstein et al., 1993). In these ‘elongation arrest’ assays, the concentration of an SRP that inhibits the synthesis of 50% of the pre-secretory protein molecules provides a sensitive measure of relative substrate binding activity. Wild-type Ffh or the Ffh G257A mutant were first mixed together with all the subunits of canine SRP except SRP54 and assembled into a ribonucleoprotein complex. The mutant protein bound to mammalian SRP RNA as well as wild-type Ffh (Figure 4A). Increasing concentrations of the chimeric SRP particles [SRP (Ffh WT), SRP (Ffh G257A)] or a particle lacking Ffh [SRP (–Ffh)] were then titrated into wheat germ translation reactions programmed with pre-prolactin mRNA and cyclin mRNA (to provide a specificity control) and the inhibition of full-length preprolactin synthesis was measured. The mutant particle displayed nearly the same level of elongation arrest activity as the wild-type particle (Figure 4B). Consistent with previous results (Bernstein et al., 1993), a particle that contained no Ffh had essentially no activity in the assay. These data suggest that the Ffh G257A mutation does not greatly impair substrate binding.

Fig. 4. The G257A mutation does not significantly impair signal peptide recognition. (A) Chimeric SRPs formed by mixing either wild-type (WT) or mutant (G257A) E.coli Ffh together with canine SRP RNA and canine SRP68/72, SRP19 and SRP9/14 proteins were purified and analyzed by SDS–PAGE. (B) SRP (Ffh WT) (squares), SRP (Ffh G257A) (diamonds) or SRP (–Ffh) (circles) was titrated into wheat germ translation reactions programmed with pre-prolactin and cyclin mRNAs and elongation arrest activity was measured.
Similarly, we found that the G257A mutation only slightly affects the GTPase activity of Ffh. Like many other GTPases, purified wild-type Ffh (either free or bound to 4.5S RNA) exhibits a detectable GTPase activity in vitro (Miller et al., 1994). To determine the effect of various GTPase domain mutations on the enzymatic activity of Ffh, we purified complexes containing 4.5S RNA and either wild-type protein or a mutant protein and measured GTP hydrolysis under steady-state conditions. All of the mutants bound to 4.5S RNA as well as wild-type Ffh (data not shown). Consistent with previous results (Powers and Walter, 1995), wild-type SRP hydrolyzed GTP with a Km of ∼1 µM and a Vmax of ∼100–200 fmol/min (Figure 5A). The Ffh G257A mutation appeared to reduce the Vmax, but only by ∼25–35%. In contrast, mutation of a conserved proline in the second element of the GTPase consensus to leucine (P142L) reduced the Vmax by >5-fold. Likewise, mutations in the first and third box of the GTPase consensus (G110S, A192D and L195P) reduced GTP hydrolysis by >10-fold (Figure 5B and data not shown). Surprisingly, although WAM121 transformed with pHDB6 derivatives containing the ffh G110S, P142L, A192D or L195P alleles failed to grow in the presence of IPTG, cells transformed with the equivalent pHDB1 derivatives (and that overproduce mutant Ffh), grew completely normally (Table I). Thus, ffh mutations that dramatically inhibit GTPase activity produce a milder phenotype than the G257A allele. Based on these results, it is very unlikely that the small reduction in catalytic activity produced by the ffh G257A mutation explains the profound growth defect.

Fig. 5. The Ffh G257A mutation only slightly affects GTPase activity but strongly inhibits the GTP-dependent interaction between SRP and FtsY. (A) SRP (5 nM) reconstituted with wild-type Ffh (WT, circles) or a mutant Ffh (G257A, squares; or P142L, triangles) was incubated with 0.3–10 µM GTP and [γ-32P]GTP for 20 min at 25°C, and GTP hydrolysis was measured. (B) SRP (5 nM) reconstituted with wild-type Ffh or a mutant Ffh (G257A, G110S, P142L, A192D or L195P) was incubated with 5 µM GTP and [γ-32P]GTP as in (A) in the presence (filled bars) or absence (open bars) of 150 nM FtsY–GST. The ratio of GTP hydrolyzed in the presence and absence of FtsY–GST is indicated. The average of two independent experiments is shown. (C) FtsY–GST (lanes 1–6) or GST (lanes 7 and 8) was bound to glutathione–agarose beads. SRP reconstituted with wild-type Ffh (lanes 1–2 and 5–8) or the G257A mutant (lanes 3 and 4) was then incubated with the beads in the presence of GMP-PMP (lanes 1–4, 7 and 8) or GDP (lanes 5 and 6). The beads were pelleted, and proteins that remained in the supernatant (S) or bound to the beads (P) were separated by SDS–PAGE.
The ffh G257A mutation severely impairs the SRP–SR interaction
Two lines of evidence suggest that the ffh G257A mutation primarily affects the SRP–SR interaction. Previous studies have shown that Ffh and FtsY reciprocally stimulate each other’s GTPase activity provided that Ffh is bound to 4.5S RNA and that both proteins bind GTP (Miller et al., 1994; Powers and Walter, 1995). To test the effect of the G257A mutation on this concerted GTPase reaction, we incubated SRPs containing either wild-type Ffh, Ffh G257A or one of several different GTPase mutants with an FtsY–GST fusion protein and measured GTP hydrolysis. The FtsY–GST fusion protein alone has negligible GTPase activity under typical reaction conditions (Miller et al., 1994; data not shown). Consistent with previous results, a combination of wild-type SRP and the FtsY–GST fusion protein hydrolyzed ∼12–15 times more GTP than SRP alone (Figure 5B). In contrast, GTPase activity was stimulated only ∼3- to 5-fold when an SRP containing the Ffh G257A mutant was mixed with the FtsY–GST fusion. Thus, the mutation inhibits the ability of one or both of the GTPases to interact with and activate the other. Interestingly, although several Ffh GTPase mutants (G110S, P142L, A192D and L195P) hydrolyzed very little GTP, a high level of GTPase activity was observed when they were mixed with the FtsY–GST fusion protein (Figure 5B). Since the basal GTPase activity was too low to measure accurately, it was not possible to determine the exact degree of GTPase stimulation. Nevertheless, in each case, the level of cross-stimulation was equal to or greater than that observed in the reaction containing wild-type SRP. These data imply that perturbation of the SRP–SR interaction is a highly specific effect that is not simply due to defects in Ffh GTPase activity.
A ‘GST pull-down’ assay demonstrated directly that the ffh G257A mutation inhibits the SRP–SR interaction. Based on the observation that E.coli SRP and FtsY form a stable complex in the presence of GMP-PNP (Miller et al., 1994), we incubated FtsY–GST fusion protein bound to glutathione–agarose beads with GMP-PNP and either wild-type SRP or SRP containing the Ffh G257A mutant, and pelleted the beads. Consistent with previous results, we found that all of the wild-type SRP was associated with the glutathione–agarose pellet (Figure 5C, lanes 1 and 2). As expected, none of the wild-type SRP was in the pellet if GDP was substituted for GMP-PNP or if GST was substituted for FtsY–GST (Figure 5C, lanes 5–8). Even in the presence of GMP-PNP, however, ∼80–90% of the mutant SRP failed to form a complex with the FtsY–GST fusion protein and remained in the supernatant (Figure 5C, lanes 3 and 4). The ffh G257A mutation appeared to inhibit the formation of stable SRP–SR complexes more severely than GTPase co-stimulation, perhaps because the direct binding assay is more stringent. In any case, the results strongly suggest that both GTP binding and communication between the Ffh N and GTPase domains are required to trigger the SRP–SR interaction.
Distortion of the FtsY N–GTPase domain interface also inhibits the SRP–SR interaction
Since SRα/FtsY orthologs contain conserved sequence motifs at the N–GTPase domain interface that are very similar to those found in SRP54/Ffh, we hypothesized that intradomain communication likewise plays an important role in protein function. Although a highly conserved ‘D/T/SAKGG’ motif is found in the GTPase domain of eubacterial and archaeal FtsY orthologs, only the second glycine is conserved in eukaryotic orthologs. As in SRP54/Ffh orthologs, this glycine is invariant and therefore probably resides at a particularly critical location. Because the loop and α-helix that contain the D/T/SAKGG motif are slightly rotated relative to the equivalent segment of Ffh (Figure 1A), however, mutation of the invariant glycine would impinge on the polypeptide backbone of the the N domain closer to the second alanine of the ALLEADV motif. In addition, larger amino acids side chains would project into the hydrophobic core of the N domain and displace the nearly invariant first leucine and the valine of the ALLEADV motif as well as a second upstream leucine (Figure 1B).
To test our hypothesis, we mutated the invariant glycine in E.coli FtsY (Gly455) to alanine, serine, valine and threonine. The mutations (G455A, G455S, G455V and G455T) were introduced into pJH15, a low copy plasmid in which ftsY expression is under the control of the trc promoter. N4156::pAra14-FtsY′ (PBAD-ftsY) transformed with a wild-type or mutant plasmid was streaked on LB agar containing either arabinose to drive expression of the chromosomal copy of ftsY or IPTG to drive expression of the plasmid-borne gene. In the presence of 20 µM IPTG, cells transformed with wild-type pJH15 produced the same amount of FtsY as control ftsY+ cells (data not shown). Cells transformed with a pJH15 derivative containing the FtsY G455A allele [pJH15(G455A)] grew as well as cells transformed with the wild-type plasmid under all conditions tested (data not shown). This result was not surprising because Gly455 in FtsY is slightly further from the N domain than Gly257 in Ffh. Cells that expressed the ftsY G455S, G455V and G455T alleles, however, showed increasingly severe growth defects. When ftsY expression was slightly reduced by lowering the IPTG concentration to 10 µM, cells transformed with pJH15(G455S) grew poorly at 37°C and were inviable at 42°C (Figure 6). Cells that expressed the G455V allele were temperature-sensitive on plates that contained either 10 or 20 µM IPTG. Finally, cells transformed with pJH15(G455T) were inviable at all temperatures.

Fig. 6. E.coli that express ftsY G455S, G455V or G455T alleles show growth defects. N4156::pAra14-FtsY′ (ftsY::PBAD-ftsY) transformed with pJH15 containing wild-type (WT) ftsY or mutant (G455S, G455V or G455T) ftsY were streaked on LB agar containing 0.2% arabinose or the indicated amount of IPTG and incubated at 42°C for 20 h (arabinose plates) or 24 h (IPTG plates) or 30°C for 38 h.
We next examined the effect of the mutations on FtsY stability and intracellular localization. N4156::pAra14-FtsY′ transformed with the cloning vector pHQ5, pJH15, pJH15(G455V) or pJH15(G455T) were grown in LB lacking arabinose to deplete endogenous FtsY. Expression of the plasmid-borne ftsY genes was then induced by the addition of 20 µM IPTG. After 1 h, cells were harvested and cell extracts were subjected to high speed centrifugation in either the presence or absence of Triton X-100. The amount of FtsY present in the extract and in the high speed supernatant and pellet fractions was then assessed by western blotting. Cells transformed with pHQ5 contained essentially no FtsY (data not shown). Cells transformed with pJH15 and pJH15(G455V) contained similar amounts of FtsY at both 30 and 42°C (Figure 7, lanes 1 and 6). Thus, the G455V mutation does not destabilize the protein at high temperature. At low temperature, most of the wild-type and mutant FtsY was found in the high speed pellet, but only about half of the protein could be solubilized with detergent (Figure 7, lanes 2–5 and 7–10). These results suggest that ∼50% of the FtsY is associated with the inner membrane and are consistent with previous data (Luirink et al., 1994). At 42°C, the level of membrane-bound FtsY did not change dramatically, although a higher percentage of the wild-type protein was found in the high speed supernatant. These observations strongly suggest that the ftsY G455V mutation does not significantly impair membrane binding at high temperature. Unlike the G455V mutant, the FtsY G455T mutant was unstable and migrated more rapidly than wild-type FtsY on SDS–PAGE (data not shown). The results suggest that the strong phenotype associated with the ftsY G455T allele is due to a significant alteration of protein structure.

Fig. 7. The FtsY G455V mutant binds effectively to the inner membrane. N4156::pAra14-FtsY′ transformed with pJH15 or pJH15 (G455V) was grown at 30°C and endogenous FtsY was depleted. IPTG (20 µM) was added and half of each culture was shifted to 42°C. After 1 h of further incubation, the amount of FtsY and CAT present in total cell extracts (T) as well as in high speed supernatants (S) and pellets (P) in the presence or absence of Triton X-100 (+TX, –TX) was determined by western blot. Lanes 1–5, cells transformed with pJH15; lanes 6–10, cells transformed with pJH15 (G455V).
Like the ffh G257A allele, the ftsY G455V mutation markedly inhibited the SRP–SR interaction. Although an FtsY (G455V)–GST fusion protein had relatively normal GTP binding activity as assessed by GTP cross-linking (Figure 8A), it was almost completely unable to bind stably to SRP in GST pull-down assays (Figure 8B, lanes 3 and 4). The severe reduction in complex formation could not have been due simply to a minor defect in nucleotide binding. Consistent with the notion that the ftsY G455V mutation hinders association with SRP, only about half as much GTP was hydrolyzed in reciprocal GTPase reactions containing SRP and the FtsY (G455V)–GST fusion protein as in control reactions containing the wild-type fusion protein (Figure 8C). Taken together, the data strongly suggest that perturbation of the N–GTPase domain interface of either Ffh or FtsY produces similar effects.

Fig. 8. The ftsY G455V mutation inhibits the GTP-dependent interaction between SRP and FtsY. (A) 150 nM wild-type FtsY–GST (circles) or FtsY (G455V)–GST (squares) was incubated with 1–30 µM GTP and [γ-32P]GTP at 25°C for 20 min, and GTP binding was measured by UV cross-linking. (B) Wild-type FtsY–GST (lanes 1 and 2) or FtsY (G455V)–GST was bound to glutathione–agarose beads. SRP was then incubated with the beads and samples were divided into supernatant (S) and pellet (P) fractions as described in the legend to Figure 5. Proteins were separated by SDS–PAGE. (C) SRP was incubated in the presence (filled bars) or absence (open bars) of wild-type FtsY–GST or FtsY (G455V)–GST as described in the legend to Figure 5 and GTPase activity was measured. The ratio of GTP hydrolyzed in the presence and absence of FtsY–GST is indicated. The average of two independent experiments is shown.
Discussion
In this report, we provide evidence that effective interaction between SRP54/Ffh and SRα/FtsY requires a precise alignment of the N and GTPase domains of both proteins. Addition of a single methyl group to an invariant glycine residue in the E.coli Ffh GTPase domain that is packed tightly against the N domain severely reduced viability. This result was striking because mutations that strongly inhibit GTPase activity produced a milder phenotype and because alteration of many highly conserved residues in Ffh, including residues near the border of the GTPase and M domain, produces no discernible effects on growth (Y.Lu, unpublished data). The genetic data suggested that the conserved glycine resides at a particularly critical position in the protein. Biochemical analysis revealed that the mutation did not grossly affect protein structure, substrate binding or the ability to bind and hydrolyze GTP. In two different assays, however, the mutation hindered SRP from interacting with its receptor. Similar results were obtained when the equivalent glycine residue in E.coli FtsY was mutated. Growth defects were observed only when amino acids larger than alanine were introduced, most probably because this residue is situated further away from the N domain. Nevertheless, a valine substitution considerably impaired association with SRP. Taken together, our results imply that GTP binding is necessary but not sufficient to promote the SRP–SR interaction. Intriguingly, recent X-ray studies have shown that the overall structures of apo-Ffh and Ffh bound to GMP-PNP are remarkably similar (Padmanabhan and Freymann, 2001). This discovery corroborates our conclusion that the SRP family of GTPases differs considerably from typical GTPases which use changes in the nucleotide-bound state to effect significant conformational changes and to drive biochemical reactions forward.
In all probability, a precise N–GTPase domain interface in SRP54/Ffh and SRα/FtsY has been preserved to facilitate interdomain communication within each protein. Although it is possible that a precise alignment of the N and GTPase domains is required to permit physical contact along an extended surface of the two proteins, it seems unlikely that the N domain evolved purely to facilitate the SRP–SR interaction. Studies on members of the Ras superfamily have demonstrated that small segments within a GTPase module are sufficient to bind effectors (Polakis and McCormick, 1993). Moreover, polypeptide segments attached to GTPase domains generally have specialized functions and can, like the α-helical domains of heterotrimeric G protein α subunits, regulate progression of the GTPase cycle. In the SRP family of GTPases, available evidence indicates that the N domain plays an important role in interactions with external ligands (Newitt and Bernstein, 1997; Millman and Andrews, 1999). In addition, X-ray analysis indicates that the Ffh N–GTPase interface is dynamic (Freymann et al., 1999). The structural studies suggest that the relative position of the N and GTPase domains changes during the targeting cycle in response to external cues and serves as an important indicator of the status of the protein.
Based on the results of this and other studies, we propose that the unique architecture of the SRP family GTPases evolved to ensure that external ligands are properly bound to SRP54/Ffh and SRα/FtsY prior to the SRP–SR interaction. At the beginning of the targeting cycle, the N domain of SRP54/Ffh facilitates signal peptide binding, possibly through an interaction between the hydrophobic residues of the ALLEADV motif and the targeting signal (Newitt and Bernstein, 1997). Targeting signal recognition might then cause a conformational change in the N domain that is transmitted across the N–GTPase domain interface (Figure 9). In this conformation, SRP54/Ffh would be activated or ‘primed’ to bind to the SR. The observation that signal peptide recognition increases the affinity of SRP54 for GTP (Bacher et al., 1996) is consistent with the idea that ligand binding produces an activated state. Likewise, interaction between SRα/FtsY and membrane components might cause a parallel conformational change that primes the SR. In bacteria, SR priming presumably would require an interaction between FtsY and membrane lipids and proteins (possibly a component of the translocon) that involves the participation of the N domain. In light of recent evidence that SRα and SRβ interact in a dynamic fashion (Legate et al., 2000) and that SRβ interacts directly with the translocon (Fulga et al., 2001), SR priming in eukaryotic cells would probably involve an interaction between the two receptor subunits. Subsequently, interaction between the primed SRP54/Ffh and SRα/FtsY GTPases would promote stable GTP binding which, in turn, would facilitate nascent chain release (and perhaps dissociation of SR from the translocon as well). GTPase priming would provide a particularly effective means of coordinating the release of nascent chains with arrival of ribosome–nascent chain complexes at the translocon. Perhaps more significantly, GTPase priming would also prevent SRP from interacting with the SR inappropriately (e.g. before a targeting signal is bound) and short-circuiting the targeting pathway. Ultimately, it should be possible to test this model by directly examining the effect of external ligand binding on the conformation and GTPase cycles of SRP54/Ffh and SRα/FtsY.

Fig. 9. Model of events that precede the SRP–SR interaction. In an initial step, SRP54/Ffh binds to targeting signals and SRα/FtsY interacts with lipids and membrane proteins (perhaps SRβ in the ER and translocon components in the bacterial inner membrane). The N domain participates in or senses each binding reaction and changes conformation. Communication of this conformational change across the N–GTPase domain interface activates or ‘primes’ SRP54/Ffh and SRα/FtsY. Interaction between the primed GTPases leads to stable GTP binding and release of the nascent chain. The bacterial SRP pathway is illustrated here.
Several distinctive features of the SRP pathway may explain the evolution of two intersecting GTPase cycles as well as the unusual properties of the GTPases. In most biochemical pathways, GTPases monitor the assembly of a single macromolecular complex, the occupancy of a receptor or the activation of another GTPase. In the SRP pathway, however, it appears that two half-reactions (binding of the signal peptide and assembly or activation of the translocon) must be monitored independently and then brought together before a translocation event can be initiated. The utilization of two GTPases that can interact physically provides an effective means of coping with this relatively unique scenario. Moreover, there may be additional constraints on the SRP pathway that arise from the combination of ligand binding and regulatory functions in a single protein. Based on available evidence (Rapiejko and Gilmore, 1997), it is conceivable that during the targeting reaction SRP54/Ffh cannot stably bind GTP and signal peptides simultaneously. This problem does not arise in many other biochemical pathways in which the receptor and signal transducer functions are housed in separate molecules. Indeed, it is intriguing that the only other GTPases that are known to have non-canonical GTPase cycles are Obg and Era (Lin et al., 1999; Sullivan et al., 2000), two widely conserved proteins that, like SRP, are of ancient origin. It is conceivable that the first GTPases utilized nucleotide-bound and nucleotide-free states to transduce signals, and that the more elaborate paradigm of GTPases that are stimulated to oscillate between GTP- and GDP-bound states by separate GAPs and GEFs emerged later in the course of evolution.
Materials and methods
Reagents, bacterial strains and media
Polyclonal antiserum against chloramphenicol acetyl transferase (CAT) was obtained from 5Prime–3Prime. Affinity-purified antibodies against Ffh and FtsY have been described (Ulbrandt et al., 1997). The bacterial strains used in this study and their genotypes are: MC4100 [F- araD139 Δ(argF-lac)U169 rpsL150 relA1 thi fib5301 deoC1 ptsF25 rbsR], WAM121 [MC4100 ara+ ffh::kan attB::(OriR6K PBAD-ffh tet)] (Phillips and Silhavy, 1992) and N4156::pAra14-FtsY′ [polA end thy gyrA ftsY::(OriColE1 PBAD-ftsY bla)] (Luirink et al., 1994). Media preparation and basic bacterial manipulations were performed using standard methods. Selective media generally contained 100 µg/ml ampicillin and 40 µg/ml chloramphenicol as required. N4156::pAra14-FtsY′ was routinely grown in the presence of 50 µg/ml ampicillin to retain the integrated plasmid.
Plasmid construction
Plasmids pHDB1 (Orip15A ffh+), pHDB6 (OriColE1 lacIQ PLAC-ffh) and a pGEX1 derivative containing a FtsY–GST fusion have been described previously (Poritz et al., 1990; Miller et al., 1994; Bernstein and Hyndman, 2001). To construct pNU251, an NdeI–BamHI fragment from pHDB1 containing ffh was cloned into the cognate sites of pET17b (Novagen). The two small PvuII fragments of pLG338 (Stoker et al., 1982) were replaced with the 1.4 kb BsaAI fragment from pACYC184 that contains the CAT gene to create pNU75. To construct pHQ5, the HindIII–EcoRV fragment from the tet gene of pNU75 was replaced with a BsaAI–HindIII fragment that contains the lacIQ gene, trc promoter and polylinker from pTRC99a (Amann et al., 1988). pJH15 was then generated by digesting pTRC-FtsY (Ulbrandt et al., 1997) with EcoRI, treating the DNA with the Klenow fragment of E.coli DNA polymerase I, digesting with HindIII and ligating the fragment containing ftsY to pHQ5 that had been digested with SmaI and HindIII. All mutations were generated using the QuikChange mutagenesis kit (Strategene).
Plasmid-based expression of ffh and ftsY and cell fractionation
WAM121 transformed with pHDB6 (or a derivative thereof) were grown overnight at 37°C in M9 medium containing 40 µg/ml l-amino acids (except methionine and cysteine), 0.2% fructose and 0.2% arabinose. Cells were washed and placed in medium containing glucose instead of arabinose at OD550 = 0.01. When cultures reached OD550 = 0.2, 200 µM IPTG was added to induce expression of plasmid-borne ffh. After a 20 min incubation, pulse–chase labeling and immunoprecipitation of Ffh from cell extracts was performed essentially as described (Ulbrandt et al., 1997).
N4156::pAra14-FtsY′ transformed with pJH15 were grown in LB containing 0.2% arabinose overnight at 30°C. Cells were washed and placed in LB lacking arabinose at OD550 = 0.02, and 20 µM IPTG was added when the cultured cells reached OD550 = 0.2. After 1 h, 10 ml of cells were poured over ice and collected by centrifugation. Cells were then resuspended in 0.5 ml of 20 mM Tris-OAc pH 7.5, 100 mM KOAc, 10 mM MgOAc, 1 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF) and frozen in liquid nitrogen. Thawed cells were sonicated for 15 s using a microtip. Unbroken cells were removed by centifugation at 3000 g for 1 min. ‘Total’ cell lysates were then divided in half. Triton X-100 (0.5%) was added to one half, and the samples were centrifuged for 30 min in a TLA 120.2 rotor at 140 000 g to obtain high speed supernatant and pellet fractions.
Purification of Ffh, reconstituted SRPs and FtsY–GST fusion proteins
To produce E.coli Ffh, BL21(DE3)pLysS (Novagen) was first transformed with either pNU251 or a derivative containing a mutant ffh gene. Overnight cultures were washed and diluted 1:50 in fresh LB and grown to OD550 = 0.8. IPTG (1 mM) was then added and cultures were incubated for another 2 h. Cell pellets were frozen in liquid nitrogen, thawed and resuspended in 1/10 culture volume of buffer A (50 mM Tris–HCl pH 7.5, 100 mM KOAc, 2 mM EDTA, 1 mM DTT) containing protease inhibitors. Triton X-100 (1%) was added, cells were sonicated briefly and cell debris was removed by centrifugation at 27 000 g for 15 min. S-Sepharose equilibrated in buffer A was then added to the cell lysate and samples were rocked at 4°C for 15 min. The resin was transferred to a column and washed twice with two column volumes of buffer B (50 mM HEPES-OAc pH 7.5, 200 mM KOAc, 0.1 mM EDTA, 1 mM DTT, 0.01% Nikkol, 10% glycerol). Ffh was then eluted with buffer C (buffer B containing 500 mM KOAc).
For SRP reconstitutions, 4.5S RNA and mammalian SRP subunits were purified as described (Poritz et al., 1990; Zopf et al., 1993). To assemble E.coli SRP, purified Ffh was added in a 2-fold molar excess to 4.5S RNA in buffer C containing 5 mM MgOAc. To assemble chimeric SRP particles, Ffh was added in a 2-fold molar excess to a mixture of all the mammalian SRP subunits except SRP54 in 50 mM HEPES-OAc pH 7.5, 500 mM KOAc, 5 mM MgOAc, 1 mM DTT, 0.01% Nikkol. Reactions were incubated on ice for 10 min and then at 37°C for 10 min. Reconstituted particles were purified using Ultrafree MC DEAE anion exchange filter units (Millipore) essentially as described (Chang et al., 1997) except that the filters were washed with a buffer containing 250 mM KOAc.
FtsY–GST fusion proteins were purified essentially as described (Smith and Johnson, 1988) except that TBS (50 mM Tris–HCl pH 7.4, 150 mM NaCl) was used in place of MTPBS.
Protease assays
The protease sensitivity of Ffh was evaluated using V8 protease (Roche) as described (Zheng and Gierasch, 1997) except that the final concentration of Ffh was 20 µg/ml and the ratio of Ffh to protease was ∼5:1. Reactions were stopped by precipitating the proteins with cold 10% trichloroacetic acid (TCA).
Ffh/FtsY functional assays
Elongation arrest assays were performed as described (Bernstein et al., 1993). The percentage elongation arrest has been defined previously (Siegel and Walter, 1985). GTPase assays for E.coli SRP and the FtsY–GST fusion protein were conducted essentially as described (Miller et al., 1994) with slight modifications. Reactions (20 µl) containing 5 nM reconstituted SRP, 150 nM FtsY–GST, 10 µCi of [γ-32P]GTP (ICN, 3000 Ci/mmol) and various amounts of non-radioactive GTP in 25 mM triethanolamine pH 7.5, 25 mM KOAc, 2.5 mM MgOAc, 5% glycerol, 1 mM DTT and 0.1% Nikkol were incubated for 20 min at 25°C. Subsequently, reactions were placed on ice and 200 µl of a 5% suspension of activated charcoal in 20 mM H3(PO)4 was added. The charcoal was then removed by centrifugation. Half (100 µl) of each supernatant was mixed with 50 µl of 20 mM NaOH and the phosphate liberated during the reaction was counted. The background counts from a reaction containing buffer only were subtracted. The incubation conditions used to measure GTP hydrolysis were also used to assess GTP binding by FtsY–GST. GTP cross-linking, analysis of proteins by SDS–PAGE and quantitation of covalently bound GTP by autoradiography were performed essentially as described (Miller et al., 1993), except that the samples were irradiated 1 cm from the light source for 15 min. GST pull-down assays were performed as described (Miller et al., 1994), except that the Nikkol concentration was 0.01%.
Gel electrophoresis and western blotting
Protein samples were analyzed by SDS–PAGE on 8–16% minigels (Novex) unless otherwise noted and visualized by Coomassie Blue staining. Western blotting was performed as described (Ulbrandt et al., 1997).
Acknowledgments
Acknowledgements
We thank Doug Freymann, Shu-ou Shan and Peter Walter for stimulating discussions, and Manu Hegde for critical reading of the manuscript.
References
- Amann E., Ochs,B. and Abel,K.J. (1988) Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene, 69, 301–315. [DOI] [PubMed] [Google Scholar]
- Bacher G., Lütcke,H., Jungnickel,B., Rapoport,T.A. and Dobberstein,B. (1996) Regulation by the ribosome of the GTPase of the signal-recognition particle during protein targeting. Nature, 381, 248–251. [DOI] [PubMed] [Google Scholar]
- Bernstein H.D. and Hyndman,J.B. (2001) Physiological basis for conservation of the signal recognition particle targeting pathway in Escherchia coli. J. Bacteriol., 183, 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein H.D., Zopf,D., Freymann,D.M. and Walter,P. (1993) Functional substitution of the signal recognition particle 54-kDa subunit by its Escherichia coli homolog. Proc. Natl Acad. Sci. USA, 90, 5229–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne H., Sanders,D.A. and McCormick,F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature, 348, 125–132. [DOI] [PubMed] [Google Scholar]
- Bourne H., Sanders,D.A. and McCormick,F. (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature, 349, 117–127. [DOI] [PubMed] [Google Scholar]
- Brown S. and Fournier,M.J. (1984) The 4.5S RNA gene of Escherichia coli is essential for cell growth. J. Mol. Biol., 178, 533–550. [DOI] [PubMed] [Google Scholar]
- Chang D.-Y., Newitt,J.A., Hsu,K., Bernstein,H.D. and Maraia,R.J. (1997) A highly conserved nucleotide in the Alu domain of SRP RNA mediates translation arrest through high affinity binding to SRP9/14. Nucleic Acids Res., 25, 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. and Stowers,L. (1995) GTPase cascades choreographing cellular behavior: movement, morphogenesis and more. Cell, 81, 1–4. [DOI] [PubMed] [Google Scholar]
- Connolly T., Rapiejko,P.J. and Gilmore,R. (1991) Requirement of GTP hydrolysis for dissociation of the signal recognition particle from its receptor. Science, 252, 1171–1173. [DOI] [PubMed] [Google Scholar]
- deGier J.W., Mansournia,P., Valent,Q.A., Phillips,G.J., Luirink,J. and von Heijne,G. (1996) Assembly of a cytoplasmic membrane protein in Escherichia coli is dependent on the signal recognition particle. FEBS Lett., 399, 307–309. [DOI] [PubMed] [Google Scholar]
- deLeeuw E., te Kaat,K., Moser,C., Menestrina,G., Demel,R., de Kruijff,B., Oudega,B., Luirink,J. and Sinning,I. (2000) Anionic phospholipids are involved in membrane association of FtsY and stimulate its GTPase activity. EMBO J., 19, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymann D.M., Keenan,R.J., Stroud,R.M and Walter,P. (1997) Structure of the conserved GTPase domain of the signal recognition particle. Nature, 385, 361–364. [DOI] [PubMed] [Google Scholar]
- Freymann D.M., Keenan,R.J., Stroud,R.M and Walter,P. (1999) Functional changes in the structure of the SRP GTPase on binding GDP and Mg2+GDP. Nat. Struct. Biol., 6, 793–801. [DOI] [PubMed] [Google Scholar]
- Fulga T.A., Sinning,I., Dobberstein,B. and Pool,M.R. (2001) SRβ coordinates signal sequence release from SRP with ribosome binding to the translocon. EMBO J., 20, 2338–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan R.J., Freymann,D.M., Stroud,R.M. and Walter,P. (2001) The signal recognition particle. Annu. Rev. Biochem., 70, 755–775. [DOI] [PubMed] [Google Scholar]
- Legate K.R., Falcone,D. and Andrews,D.W. (2000) Nucleotide-dependent binding of the GTPase domain of the signal recognition particle receptor β-subunit to the α-subunit. J. Biol. Chem., 275, 27439–27446. [DOI] [PubMed] [Google Scholar]
- Lin B., Covalle,K.L. and Maddock,J.R. (1999) The Caulobacter crescentus CgtA protein displays unusual guanine nucleotide binding and exchange properties. J. Bacteriol., 181, 5825–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luirink J., ten Hagen-Jongman,C.M., van der Weijden,C.C., Oudega,B., High,S. and Dobberstein,B. (1994) An alternative protein targeting pathway in Escherichia coli: studies on the role of FtsY. EMBO J., 13, 2289–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.D., Wilhelm,H., Gierasch,L., Gilmore,R. and Walter,P. (1993) GTP binding and hydrolysis by the signal recognition particle during initiation of protein translocation. Nature, 366, 351–354. [DOI] [PubMed] [Google Scholar]
- Miller J.D., Bernstein,H.D. and Walter,P. (1994) Interaction of E.coli Ffh/4.5S ribonucleoprotein and FtsY mimics that of mammalian signal recognition particle and its receptor. Nature, 367, 657–659. [DOI] [PubMed] [Google Scholar]
- Millman J.S. and Andrews,D.W. (1999) A site-specific, membrane-dependent cleavage event defines the membrane binding domain of FtsY. J. Biol. Chem., 274, 33227–33234. [DOI] [PubMed] [Google Scholar]
- Millman J.S., Qi,H.-Y., Vulcu,F., Bernstein,H.D. and Andrews,D.W. (2001) FtsY binds to the Escherichia coli inner membrane via interactions with phosphatidylethanolamine and membrane proteins. J. Biol. Chem., 276, 25982–25989. [DOI] [PubMed] [Google Scholar]
- Montoya G., Svensson,C., Luirink,J. and Sinning,I. (1997) Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature, 385, 365–368. [DOI] [PubMed] [Google Scholar]
- Moser C., Mol,O., Goody,R.S. and Sinning I. (1997) The signal recognition particle receptor of Escherichia coli (FtsY) has a nucleotide exchange factor built into the GTPase domain Proc. Natl Acad. Sci. USA, 94, 11339–11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Haefelin C., Schafer,U., Müller,M. and Koch,H.G. (2000) SRP-dependent co-translational targeting and SecA-dependent translocation analyzed as individual steps in the export of a bacterial protein. EMBO J., 19, 6419–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newitt J.A. and Bernstein,H.D. (1997) The N-domain of the signal recognition particle 54-kDa subunit promotes efficient signal sequence binding. Eur. J. Biochem., 245, 720–729. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S. and Freymann,D.M. (2001) The conformation of bound GMPPNP suggests a mechanism for gating the active site of the SRP GTPase. Structure, 9, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G.J. and Silhavy,T.J. (1992) The E.coli ffh gene is necessary for viability and efficient protein export. Nature, 359, 744–746. [DOI] [PubMed] [Google Scholar]
- Polakis P. and McCormick,F. (1993) Structural requirements for the interaction of p21ras with GAP, exchange factors and its biological effector target. J. Biol. Chem., 268, 9157–9160. [PubMed] [Google Scholar]
- Poritz M.A., Bernstein,H.D., Strub,K., Zopf,D., Wilhelm,H. and Walter,P. (1990) An E.coli ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle. Science, 250, 1111–1117. [DOI] [PubMed] [Google Scholar]
- Powers T. and Walter,P. (1995) Reciprocal stimulation of GTP hydrolysis by two directly interacting GTPases. Science, 269, 1422–1424. [DOI] [PubMed] [Google Scholar]
- Rapiejko P.J. and Gilmore,R. (1997) Empty site forms of the SRP54 and SRα GTPases mediate targeting of ribosome–nascent chain complexes to the endoplasmic reticulum. Cell, 89, 703–713. [DOI] [PubMed] [Google Scholar]
- Romisch K., Webb,J., Lingelbach,K., Gausepohl,H and Dobberstein,B. (1990) The 54-kD protein of signal recognition particle contains a methionine-rich RNA binding domain. J. Cell Biol., 111, 1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A. and Bibi,E. (1997) FtsY, the prokaryotic signal recognition particle receptor homologue, is essential for biogenesis of membrane proteins. J. Biol. Chem., 272, 2053–2055. [DOI] [PubMed] [Google Scholar]
- Siegel V. and Walter,P. (1985) Elongation arrest is not a prerequisite for secretory protein translocation across the microsomal membrane. J. Cell Biol., 100, 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B. and Johnson,K.S. (1988) Single-step purification of polypeptides in Escherichia coli as fusions with glutathione S-transferase. Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Stoker N.G., Fairweather,N.F. and Spratt,B.G. (1982) Versatile low-copy-number plasmid vectors for cloning in Escherichia coli. Gene, 18, 335–41. [DOI] [PubMed] [Google Scholar]
- Sullivan S.M., Mishra,R., Neubig,R.R. and Maddock,J.R. (2000) Analysis of guanine nucleotide binding and exchange kinetics of the Escherichia coli GTPase Era. J. Bacteriol., 182, 3460–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrandt N.D., Newitt,J.A. and Bernstein,H.D. (1997) The E.coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell, 88, 187–196. [DOI] [PubMed] [Google Scholar]
- Zheng N. and Gierasch,L.M. (1997) Domain interactions in E.coli SRP: stabilization of M domain by RNA is required for effective signal sequence modulation of NG domain. Mol. Cell, 1, 79–87. [DOI] [PubMed] [Google Scholar]
- Zopf D., Bernstein,H.D., Johnson,A.E. and Walter,P. (1990) The methionine-rich domain of the 54 kd protein subunit of the signal recognition particle contains an RNA binding site and can be crosslinked to a signal sequence. EMBO J., 9, 4511–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zopf D., Bernstein,H.D. and Walter,P. (1993) GTPase domain of the 54-kD subunit of the mammalian signal recognition particle is required for protein translocation but not for signal sequence binding. J. Cell Biol., 120, 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]