

Abstract
Thioredoxin reductase (TrxR) is an essential enzyme required for the efficient maintenance of the cellular redox homeostasis, particularly in cancer cells that are sensitive to reactive oxygen species. In mammals, distinct isozymes function in the cytosol and mitochondria. Through an intricate mechanism, these enzymes transfer reducing equivalents from NADPH to bound FAD and subsequently to an active-site disulfide. In mammalian TrxRs, the dithiol then reduces a mobile C-terminal selenocysteine-containing tetrapeptide of the opposing subunit of the dimer. Once activated, the C-terminal redox center reduces a disulfide bond within thioredoxin. In this report, we present the structural data on a mitochondrial TrxR, TrxR2 (also known as TR3 and TxnRd2). Mouse TrxR2, in which the essential selenocysteine residue had been replaced with cysteine, was isolated as a FAD-containing holoenzyme and crystallized (2.6 Å; R = 22.2%; Rfree = 27.6%). The addition of NADPH to the TrxR2 crystals resulted in a color change, indicating reduction of the active-site disulfide and formation of a species presumed to be the flavin–thiolate charge transfer complex. Examination of the NADP(H)-bound model (3.0 Å; R = 24.1%; Rfree = 31.2%) indicates that an active-site tyrosine residue must rotate from its initial position to stack against the nicotinamide ring of NADPH, which is juxtaposed to the isoalloxazine ring of FAD to facilitate hydride transfer. Detailed analysis of the structural data in conjunction with a model of the unusual C-terminal selenenylsulfide suggests molecular details of the reaction mechanism and highlights evolutionary adaptations among reductases.
Keywords: disulfide, flavoprotein, selenocysteine, selenoprotein
Thioredoxins are the major cellular protein disulfide reductases and are responsible for the regulation of numerous biochemical processes within the cell (1). These proteins are maintained in a reduced state by thioredoxin reductases (TrxR), homodimeric f lavoproteins that catalyze the NADPH-dependent reduction of thioredoxins (2, 3).
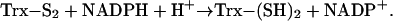 |
Two forms of TrxRs have evolved with related but distinct modes of catalysis (2–5). Low-Mr TrxRs (Mr ≈ 35 kDa) are typically found in prokaryotes, archaea, plants, and lower eukaryotes, whereas high-Mr TrxRs (Mr ≈ 55 kDa) are observed in higher eukaryotes. To date, only the green algae Chlamydomonas reinhardtii has been shown to contain both a low- and a high-Mr TrxR (6).
The general features of catalysis are retained in both low- and high-Mr TrxR (2, 4). TrxR transfers reducing equivalents from NADPH to its bound FAD, ultimately leading to the reduction of an active-site disulfide. In low-Mr TrxRs, the catalytic cycle requires a large conformational change after dithiol activation (4, 7, 8). In high-Mr TrxR, the active-site dithiol reduces a third redox active center in the highly mobile C terminus of the opposing subunit. This third group is responsible for the reduction of the disulfide bond within thioredoxin. Its nature is species-specific and ranges from a C-X-X-X-X-C disulfide in Plasmodium falciparum (9) to a vicinal disulfide in Drosophila melanogaster (10) or a vicinal selenenylsulfide in mammalian TrxRs (11, 12).
In mammalian systems, three isozymes of high-Mr TrxRs have been identified: a cytosolic (TrxR1) (11, 12), a mitochondrial (TrxR2) (13), and an isozyme highly expressed in testes (14, 15). Targeted disruption of either TrxR1 (16) or TrxR2 (17) genes results in an embryonic lethal phenotype, and TrxR1 and TrxR2 appear to have nonredundant functions (16–19). TrxR1 null embryos are affected primarily by compromised cell proliferation (16), whereas TrxR2 null embryos suffer from severe anemia and improper heart development (17). Splice variants of TrxR1 and TrxR2 have also been identified, including a variant that would result in the targeting of TrxR2 to the cytosol (20–24). However, the biological implications of these variations have not been resolved and would benefit from additional biochemical and structural characterizations of the enzymes.
Numerous biochemical and computational studies have added to our understanding of the catalytic mechanism of high-Mr TrxR (2–5, 15, 25–28). Reduction of the enzyme by a single equivalent of NADPH leads to several two-electron reduced species (EH2), with a thiolate–flavin charge transfer complex predominating. Addition of a second equivalent of NADPH leads to a four-electron reduced enzyme (EH4) in which both the active-site disulfide and the selenosulfide-containing C-terminal tail of the opposing subunit are likely reduced. Once reduced to the EH4 state, the enzyme catalyzes a disulfide exchange between the activated C-terminal tail and oxidized thioredoxin. To date, the molecular details of selenenylsulfide/disulfide exchange have not been determined.
Recently, the first x-ray structure of a high-Mr TrxR, rat cytosolic TrxR1, was described (29). The overall topology of this enzyme is similar to that of other pyridine nucleotide disulfide oxidoreductases, particularly glutathione reductases (30–35), and is in agreement with earlier modeling studies of high-Mr TrxRs (15). Interestingly, all of the residues in glutathione reductase responsible for substrate recognition are structurally conserved in rTrxR1, even though rTrxR1 cannot directly reduce oxidized glutathione. The authors (15) suggested that the mobile selenenylsulfide containing a C-terminal tail not only serves as a third redox active group, but it also blocks oxidized glutathione from binding to the enzyme. In addition, the structure confirmed the obligatory “head-to-tail” arrangement of high-Mr TrxR, with the redox-active C-terminal tail of one subunit interacting with the active site of the opposing subunit (36, 37). Unfortunately, the observed conformation of the C-terminal tail precluded direct interaction with the active-site disulfide/dithiol group, suggesting an additional catalytically competent structural arrangement must exist.
To further investigate the mechanisms of mammalian TrxR enzymes, we have determined the structure of a mitochondrial TrxR. In this report, the structural features of mouse FAD-containing TrxR2 in the absence and presence of NADPH are described. The conformational changes that accompany pyridine nucleotide binding and reduction of the enzyme are revealed by comparative inspection of these two structures. Comparisons between mitochondrial and cytosolic isoenzymes suggest potential differences in enzymatic activities and provide additional insights into the catalytic mechanism of high-Mr TrxRs.
Materials and Methods
Data Collection and Structure Determination. Mature mouse TrxR Sec-523-Cys (mTrxR2) was expressed in Escherichia coli, purified by affinity chromatography, and crystallized as described in Supporting Text, which is published as supporting information on the PNAS web site. Diffraction data for mTrxR2 holoenzyme and the mTrxR2–NADP(H) complex were collected by using radiation produced by a Rigaku (Tokyo) MicroMax-007 x-ray generator fitted with confocal blue optics and an R axis IV++ image plate system. Crystals were maintained under cryogenic conditions by an X-stream cooling system, and data were analyzed with the crystal clear software package (Molecular Structure, The Woodlands, TX) (38). The structure of mTrxR2 with its bound FAD was determined by molecular replacement by using the crystallography and nmr system software package (39) and rat TrxR1 (1H6V) (29) as the search molecule (≈55% sequence identity). An unambiguous solution was obtained with an initial R of 39.4% (Rfree = 40.6%), after rigid body refinement. The resulting phase information produced a readily interpretable electron density map containing significant positive density within the active site of the enzyme, corresponding to the omitted FAD molecule (not shown). The mTrxR2–NADP(H) complex was isomorphous with the mTrxR2–FAD structure, and the same Rfree test set (10%) was used in the refinement of each structure.
Model Building and Refinement. Models were refined by using the crystallography and nmr system (39) and rebuilt after each round of refinement by using the program o (40). Ambiguous regions of the electron density map were evaluated by using 2Fo–Fc simulated annealing omit maps. As the model neared completion, bound cofactors and water molecules obeying proper hydrogen-bonding constraints with electron densities >1.0 σ on a 2Fo–Fc map and 3.0 σ on an Fo–Fc map were also included. Model geometry was monitored by using molprobity (41), and Figs. 1, 2, 3, 4, 5 were generated by using the program chimera (University of California, San Francisco) (42).
Fig. 1.

Ribbon representation of the overall structure of the mTrxR2 holoenzyme. Subunit A is shown in dark colors, and subunit B is shown in light colors. The protein is comprised of three domains: a FAD-binding domain (yellow), an NADP(H)-binding domain (green), and an interface domain (blue). Bound FAD molecules are represented as space-filling models with carbon atoms colored in gray, nitrogen atoms in blue, oxygen atoms in red, and phosphorous atoms in cyan. Side chains of Cys-483 in each subunit, located at the dimer interface, are shown in space-filling models with sulfur atoms colored in yellow (center).
Fig. 2.

Structure of mTrxR2 in complex with NADP(H). The ribbon representation of mTrxR2 is shown in an orientation similar to Fig. 1. FAD and NADP(H) are shown as space-filling models and are colored yellow and green, respectively. The ribbon is colored according to the average B factor of its corresponding residue. Values range from 20 (dark blue) to 100 (dark red) Å2, with an average B factor for the model of 61.7 Å2 (white).
Fig. 3.

Difference Fourier electron density map of the NADP(H)-binding site. Shown in the stereodiagram is the difference map contoured at ≈3 σ, super-imposed with the final mTrxR2–NADP(H) model. Carbon atoms are colored in green, nitrogen atoms in blue, oxygen atoms in red, and phosphorous atoms in cyan. For comparison, the final model of the mTrxR2 holoenzyme is also shown (light blue).
Fig. 4.

Comparison of the active sites of mTrxR2 and rTrxR1. Models of mTrxR2 and rTrxR1 were superimposed and are shown as stick representations by using the color scheme described in Fig. 3. (A) NADP(H)-binding site. In mTrxR2 (green), the nicotinamide ring of NADP(H) is stacked above the isoalloxazine ring of FAD, but it is not observed in rTrxR1(blue), because its binding site is occupied by the side chain of Tyr-200. (B) Region adjacent to the active-site disulfide/dithiol pair. Amino acid side chains of mTrxR2 are labeled with the corresponding residues from rTrxR1 given in parentheses. An asterisk denotes an amino acid residue from the opposing subunit of the dimer. Please note that, for FAD, only the isoalloxazine ring is presented. Thr-368 of mTrxR2 and Thr-339 of rTrxR1 adopt nearly identical conformation and have been omitted for clarity.
Fig. 5.

Model of the C terminus of mTrxR2. Continuous density was not observed for the final four residues of the mTrxR2–NADP(H) structure, confirming its highly mobile nature. Thus, residues G521, C522, U523, and G524 were modeled into the active site. In the stereodiagram, the selenium atom is colored in pink, nitrogen atoms in blue, oxygen atoms in red, and sulfur atoms in yellow. Carbon atoms from subunit A are colored in green, and those from subunit B are in blue and are designated with asterisks. Modeled residues are labeled in red. For clarity, only the nicotinamide ring of NADP(H) and the isoallozaine ring of FAD are presented.
Modeling of the C-Terminal Tail of mTrxR2. The C-terminal tail of mTrxR2 is highly mobile, consistent with its role in the transfer of reducing equivalents from deep within the enzyme molecule to an active-site disulfide in thioredoxin. The electron density for both mTrxR2 structures was quite poor beyond Thr-518. However, weak backbone density for Val-519 and Thr-520 was observed in the mTrxR2–NADP(H) complex and suggested an orientation of the C-terminal tail of mTrxR2. Vicinal disulfide bonds are relatively rare in the available protein structures, and the vast majority are found in type VIII β turns (43). To model the selenenylsulfide in oxidized mTrxR2, we used the vicinal disulfide turn motif within methanol dehydrogenase as a template (44). This prototype motif allowed us to model the relative positions of Gly-521, Cys-522, Sec-523, and Gly-524, and the absolute placement of the C-terminal tail was done manually, optimizing interactions within the active site of mTrxR2 and maintaining reasonable geometric constraints.
Results and Discussion
Structure Determination and Model Quality. The structure of mouse TrxR2 was determined by molecular replacement by using the related rat cytosolic TrxR1 as a probe (29). A readily interpretable electron density map was obtained, and density was clearly observed for the protein-bound FAD molecule (data not shown). To verify the solution, a simulated annealing omit map was constructed, omitting residues within 5 Å of the proposed FAD-binding site. Strong positive density was observed for FAD, as well as the additional atoms omitted from the map calculation, indicating a correct molecular replacement solution (Fig. 6, which is published as supporting information on the PNAS web site).
mTrxR2 crystals belong to spacegroup I4122 and contain single subunit of the dimeric enzyme in the asymmetric unit. Statistics for the final mTrxR2–FAD model are given in Table 1. Ramachandran plot analysis by molprobity (41) indicated that the final model had 91% of its residues in the favored region and >99% in the allowed region. The model had an overall B factor of 57.2 Å2, reflecting high overall thermal motion within the crystal. The twofold symmetry of the dimeric enzyme corresponds to a crystallographic axis and contains extensive intersubunit contacts (Fig. 1). The N terminus of the mTrxR2 and the final six residues of the polypeptide chain are not observed in the electron density map. Furthermore, three regions of the model, corresponding to residues 158–182, 282–307, and 321–340, exhibit considerable motion as evidenced by consecutive B factors >80 Å2. These regions had similar mobility in the structure of mTrxR2 in complex with NADP(H), as discussed below (Fig. 2). Considerably fewer crystal packing contacts are observed at the extremes of the NADP(H) and FAD-binding domains and may account for the greater thermal motion observed in these regions. Although significant electron density exists for these surface-exposed residues, the exact atom placements should be evaluated with care.
Table 1. Data collection and refinement statistics for the mTrxR2 structures.
| mTrxR2 | mTrxR2 + NADP(H) | |
|---|---|---|
| Data collection statistics | ||
| Space group | I4122 | I4122 |
| Cell dimensions, Å | 108.83, 108.83, 204.95 | 109.07, 109.07, 204.67 |
| Resolution, Å | 37.40-2.6 | 32.7-3.0 |
| Number of unique reflections | 19,370 | 12,754 |
| Average redundancy | 15.9 (16.0) | 16.3 (16.7) |
| 〈I/σI〉 | 18.7 (6.5) | 17.8 (6.6) |
| Completeness, % | 100 (100) | 100 (100) |
| Rmerge, % | 8.6 (44.0) | 11.4 (43.1) |
| Refinement statistics | ||
| Number of atoms | 3,764 | 3,807 |
| Water molecules | 27 | 16 |
| Rfactor, % | 22.2 (31.8) | 24.1 (31.8) |
| Rfree, % | 27.6 (36.0) | 31.2 (37.1) |
| Overall B factor, Å2 | 57.3 | 61.7 |
| B factor (solvent), Å2 | 42.1 | 28.9 |
| rms deviation from ideal values | ||
| Bond lengths, Å | 0.007 | 0.008 |
| Bond angles, ° | 1.4 | 1.5 |
| Estimated error (Luzzati), Å | 0.33 | 0.37 |
Values for the highest-resolution data shell are given in parentheses.
Overall Structure. The structure of mTrxR2 is comparable to that of other pyridine nucleotide disulfide reductases (30–34). As described for rTrxR1 (29), mammalian TrxR is comprised of three domains: a FAD-binding domain (mTrxR2 residues 35–190, 322–392), an NADPH-binding domain (mTrxR2 residues 191–321), and an interface domain (mTxrR2 residues 393–524) (Figs. 1 and 2). Comparison of mTrxR2 with the related rTrxR1 reveals that the two enzymes have very similar structures (rms deviation for Cα = 0.79 Å). A significant difference is the presence of a surface-exposed intersubunit disulfide bond between Cys-483 in the two subunits of mTrxR2. This cysteine is highly conserved among mammalian TrxRs and is located near the end of a helix (α11 in rTrxR1) in the interface domain (Fig. 1). A nonreducing SDS gel of mTrxR2 indicates that only ≈10% of the protein is involved in an intersubunit disulfide bond (data not shown), suggesting that the crystallization conditions may promote this disulfide bond formation. Interestingly, the corresponding cysteine in rTrxR1, Cys-458, is found in a similar position but in a reduced state (29). An intriguing possibility is that Cys-483 represents a third dithiol/disulfide pair in the enzyme with as-yet-unidentified function.
NADPH-Binding Site. To identify the cofactor-binding site in the enzyme, mTrxR2 holoenzyme crystals were transferred to an artificial mother liquor containing NADPH. A striking color change was observed: the yellow mTrxR2 crystals became a deep red (Fig. 7, which is published as supporting information on the PNAS web site), suggesting the formation of a charge-transfer complex with the addition of NADPH. As part of the proposed catalytic mechanism, reduction of the active-site disulfide results in the formation of a charge-transfer complex between FAD and an active-site thiolate (2, 4, 25–27). It is important to note that the excess NADPH added to the crystal can displace the resulting oxidized cofactor without disruption of the charge-transfer complex (25). Therefore, we cannot unambiguously state whether NADP+ or NADPH is bound in the active site and thus designate the cofactor as NADP(H). Furthermore, to account for the charge-transfer complex, the active-site cysteine residues of mTrxR2, Cys-86, and Cys-91 were modeled in the reduced state, such that the sulfur of Cys-91 is in close proximity to C4A of the flavin ring (≈3.4 Å).
Because of the moderate resolution of the structure (3.0 Å), an examination of the electron density per se cannot exclude the possibility of a disulfide between Cys-86 and Cys-91. However, modeling of a disulfide bond between Cys-86 and Cys-91 in the mTrxR2–NADP(H) complex results in several difference peaks in the corresponding region of the Fo–Fc map, suggesting that the modeled disulfide does not agree well with the x-ray data (data not shown). Furthermore, comparison of the mTrxR2 holoenzyme and mTrxR2–NADP(H) complex by examination of a difference Fourier electron density map suggests modest differences in the placement of Cys-86 and Cys-91, consistent with reduction of the disulfide (data not shown). The final model of the mTrxR2–NADP(H) complex is shown in Fig. 2, and model statistics are provided in Table 1.
Overall, the mTrxR2–NADP(H) and the mTrxR2 holoenzyme structures are very similar, with rms deviation for Cα = 0.45 Å. As with the oxidized mTrxR2 structure, residues at the dimer interface and within the core of the active site exhibit considerably lower B factors than residues at the extreme edges of the FAD- and NADPH-binding domains (Fig. 2). These highly mobile regions do not appear to have a direct role in catalysis and correspond to more divergent regions of high-Mr TrxR primary structure. To reduce the bias introduced by a direct comparison of the mTrxR2 holoenzyme and mTrxR2–NADP(H) models, a difference Fourier electron density map was calculated by using the phases from the mTrxR2 holoenzyme. This Fo–Fo map was used to assess the structural changes in mTrxR2 on addition of NADPH (Fig. 3). Continuous electron density was observed for the NADP(H) cofactor, although the quality of the map in the region of the nicotinamide ring was somewhat degraded by the placement of Tyr-228 in the oxidized mTrxR2 model. The positions of the isoalloxazine rings from each complex were nearly identical, and both ring systems were essentially planar.
Several significant displacements are observed in the mTrxR2–NADP(H) protein structure. Two arginine residues, Arg-249 and Arg-254, reorient to interact with the 2′-phosphate group of NADPH and likely confer the enzyme's specificity for NADPH relative to NADH (29). In addition, the hydroxyl group of Ser-227 must adjust to allow binding of one of the bridging phosphate groups of NADPH. Last, the Fo–Fo map indicates that the side chain of Tyr-228 must rotate to accommodate NADPH. Thus, to orchestrate this critical catalytic event, Tyr-228, which normally shields the flavin ring of FAD from solvent in the absence of NADPH, must reposition itself to stack against the nicotinamide ring upon cofactor binding. A conserved tyrosine residue in human glutathione reductase, Tyr-197, has been shown to serve a comparable function (34, 45–47).
The importance of such a rotation of the conserved tyrosine residue upon pyridine nucleotide binding is illustrated by examination of the rTrxR1 structure (29). Density is not observed for the nicotinamide ring of NADP+ in the oxidized rTrxR1–NADP+ complex. The equivalent tyrosine residue, Tyr-200, is arranged to shield the flavin ring from solvent (Fig. 4A, blue model) and thus precludes efficient binding of NADP+. Only the adenosine portions of each cofactor have similar orientations in the rTrxR1 and mTrxR2 structures. The bridging phosphate groups and the nicotinamide ribose of rTrxR1-bound NADP+ are displaced significantly relative to the corresponding atoms in the mTrxR2–NADP(H) structure. Without reorientation of the conserved tyrosine residue, the nicotinamide and isoalloxazine rings cannot stack in the position necessary for hydride transfer.
Active Site of mTrxR2. The overall architecture of the mTrxR2 active site is comparable to that of rTrxR1 (Fig. 4B) (29) and human glutathione reductase (31, 34, 46). In the mTrxR2–NADP(H) complex, the active-site disulfide/dithiol pair is observed in the reduced state, whereas this active-site redox center is found in the oxidized state in both the mTrxR2 holoenzyme (Fig. 1) and the rTrxR1–NADP+ complex (Fig. 4B). In addition to the active-site disulfide/dithiol functional group, a histidine–glutamate pair is located in similar orientations in these enzymes (H497*-E502* in mTrxR2; an asterisk designates an amino acid from the opposing subunit). Mutagenesis studies have demonstrated that both cysteine residues, as well as the active-site histidine residue, are essential for efficient catalysis (27). Other functional groups of mTrxR2 that face into the enzyme active site include Lys-56, His-143, Glu-503*, and Lys-506*. Glu-503* and Lys-506* are positioned to form a salt bridge and may stabilize the placement of Glu-502*. Such an interaction is absent in human glutathione reductase and rTrxR1, but other hydrogen bond networks maintain the tertiary structure in this region. The epsilon amino group of Lys-56 is located ≈3.1 Å from ND1 of His-143 and may help position the imidazole ring. Interestingly, a tyrosine residue replaces His-143 in both human glutathione reductase (Tyr-114) and rTrxR1 (Tyr-116; Fig. 4B). This tyrosine residue has been implicated in catalysis (47), as discussed below, and thus substitution with a histidine residue may modulate the catalytic activity of mTrxR2 relative to these homologues.
Modeling of the Redox-Active C-Terminal Tail. The molecular details of how reducing equivalents are transferred from the disulfide/dithiol pair deep within the active site, to the mobile C-terminal tail containing the Gly-Cys-Sec-Gly motif, and ultimately to oxidized thioredoxin remain unresolved. In the structure of the rat cytosolic isozyme (29), the C terminus was arranged in such a conformation that the selenocysteine residue could not interact with the active-site disulfide/dithiol, suggesting that an alternate conformation for the C terminus must exist. In the current study, the C-terminal tail of mTrxR2 exhibits considerable conformational flexibility, as evidenced by the high B factors of its residues. Furthermore, the last four residues are not observed in the electron density.
The Gly-Cys-Sec-Gly motif of the mTrxR2-NADP(H) structure has been modeled into the active site of the enzyme (Fig. 5). A suitable template for the selenenylsulfide was identified, and the C terminus of the enzyme was positioned in the active site of the opposing subunit, optimizing interactions and minimizing geometric strains. Possible conformations were limited by the observed position of residues 518–520 of the mTrxR2–NADP(H) structure. Although additional conformations are certainly possible given the highly mobile nature of this region of the protein (29), the model is consistent with several lines of biochemical evidence, as discussed below, and is a reasonable starting point for biochemical studies aimed at elucidating the precise mechanism of the enzyme. The eight-membered ring structure of the selenenylsulfide is juxtaposed to Cys-86 of the active-site disulfide/dithiol (Fig. 5). The selenium atom of selenocysteine 523* is located nearly equidistant from the sulfur of Cys-86 and the epsilon nitrogen of His-497*. Similarly, the sulfur of Cys-522* is adjacent to the epsilon nitrogen of His-143. Last, the carboxylate of Gly-524* may interact with the positively charged side chain of Lys-506* (not shown).
Implications for Catalysis. The precise mechanism of selenenylsulfide/disulfide exchange has not yet been determined for mammalian TrxRs, but studies of D. melanogaster (DmTrxR1) (26, 48, 49) and P. falciparum (27, 37, 50) high-Mr TrxRs provide considerable insight. DmTrxR1 exhibits significant sequence homology to mammalian TrxRs but is not a selenoprotein (26, 48). It exhibits the salient features of catalysis described for selenocysteine-containing TrxRs but instead has a Ser-Cys-Cys-Ser motif at its C terminus. Bauer et al. (26) have identified Cys-57 and Cys-490* (equivalent to Cys-86 and Sec-523* in mTrxR2) as the two thiol groups involved in dithiol/disulfide exchange (26). Similarly, studies with P. falciparum TrxR have suggested that Cys 540* (comparable to Sec-523* in mTrxR2) is the C-terminal interchange thiol (27). The presented model of the C-terminal tail of mTrxR2 within the enzyme active site is consistent with Sec-523* interacting with active site dithiol/disulfide via Cys-86. After reduction of the selenenylsulfide, the resulting selenolate likely attacks the disulfide of oxidized thioredoxin (29), although this has not yet been experimentally confirmed.
This placement of the redox active C-terminal tail suggests several active-site residues that may facilitate catalysis (Fig. 5). An active-site glutamate–histidine pair is conserved in high-Mr TrxRs and is thought to promote reduction of the selenenylsulfide by stabilizing the resulting negative charge on the selenolate (28). Mutagenesis studies involving the analogous glutamate–histidine pair in P. falciparum TrxR have demonstrated that this histidine is required for efficient catalysis (27). In the mTrxR2 structure in which the C-terminal selenenylsulfide has been modeled (Fig. 5), the epsilon nitrogen of His 497* is located nearly equidistant from the sulfur of Cys-86 and the selenium of Sec-523* and is consistent with His-497* being involved in catalysis.
A second active-site histidine is observed in mTrxR2 that may contribute to catalysis. The epsilon nitrogen of His-143 is located ≈3 Å from the modeled sulfur of Cys-522*, and its imidazole ring appears to be oriented by an adjacent lysine residue, Lys-56 (Fig. 5). Interestingly, His-143 is replaced with a tyrosine residue in rTrxR1 (Tyr 116; Fig. 4B) and glutathione reductase (Tyr-114). Site-directed mutagenesis studies in which Tyr-114 of glutathione reductase has been replaced with a leucine residue indicate that Tyr-114 is indeed involved in catalysis (47). The Y114L mutant exhibits a nearly 7-fold reduction in specific activity, and although not required for catalysis, Tyr-114 contributes to enzymatic efficiency. This observation, in concert with the proposed model, suggests that His-143 may be involved in the mechanism of mTrxR2. However, additional site-directed mutagenesis and kinetic studies will be required to assess the exact impact of His-143 on catalysis.
The thioredoxin-binding site of TrxR is thought to be located in the cleft between the FAD-binding domain and the dimer interface domain, near the C terminus of the enzyme (Figs. 1 and 2). In the absence of a high-Mr TrxR/thioredoxin complex structure, modeling studies have suggested interactions that mediate substrate recognition by TrxR (15, 28, 29). For rTrxR1, several amino acid side chains in the region comprised of residues 115–124 and the C-terminal tail were suggested to interact with thioredoxin (29). The corresponding regions in mTrxR2 (residues 142–151) have similar structural motifs but do exhibit some sequence variability. These differences may reflect different recognition residues on their respective thioredoxin substrates. However, without detailed structural information about the protein–protein complex, it remains difficult to ascribe function to particular residues.
Conclusion
Comparisons of mTrxR2 structures in the presence and absence of NADPH reveal several structural rearrangements that occur upon binding of the reduced pyridine nucleotide and suggest that an active-site tyrosine residue, Tyr-228, is essential for optimal placement of the nicotinamide ring of NADPH. Although mTrxR2 maintains the overall topology observed in cytosolic rat TrxR, several key active-site residues have been replaced. These substitutions probably impact enzymatic activity, but the importance of specific active-site residues to catalytic efficiency will need to be evaluated by site-directed mutagenesis and kinetic studies. In addition, a potential redox regulatory center has been identified that involves Cys-483 residues on opposing subunits. Collectively, these insights provided by examination of the reported crystal structures will assist in the design of further mechanistic studies of high-Mr TrxRs.
Supplementary Material
Acknowledgments
We thank Dr. Hideaki Moriyama and Dr. Kohei Homma of the University of Nebraska Structural Biology Core Facility for their contributions through the maintenance of the x-ray and computational resources, and Dr. Melanie A. Simpson (University of Nebraska) for thoughtful discussions regarding the manuscript. The project described was supported by National Institutes of Health Grants GM065204 (to V.N.G.) and P20 RR-17675 and by the Nebraska Research Initiative's funding of the Nebraska Center for Structural Biology.
Author contributions: E.I.B., A.A.T., V.N.G., and J.J.B. designed research; E.I.B., A.A.T., and J.J.B. performed research; A.A.T. and V.N.G. contributed new reagents/analytic tools; E.I.B., V.N.G., and J.J.B. analyzed data; and E.I.B., A.A.T., V.N.G., and J.J.B. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: TrxR, thioredoxin reductase.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank (PDB ID codes 1ZKQ and 1ZDL).
References
- 1.Holmgren, A. (1985) Annu. Rev. Biochem. 54, 237–271. [DOI] [PubMed] [Google Scholar]
- 2.Gromer, S., Urig, S. & Becker, K. (2004) Med. Res. Rev. 24, 40–89. [DOI] [PubMed] [Google Scholar]
- 3.Arner, E. S. & Holmgren, A. (2000) Eur. J. Biochem. 267, 6102–6109. [DOI] [PubMed] [Google Scholar]
- 4.Williams, C. H., Arscott, L. D., Muller, S., Lennon, B. W., Ludwig, M. L., Wang, P. F., Veine, D. M., Becker, K. & Schirmer, R. H. (2000) Eur. J. Biochem. 267, 6110–6117. [DOI] [PubMed] [Google Scholar]
- 5.Gromer, S., Wissing, J., Behne, D., Ashman, K., Schirmer, R. H., Flohe, L. & Becker, K. (1998) Biochem. J. 332, 591–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Novoselov, S. V. & Gladyshev, V. N. (2003) Protein Sci. 12, 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams, C. H., Jr. (1995) FASEB J. 9, 1267–1276. [DOI] [PubMed] [Google Scholar]
- 8.Lennon, B. W., Williams, C. H., Jr., & Ludwig, M. L. (2000) Science 289, 1190–1194. [DOI] [PubMed] [Google Scholar]
- 9.Muller, S., Gilberger, T. W., Farber, P. M., Becker, K., Schirmer, R. H. & Walter, R. D. (1996) Mol. Biochem. Parasitol. 80, 215–219. [DOI] [PubMed] [Google Scholar]
- 10.Kanzok, S. M., Fechner, A., Bauer, H., Ulschmid, J. K., Muller, H. M., Botella-Munoz, J., Schneuwly, S., Schirmer, R. & Becker, K. (2001) Science 291, 643–646. [DOI] [PubMed] [Google Scholar]
- 11.Gasdaska, P. Y., Gasdaska, J. R., Cochran, S. & Powis, G. (1995) FEBS Lett. 373, 5–9. [DOI] [PubMed] [Google Scholar]
- 12.Gladyshev, V. N., Jeang, K. T. & Stadtman, T. C. (1996) Proc. Natl. Acad. Sci. USA 93, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasdaska, P. Y., Berggren, M. M., Berry, M. J. & Powis, G. (1999) FEBS Lett. 442, 105–111. [DOI] [PubMed] [Google Scholar]
- 14.Sun, Q. A., Wu, Y., Zappacosta, F., Jeang, K. T., Lee, B. J., Hatfield, D. L. & Gladyshev, V. N. (1999) J. Biol. Chem. 274, 24522–24530. [DOI] [PubMed] [Google Scholar]
- 15.Sun, Q. A., Kirnarsky, L., Sherman, S. & Gladyshev, V. N. (2001) Proc. Natl. Acad. Sci. USA 98, 3673–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakupoglu, C., Przemeck, G. K., Schneider, M., Moreno, S. G., Mayr, N., Hatzopoulos, A. K., de Angelis, M. H., Wurst, W., Bornkamm, G. W., Brielmeier, M., et al. (2005) Mol. Cell. Biol. 25, 1980–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conrad, M., Jakupoglu, C., Moreno, S. G., Lippl, S., Banjac, A., Schneider, M., Beck, H., Hatzopoulos, A. K., Just, U., Sinowatz, F., et al. (2004) Mol. Cell. Biol. 24, 9414–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nalvarte, I., Damdimopoulos, A. E., Nystom, C., Nordman, T., Miranda-Vizuete, A., Olsson, J. M., Eriksson, L., Bjornstedt, M., Arner, E. S. & Spyrou, G. (2004) J. Biol. Chem. 279, 54510–54517. [DOI] [PubMed] [Google Scholar]
- 19.Patenaude, A., Ven Murthy, M. R. & Mirault, M. E. (2004) J. Biol. Chem. 279, 27302–27314. [DOI] [PubMed] [Google Scholar]
- 20.Sun, Q. A., Zappacosta, F., Factor, V. M., Wirth, P. J., Hatfield, D. L. & Gladyshev, V. N. (2001) J. Biol. Chem. 276, 3106–3114. [DOI] [PubMed] [Google Scholar]
- 21.Damdimopoulos, A. E., Miranda-Vizuete, A., Treuter, E., Gustafsson, J. A. & Spyrou, G. (2004) J. Biol. Chem. 279, 38721–38729. [DOI] [PubMed] [Google Scholar]
- 22.Miranda-Vizuete, A. & Spyrou, G. (2002) Mol. Cells 13, 488–492. [PubMed] [Google Scholar]
- 23.Rundlof, A. K., Janard, M., Miranda-Vizuete, A. & Arner, E. S. (2004) Free Radic. Biol. Med. 36, 641–656. [DOI] [PubMed] [Google Scholar]
- 24.Su, D. & Gladyshev, V. N. (2004) Biochemistry 43, 12177–12188. [DOI] [PubMed] [Google Scholar]
- 25.Arscott, L. D., Gromer, S., Schirmer, R. H., Becker, K. & Williams, C. H., Jr. (1997) Proc. Natl. Acad. Sci. USA 94, 3621–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer, H., Massey, V., Arscott, L. D., Schirmer, R. H., Ballou, D. P. & Williams, C. H., Jr. (2003) J. Biol. Chem. 278, 33020–33028. [DOI] [PubMed] [Google Scholar]
- 27.Gilberger, T. W., Walter, R. D. & Muller, S. (1997) J. Biol. Chem. 272, 29584–29589. [DOI] [PubMed] [Google Scholar]
- 28.Brandt, W. & Wessjohann, L. A. (2005) ChemBioChem 6, 386–394. [DOI] [PubMed] [Google Scholar]
- 29.Sandalova, T., Zhong, L., Lindqvist, Y., Holmgren, A. & Schneider, G. (2001) Proc. Natl. Acad. Sci. USA 98, 9533–9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattevi, A., Obmolova, G., Sokatch, J. R., Betzel, C. & Hol, W. G. (1992) Proteins 13, 336–351. [DOI] [PubMed] [Google Scholar]
- 31.Schulz, G. E., Schirmer, R. H., Sachsenheimer, W. & Pai, E. F. (1978) Nature 273, 120–124. [DOI] [PubMed] [Google Scholar]
- 32.Zhang, Y., Bond, C. S., Bailey, S., Cunningham, M. L., Fairlamb, A. H. & Hunter, W. N. (1996) Protein Sci. 5, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bond, C. S., Zhang, Y., Berriman, M., Cunningham, M. L., Fairlamb, A. H. & Hunter, W. N. (1999) Structure Fold. Des. 7, 81–89. [DOI] [PubMed] [Google Scholar]
- 34.Karplus, P. A. & Schulz, G. E. (1987) J. Mol. Biol. 195, 701–729. [DOI] [PubMed] [Google Scholar]
- 35.Sarma, G. N., Savvides, S. N., Becker, K., Schirmer, M., Schirmer, R. H. & Karplus, P. A. (2003) J. Mol. Biol. 328, 893–907. [DOI] [PubMed] [Google Scholar]
- 36.Zhong, L., Arner, E. S. & Holmgren, A. (2000) Proc. Natl. Acad. Sci. USA 97, 5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krnajski, Z., Gilberger, T. W., Walter, R. D. & Muller, S. (2000) J. Biol. Chem. 275, 40874–40878. [DOI] [PubMed] [Google Scholar]
- 38.Pflugrath, J. W. (1999) Acta Crystallogr. D 55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- 39.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 40.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 41.Lovell, S. C., Davis, I. W., Arendall, W. B., 3rd, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003) Proteins 50, 437–450. [DOI] [PubMed] [Google Scholar]
- 42.Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004) J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 43.Carugo, O., Cemazar, M., Zahariev, S., Hudaky, I., Gaspari, Z., Perczel, A. & Pongor, S. (2003) Protein Eng. 16, 637–639. [DOI] [PubMed] [Google Scholar]
- 44.Xia, Z., Dai, W., Zhang, Y., White, S. A., Boyd, G. D. & Mathews, F. S. (1996) J. Mol. Biol. 259, 480–501. [DOI] [PubMed] [Google Scholar]
- 45.Pai, E. F., Karplus, P. A. & Schulz, G. E. (1988) Biochemistry 27, 4465–4474. [DOI] [PubMed] [Google Scholar]
- 46.Karplus, P. A. & Schulz, G. E. (1989) J. Mol. Biol. 210, 163–180. [DOI] [PubMed] [Google Scholar]
- 47.Krauth-Siegel, R. L., Arscott, L. D., Schonleben-Janas, A., Schirmer, R. H. & Williams, C. H., Jr. (1998) Biochemistry 37, 13968–13977. [DOI] [PubMed] [Google Scholar]
- 48.Gromer, S., Johansson, L., Bauer, H., Arscott, L. D., Rauch, S., Ballou, D. P., Williams, C. H., Jr., Schirmer, R. H. & Arner, E. S. (2003) Proc. Natl. Acad. Sci. USA 100, 12618–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacob, J., Schirmer, R. H. & Gromer, S. (2005) FEBS Lett. 579, 745–748. [DOI] [PubMed] [Google Scholar]
- 50.Wang, P. F., Arscott, L. D., Gilberger, T. W., Muller, S. & Williams, C. H., Jr. (1999) Biochemistry 38, 3187–3196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}