

Abstract
In higher eukaryotes, the ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) checkpoint kinases play distinct, but partially overlapping, roles in DNA damage response. Yet their interrelated function has not been defined for telomere maintenance. We discover in Drosophila that the two proteins control partially redundant pathways for telomere protection: the loss of ATM leads to the fusion of some telomeres, whereas the loss of both ATM and ATR renders all telomeres susceptible to fusion. The ATM-controlled pathway includes the Mre11 and Nijmegen breakage syndrome complex but not the Chk2 kinase, whereas the ATR-regulated pathway includes its partner ATR-interacting protein but not the Chk1 kinase. This finding suggests that ATM and ATR regulate different molecular events at the telomeres compared with the sites of DNA damage. This compensatory relationship between ATM and ATR is remarkably similar to that observed in yeast despite the fact that the biochemistry of telomere elongation is completely different in the two model systems. We provide evidence suggesting that both the loading of telomere capping proteins and normal telomeric silencing requires ATM and ATR in Drosophila and propose that ATM and ATR protect telomere integrity by safeguarding chromatin architecture that favors the loading of telomere-elongating, capping, and silencing proteins.
Keywords: Mre11 and Nbs1, telomerase-independent mechanism, telomere capping, telomeric silencing
ADNA double-strand break (DSB) is one of the most potent genotoxic agents, and eukaryotic cells have evolved an elaborate system to detect and repair DSBs. However, the natural ends of linear chromosomes are normally protected from such repair to prevent chromosome end-to-end fusions that could have devastating consequence in terms of genome instability. Therefore, it is rather counterintuitive that many of the repair/checkpoint proteins have been shown to both promote fusion between internal broken ends and prevent fusion between telomeres (1). Two of these proteins are the ataxia telangiectasia mutated (ATM) protein and the ATM and Rad3-related (ATR) protein. They belong to a family of phosphatidylinositol 3-kinase-like protein kinases that have important functions in various aspects of cell-cycle regulation (2, 3). ATM and ATR are central to almost all DNA damage response from yeast to human. We have significant understanding of how they transduce the signal of DNA damage. However, their precise roles in maintaining telomere homeostasis have not been well studied, even though one of the most common defects in ATM-deficient cells is abnormal telomeres (4–10).
Studies in both budding and fission yeasts have provided most of our understanding of the telomere-maintaining function of ATM or ATR. In Saccharomyces cerevisiae, both tel1 (atm) and mec1 (atr) mutants have shortened, but nevertheless stable, telomeres (11). A tel1 mec1 double mutant, on the other hand, has a much more severe telomere dysfunction. Cells enter senescence experiencing great loss of telomeric repeats, frequent telomere fusions, and gross genome rearrangements (11–13). A similar situation exists in Schizosaccharomyces pombe (14–16). Therefore, ATM and ATR are partially redundant for telomere maintenance in both yeasts.
In higher eukaryotes, the mechanism by which ATM maintains telomere integrity is largely unclear. A possible role of ATR in telomere maintenance has not been well studied either, let alone the possibility of functional redundancy between metazoan ATM and ATR. Such studies bear particular importance given that functional divergence has been shown between the yeast and mammalian ATM and ATR proteins. For example, the yeast ATM homologs play minor roles in cell-cycle checkpoint control compared with their mammalian counterparts (2, 3). Additionally, mammalian ATM and ATR have acquired new functions in cell-cycle control and apoptosis by modulating the tumor suppressor p53. It remains untested whether they have also acquired an additional telomere-protecting function that is specific for the development of a multicellular organism.
We and others have shown that mutations in the telomere fusion gene (tefu), which encodes the Drosophila ATM homolog, lead to telomere fusions and consequent genome instability (7–10, 17). In addition, the Mre11 and Rad50 proteins, which work intimately with ATM in DNA damage response and repair, have been shown to serve in Drosophila telomere protection (7, 18, 19). These results are perhaps intriguing considering that normal Drosophila telomeres are not elongated by a telomerase. The end of a Drosophila chromosome is enriched with three terminal-specific retrotransposable elements: HeT-A, TART, and TAHRE (20, 21). Telomere shortening caused by incomplete end replication is counteracted by de novo transposition of a new element to the end. Furthermore, terminally deleted chromosomes have been stably maintained in which other heterogeneous sequences constituted the end of a chromosome (22, 23, 24). These findings suggest that the end of a Drosophila chromosome could consist of essentially any sequence and that the function of a Drosophila telomere is independent of its sequence, unlike the situations in yeast and mammals. In addition, the telomere-maintaining function of ATM, Mre11, and Rad50 is evolutionarily conserved and could be independent of a telomerase.
In this study, we extend our analysis to Drosophila ATR and test its genetic interaction with ATM at telomeres. We show in a metazoan that ATM and ATR control partially redundant telomere-protecting mechanisms, a situation remarkably similar to that found in yeast. Therefore, not only is this redundancy between ATM and ATR conserved throughout evolution, it also appears intrinsic to telomere as a special chromosome structure regardless of how it is elongated. Furthermore, we show that the Nijmegen breakage syndrome (Nbs) protein, which functions as a complex with Mre11 and Rad50, is essential for telomere protection in Drosophila.
Materials and Methods
Drosophila Genetics. The stocks used in this study are listed in Supporting Text, which is published as supporting information on the PNAS web site. Mutations on chromosomes 2 and 3 were kept over balancer chromosomes with the Tubby larval marker. For the mei-4129D tefu double mutant, the mei-4129D mutation was kept in males only, aided by the use of females with attached X chromosomes. For the mei-4129D mre11 double mutant, mei-4129D was kept over an FM7 chromosome marked with an actin-driven GFP transgene, and mre11 was kept over a CyO balancer with actin-GFP. The double homozygotes were selected for the lack of fluorescence produced by actin-GFP. The mei-41RT1 and mei-41D9 mutations were kept as homozygotes.
Cytology. DAPI-stained mitotic cells were prepared as described (25). HP1/ORC-associated protein (HOAP) immunostaining on mitotic cells was performed as described (26). HP1 staining on polytene chromosomes was performed with the C1A9 antibody, a generous gift from L. Wallrath (University of Iowa, Iowa City). At least 20 telomeres were examined for each genotype.
RNA Dot Blots. We used only female larvae to avoid possible interference from transcripts originated from HeT-A sequences internal to the male Y chromosome (27). Total RNA was extracted by using the Total RNA extraction kit from Amersham Pharmacia and purified by using the Qiagen (Valenica, CA) RNeasy kit with an on-column DNase digestion step. The “HeT-A” probe was PCR-amplified from the “orf” region with the primers 5′-gatcttctccgttctacctc and 5′-aactcataggctgctcgtct. The “HeT-A#2” probe was amplified from the same region with the primers 5′-catcatcagacatctccacc and 5′-gaggcctttttgtgagttgg. The probe for ras64B was amplified with the primers 5′-ttggacattttggacacgga and 5′-gcacttgttacccaccatcagc.
Results
ATR Functions to Partly Compensate for the Loss of ATM at the Telomere. We showed earlier that mitotic cells from ATM-deficient animals (tefu-/-) had an average of three end-to-end fusions, which represents 6 of 32 telomeres in a cell after replication. A similar frequency was also observed for our mre11 mutant (7). Because a much higher fusion frequency has been observed in the caravaggio1 (cav1) mutant, which is deficient for the HOAP telomere capping protein (26, 28), we reasoned that in tefu and mre11 mutants there is residual telomere capping activity so that some of the ends are protected. We set out to test whether ATR regulates this residual capping activity. To assess the severity of telomere fusion, we examined DAPI-stained mitotic chromosomes from larval neuroblasts. Some representative chromosome configurations are shown in Fig. 1. We classify telomere fusions into three classes: single, double, and “other” fusions, which are defined in Fig. 1. The average telomere fusion frequencies for various mutants are shown in Fig. 2.
Fig. 1.

Telomere fusions in various mutants. DAPI-stained mitotic nuclei from third instar larval neuroblasts. (A) A WT female nucleus with all of the chromosomes labeled. (B)A grp1 tefu nucleus. Two fusions between chromosomes 2 and 4 (arrow) are defined as a double fusion. The two chromosome 4s are fused, which is defined as an other fusion because one cannot unequivocally define it as either single or double fusion. An unknown fragment (indicated by?) is fused with the short arm of an X chromosome (arrowhead). (C)A Su(var)2–504/05 tefu nucleus. A single fusion between the two sisters of an X chromosome is indicated by an arrowhead. Five double fusions (X-2, 3–3, 3–2, 2–4, and between the short and long arms of an X) are also present. (D) A mus304D1/D3 tefu nucleus. Fusions present are: five single nonsister fusions (arrows), a double X-2 fusion, and an other fusion between a 4 and the short arm of an X. (E)A mei-4129D tefu nucleus. Fusions present are: several double fusions (arrows) forming two chromosome “chains,” an intra-Y fusion (the ring), and two sister fusions (arrowheads). Note that one arm of chromosome 3 is longer than the same arm from its homolog indicative of a rearrangement. (F)A cav1 nucleus. Fusions present are: two 2–3, two X-4, and one intra-3 (the ring) double fusions and two X-4 other fusions. (G) A mei-41RT1 nbs1 nucleus. Fusions present are: two 2–2 (the ring) and one 3–3 double fusions, an X sister (arrowhead), and one 3–3 (arrow) single fusion. (H)A mei-41RT1 tefu nucleus. Double fusions among chromosomes 2 and 3 led to chromosome entanglement (arrowhead). The two Xs are entangled (arrow) accompanied by two sister fusions. (I) A mei-4129D mre11 nucleus showing numerous chromosome fragments. (J) A mus304D1/D3 tefu nucleus in anaphase with seven bridges between the two separating nuclei (arrow). (K) A mei-4129D tefu polyploid nucleus with all chromosome ends fused. (L) A tetraploid mei-41RT1 tefu nucleus. The four chromosome 2s formed a cluster by multiple fusions. The four 3s and the two Ys are clustered by multiple fusions. The two Xs and two 4s formed a ring structure.
Fig. 2.

Telomere fusion frequencies in various mutants. The graph depicts the average frequencies for different types of fusion and their sums, which are indicated by the numbers next to the horizontal bars with the number of nuclei scored in parentheses. Even though a double fusion is likely derived from the replication of a single G1 fusion, we cannot rule out that some double fusions were the result of independent fusions occurring in G2. We count each double fusion as two fusions for frequency calculations. Even with a double fusion counted as a single G1 event, our conclusions are fully supported by statistical analyses (data not shown). In the Genotype column, the common non-Drosophila nomenclatures are provided in parentheses. *, Data from ref. 7; **, data from ref. 23. LP, late pupal; EP, early pupal; L3, late larval; nd, not done.
The Drosophila ATR homolog is encoded by the mei-41 gene (29, 30). The loss of Mei-41 alone does not lead to telomere fusion (ref. 31 and unpublished data). We generated double mutants of a tefu loss-of-function allele (7) and three alleles of mei-41 (with severity referring to cell cycle checkpoint and female fertility defects): the D9 weak allele (30, 32), the RT1 strong allele (29, 33), and the 29D null allele (30). As shown in Fig. 2, tefu mei-41 double mutants had significantly more telomere fusion on average than the tefu single mutant. In many of the double mutant nuclei, most chromosome ends were fused (Fig. 1E). The severity of telomere fusion correlates well with the strength of the mutant alleles of mei-41. The tefu mei-41D9 combination resulted in close to five fusions per nucleus, whereas the stronger tefu mei-4129D and tefu mei-41RT1 combinations led to seven fusions per cell, a fusion frequency that matches the one observed in cav1 (ref. 26 and Figs. 1F and 2). In addition, 19% of the tefu mei-41RT1 mitotic nuclei (n = 142) and 13% of the tefu mei-4129D nuclei (n = 85) were polyploid. Remarkably, most chromosome ends were fused in these polyploid nuclei (Fig. 1 K and L). The frequency of polyploid nuclei correlates with the frequency of telomere fusion. For the weaker tefu mei-41D9 combination, only 1.8% (n = 112) of the nuclei were polyploid. In the tefu single mutant, <1% of the nuclei were polyploid (unpublished data). We envision that multiple mitotic bridges (Fig. 1J) could prevent cytokinesis, and defective cell cycle checkpoints (34) could allow these abnormal cells to continue cycling, leading to the accumulation of polyploid cells. Chromosome breaks were abundant, and in the most extreme cases, led to genome fragmentation (Fig. 1I). These breaks could be caused by breakage of chromosome bridges (7). A mutation in mei-41 could have a compounding effect because it causes an abnormally high frequency of spontaneous breaks (31). Chromosome rearrangements were also abundant, which could result from fusions between double-strand breaks (DSB) or between a DSB and a telomere (Fig. 1 E, H, and I). Lastly, chromosomes of the double mutants were often tangled with each other, similar to concatemers of circular DNA (Fig. 1H). We suggest that entanglement could be caused by physical exchanges between two chromosomes, homologous or not, both of which are involved in telomere fusion.
In summary, the loss of both ATM and ATR led to much more severe telomere defects than the loss of either protein individually. In the double mutants, most, if not all, chromosome ends were uncapped and susceptible to fusion. This severe telomere dysfunction led to abnormal development of the animals. The tefu mei-41RT1 and tefu mei-4129D double mutants developed more slowly than WT animals and arrested as third instar larvae for several days before death. There were fewer mitotic cells in the proliferating tissues of the double mutants (34).
The ATM-Controlled Telomeric Pathway Includes Mre11 and Nbs. In S. cerevisiae, the Mre11–Rad50–Xrs2 complex functions in the same telomere-protecting pathway as Tel1 (35). The functional homolog of Xrs2 is encoded by the nbs gene in higher eukaryotes, which is mutated in the human disease Nbs (36). We set out to test whether Drosophila Nbs functions in telomere maintenance.
We discovered that the previously identified lethal (3)67BDp1 mutation was a small deletion of the nbs-coding region (ref. 37 and Supporting Text). The mutation caused pupal lethality, and an average 1.9 telomere fusions per nucleus (Fig. 2), suggesting an essential role of Drosophila Nbs in telomere protection. We suggest renaming the mutation nbs1. In addition, an mre11 nbs1 double mutant has a fusion frequency similar to that of the mre11 single mutant (data not shown), suggesting that Nbs and Mre11 are in the same telomere protection pathway.
We suggested earlier that Drosophila ATM and Mre11 are in the same telomere-protecting pathway because a tefu mre11 double mutant has telomere defects similar to either single mutant in severity (7). By similar double mutant analyses, we conclude that Nbs is in the same pathway as ATM (unpublished data). Based on our earlier results, we predict that an mre11 mei-41 or an nbs mei-41 double mutation would behave similarly as a tefu mei-41 double mutation in causing telomere fusion. Indeed, an average mre11 mei-4129D cell had six fusions, and 18% of the double mutant nuclei were polyploid (n = 89). Both frequencies are similar to those observed for the tefu mei-4129D double mutant. Noticeably, more mre11 mei-41 nuclei showed chromosome fragmentation than tefu mei-41 nuclei (Fig. 1I and unpublished data), which is consistent with our earlier results suggesting that Mre11 has a bigger role than ATM in preventing chromosome breakage (7, 34). We also generated nbs1 mei-41RT1 and nbs1 mei-4129D double mutants. Both combinations led to a synergistic increase of fusion rates (Figs. 1G and 2 and data not shown). An nbs1 mei-41RT1 nucleus had >5 fusions on average, 2.5 times as many as the nbs1 single mutant. Over 12% of the double mutant nuclei were polyploid (n = 106). These results suggest that Drosophila Mre11 and Nbs are in a telomere-protecting pathway partially redundant to the one controlled by ATR.
ATM and ATR Have Different Targets at Telomeres Versus at DNA Damage Sites. In mammalian cells, the Chk2 and Chk1 kinases are the major downstream signal transducers for the damage response pathways controlled by ATM and ATR, respectively (38, 39). We examined whether these downstream kinases participate in telomere protection along with their upstream regulators. The Drosophila Grapes (Grp) protein is homologous to Chk1 (40, 41). We generated grp mei-4129D, grp tefu, and grp mre11 double mutants. Flies deficient for either Grp singly or both Grp and Mei-41 developed normally, and the males were fertile, suggesting no loss of telomere protection. On the other hand, grp tefu or grp mre11 double mutants remained pupal lethal, but we did not observe an increase of telomere fusion for either double mutant (Figs. 1B and 2). Therefore, Chk1 does not act downstream of ATR to regulate telomere protection in Drosophila. The Drosophila Chk2 homolog is encoded by the dmchk2/mnk/loki gene (42). We generated mnkp6 mei-4129D, mnkp6 tefu, and mnkp6 mre11 double mutants. mnkp6 mei-4129D mutants developed normally and the males were fertile, suggesting no loss of telomere protection. In addition, mnkp6 tefu or mnkp6 mre11 double mutants remained pupal lethal, consistent with the fact that no increase in telomere fusion was observed for either double mutant (Fig. 2 and data not shown). Taken together, our results suggest that a different set of factors acts downstream of ATM and ATR at the sites of DNA damage versus at the telomeres.
We also discovered that homologous recombination pathways controlled by Rad51 or Rad54 do not contribute to the ATM- or ATR-controlled telomere-protecting pathway in Drosophila (Fig. 2 and Supporting Text).
ATR-Interacting Protein (ATRIP) Acts with ATR to Protect Telomeres. ATR interacts with a partner, ATRIP, at the sites of DNA damage (43). A similar partner relationship has been suggested for the Drosophila homologs in mitotic cells (33). However, results from the previous section raise the possibility that ATRIP may not necessarily be the partner for ATR at telomeres. We set out to test this hypothesis.
In Drosophila, the mutagen-sensitive 304 (mus304) gene encodes the functional homolog of ATRIP (33). The fact that either mus304 single or mei-41 mus304 double mutant animals have essentially normal development suggests that the loss of ATR and/or ATRIP does not lead to frequent telomere fusions. We generated tefu mus304 (atm atrip) double mutants to test whether Mus304, like ATR, can compensate for the loss of ATM. We used the D3 weak and the D1 null alleles of mus304. The mus304D3 mutation only led to a small increase in fusion frequency when combined with tefu (Fig. 2). For the stronger mus304D1/D3 tefu combination, we observed 5.35 fusions on average (Figs. 1D and 2). For mus304D1/D1 tefu, the strongest combination, we observed 7.12 fusions per nucleus. In addition, close to 20% of these nuclei were polyploid (n = 86). These results indicate a synergistic interaction between the tefu and mus304 mutations. We also generated an mre11 mus304D1 double mutant and observed an average of 5.44 fusions per nuclei, also indicating a synergistic interaction between mre11 and mus304. Taken collectively, our results suggest that ATRIP works in concert with ATR for telomere protection similar to their relationship at sites of DNA damage.
Loading of Telomere Capping Proteins Is Facilitated by ATM and ATR. The normal ends of Drosophila chromosomes are thought to be elongated by transposition of mainly the HeT-A retrotransposons (20, 22). Because we did not observe extensive loss of telomeric HeT-A transposons in any of our mutants with telomere fusions (ref. 7 and Fig. 5, which is published as supporting information on the PNAS web site), the roles of ATM and ATR at Drosophila telomeres are not likely to maintain telomere lengths by regulating HeT-A transposition. Because telomere fusion was the most significant telomeric defect that we observed in these mutants, we propose that ATM and ATR facilitate the loading of telomere capping proteins in Drosophila.
HOAP has been suggested to be part of a capping complex in Drosophila (26). We showed earlier that ATM is not essential for HOAP binding to telomeres in diploid cells (7). We hypothesized that ATR might compensate for the loss of ATM by facilitating HOAP binding to tefu telomeres. We tested this hypothesis by immunolocalizing HOAP in mitotic cells from various tefu and mei-41 mutants (Figs. 2 and 3). From three tefu mei-4129D animals, 2 of 60 nuclei had a weak HOAP signal on one chromosome end. For the rest of the nuclei, HOAP was completely absent from the chromosomes. In Fig. 3 E and F, a tefu mei-4129D nucleus was shown that had a few fusion-free ends, yet no HOAP was present there. On the other hand, from three tefu single mutant animals, 119 of 128 nuclei have HOAP on most chromosome ends not fused (ref. 7 and data not shown). We suggest that either ATM or ATR is sufficient for HOAP localization to mitotic telomeres. This hypothesis is further supported by telomere fusion analyses on the tefu cav1 and mei-41RT1 cav1 double mutants: they have similar extents of telomere fusion as the cav1 single mutant (unpublished data). Interestingly, we observed that HOAP was localized normally to most free ends in both tefu mei-41D9 and tefu mus304D1/D3 cells (Fig. 3 G and H). In contrast, telomeric HOAP signals were observed in 6 of 64 nuclei from four tefu mus304D1 animals. Because we used a weaker mutant allele in the first two cases, we suggest that in the absence of ATM little ATR/ATRIP activity is sufficient for HOAP loading.
Fig. 3.

HOAP localization in various mutants. The genotypes are given to the left. (A, C, E, and G) DAPI-stained mitotic nuclei with chromosomes labeled except for E. (B, D, F, and H) Merged views of DAPI and anti-HOAP antibody staining (red). In C and D, the arrow indicates a 3–3 double fusion with no HOAP signal. The arrowhead indicates an X sister fusion with a HOAP signal at the junction. In F, the antibody signals are overexposed to indicate the lack of any telomeric signals. In G and H, the arrow indicates a 2–4 double fusion with HOAP signals at the junctions.
We and others have showed that mutations in mre11 or rad50 led to variable loss of HOAP at mitotic telomeres (7, 18). We observed that a defective Nbs, the third component of the complex, had a similar effect: only 41% of the nuclei from three nbs1 animals had HOAP signals on more than one end (n = 117). mei-41 mutations further reduced HOAP's ability to bind telomeres: weak HOAP signals on no more than one chromosome end were observed in 4 of 75 nuclei from five mre11 mei-4129D animals and in 3 of 45 nuclei from three nbs1 mei-41RT1 animals. Both cases are similar to the situation in tefu mei-4129D. Therefore, Mre11 and Nbs similarly regulate HOAP loading to telomeres and ATR can partially compensate for the loss of these factors.
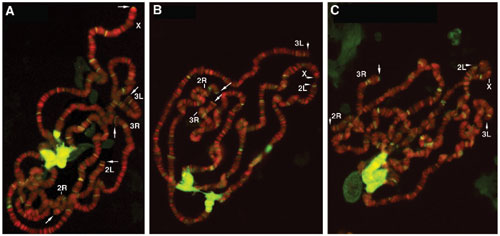
The other Drosophila protein implicated in telomere capping is the conserved heterochromatin protein 1 (HP1; ref. 44), which is encoded by the Su(var)2–5 gene. A tefu Su(var)2–504/05 double mutant had a similar degree of telomere fusion as the Su(var)2–504/05 single mutant (Fig. 1C and unpublished data), indicating an epistatic relationship between ATM and HP1 in telomere capping. To monitor HP1's ability to bind to telomeres, we immunolocalized HP1 to telomeres of polytene chromosomes from larval salivary glands and observed greatly reduced HP1 and K9-methylated histone H3 levels in both tefu and mei-4129D tefu mutants (Figs. 6 and 7, which are published as supporting information on the PNAS web site). We are more interested in studying HP1's telomeric loading in proliferating cells because telomere fusions are more abundantly observed in these cells (data not shown). However, our effort to consistently localize HP1 to mitotic telomeres has been hampered by high backgrounds in immunostaining experiments. Therefore, further investigation is needed to identify a positive influence of ATM or ATR on the binding of HP1 to diploid telomeres.
ATM and ATR Control Partially Redundant Pathways for Telomeric Silencing. Because the transcriptional profile at telomeric regions has been widely used as an indicator of the structural integrity of telomeres, we examined the effect of Drosophila atm and atr mutations on telomeric transcription to provide further evidence that ATM and ATR act redundantly for telomere maintenance. In both fission and budding yeast, certain atr mutations alleviate telomeric silencing of adjacent transgenes (15, 45). It was previously reported that a tefu mutation variably suppressed the silencing of telomeric reporter genes in Drosophila (8). We tested the mei-4129D mutation and observed no effect on the silencing of the same reporter insertions (unpublished data). Because most of these Drosophila telomeric silencing lines have a transgene inserted at subtelomeric regions (46), it is possible that this reporter gene assay may not detect a larger effect of atm or atr mutations on transcription from more telomere-proximal regions. It was shown earlier that telomeric HeT-A transposons are silenced transcriptionally, and this silencing depends on HP1 (47, 48). This observation prompted us to examine HeT-A transcription in tefu and mei-41 mutants. We recovered total RNA from various lines and performed RNA dot-blot analyses on HeT-A transcription by using the ras64B transcript as a control. As shown in Fig. 4, HeT-A transcript is low in the WT control, but greatly elevated in Su(var)2–504/05 mutants, both of which are consistent with previous reports. The mei-41RT1 mutation did not lead to derepression of HeT-A, whereas the tefu mutation resulted in a moderate increase of HeT-A RNA. More importantly, the double mutant caused a synergistic increase of HeT-A transcripts, which is consistent with a compensatory relationship between ATM and ATR. The degree of HeT-A derepression in the tefu mei-41RT1 double mutant was lower than that in Su(var)2–504/05, which could be caused by small amounts of HP1 binding to the telomeres in the absence of ATM and ATR. In summary, ATM and ATR act redundantly to prevent telomere fusion and silence telomeric transposon arrays in Drosophila.
Fig. 4.

HeT-A derepression by atm and atr mutations. RNA dot-blot analyses from three separate trials are shown. Genotypes are: +, w1118; atm, tefu; atr, mei-41RT1; hp1, Su(var)2–504/05; hoap, cav1. The probes used are indicated at the bottom. In A, 2.5 μg of RNA was loaded per dot. In B and C, 5 μg of RNA was loaded per dot. For each genotype, three separate dots were loaded with RNA for trial A, one for trial B, and two for trial C.
Discussion
For the conserved ATM and ATR kinases, their interrelated function for telomere maintenance has not been defined for complex organisms. In this study, we genetically defined in Drosophila two partially redundant pathways, regulated by ATM and ATR, respectively, that ensure complete protection of Drosophila telomeres. We further suggest that Mre11 and Nbs belong to the same ATM-regulated pathway, whereas ATRIP participates in the ATR-controlled pathway. This conclusion would be consistent with the pathway components defined in yeasts (13, 16). Based on the facts that Drosophila tefu mutants have widespread telomere fusions that eventually lead to lethality, whereas mei-41 mutants are viable with no apparent telomeric defects, we propose that ATM is more important in regulating telomere protection, with ATR playing a backup role. In mei-41 mutants, ATM may fully compensate ATR's absence on telomeres to ensure complete capping throughout the cell cycle. On the other hand, ATR in tefu mutants may partially compensate for the loss of ATM because it is a less efficient regulator of capping as suggested earlier. This partial redundancy leads to the fusion of some telomeres.
ATM, ATR, and their cofactors are known to control multiple checkpoints in response to DNA damage and abnormal telomeres. Perhaps cells with uncapped telomeres are allowed to continue cycling because of defective checkpoints and that leads to the fusion of these telomeres. We consider this possibility unlikely based on several observations. First, in cav1 mutant, telomere fusion occurs at a high rate even in the presence of a full complement of checkpoint genes. In addition, we did not observe an exacerbated telomere dysfunction in either the cav1 tefu (atm) or the cav1 mei-41 (atr) double mutant. Furthermore, mutations in Drosophila chk1 or chk2 did not affect existing telomere defects in either tefu or mre11 mutant, suggesting that checkpoints jointly controlled by these effectors and the respective upstream kinases do not normally respond to dysfunctional telomeres. Therefore, the contribution to the telomere dysfunction from checkpoint defects is likely small in the cases that we studied.
Our cytological analyses suggest that one of the functions of ATM and ATR at Drosophila telomeres is to facilitate the loading of telomere capping and silencing proteins, such as HOAP and HP1, and they do so in a partially redundant fashion. In the case of HOAP's binding to mitotic telomeres, either ATM or ATR is sufficient for its normal loading. When both kinases are absent, HOAP can no longer be detected at the telomeres. It was interesting that there were more telomere fusions in either tefu mei-41D9 or tefu mus304D1/D3 than in either mre11 or nbs1 cells (Fig. 2), yet normal HOAP signals were detected on fusion-free telomeres for only the first two genotypes. It is known that the presence of HOAP at chromosome ends is not sufficient to prevent fusion (e.g., Fig. 3 D and G and refs. 7 and 26), which suggests that the loss of other capping proteins could also lead to fusion. That may be the case in tefu mei-41D9 and tefu mus304D1/D3 cells. It is also known that the absent of HOAP at any particular telomere does not necessarily lead to fusion, suggesting other capping proteins can sometimes compensate for HOAP's function. That idea, on the other hand, may apply to the situation in cells deficient for the Mre11 complex.
The mechanism by which ATM and ATR maintain telomere integrity remains largely unclear. Because the conserved kinase domain of both ATM and ATR is essential for their normal telomere function in S. cerevisiae (4, 49), ATM and ATR likely exert their function by protein phosphorylation. We showed that neither Chk1 nor Chk2 was involved in telomere protection in Drosophila, a situation similar to S. pombe (15, 16), which suggests that ATM and ATR modify a different set of proteins at telomeres. As we proposed earlier, ATM and ATR may regulate HOAP's capping activity by directly phosphorylating HOAP (7). However, we recovered no evidence that HOAP is phosphorylated in WT cells (Fig. 8, which is published as supporting information on the PNAS web site). Therefore, ATM and ATR may indirectly regulate HOAP's ability to bind telomeres, perhaps by modulating telomeric structure. Our results support the hypothesis that ATM and ATR have a conserved function at the telomeres that is independent of telomerase (6, 11, 15). Because Drosophila telomeres are not elongated by a telomerase, the fly may be an excellent system for studying the roles of ATM and ATR in telomere protection, uncoupled from their roles in telomere elongation.
Supplementary Material
Acknowledgments
We thank Drs. Michael Lichten, Howard Nash, and Carl Wu for insightful comments on the manuscript; Cassandra Rauser and Germana Colazzo for technical assistance; and the laboratories of Drs. Michael Brodsky (University of Massachusetts, Worcester), James Kennison (National Institutes of Health), Ruth Lehman (New York University, New York), Trudi Schupbach (Princeton University, Princeton, NJ), Tin Tin Su (University of Colorado, Boulder), William Sullivan (University of California, Santa Cruz), and Lori Wallrath for stocks and reagents. X.B., D.S., and Y.S.R. are supported by the intramural research program of the National Cancer Institute. R.B. and R.K. are supported by National Institutes of Health Grant GM059765.
Author contributions: X.B., S.P., R.K., and Y.S.R. designed research; X.B., D.S., L.F., R.B., and Y.S.R. performed research; X.B., S.P., R.K., and Y.S.R. analyzed data; and X.B., S.P., R.K., and Y.S.R. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ATM, ataxia telangiectasia mutated; ATR, ATM and Rad3-related; ATRIP, ATR-interacting protein; Nbs, Nijmegen breakage syndrome; tefu, telomere fusion gene; HOAP, HP1/ORC-associated protein; cav1, caravaggio1; Grp, Grapes; mus304, mutagen-sensitive 304; HP1, heterochromatin protein 1.
References
- 1.d'Adda di Fagagna, F., Teo, S. H. & Jackson, S. P. (2004) Genes Dev. 18, 1781-1799. [DOI] [PubMed] [Google Scholar]
- 2.Abraham, R. T. (2001) Genes Dev. 15, 2177-2196. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh, Y. (2003) Nat. Rev. Cancer 3, 155-168. [DOI] [PubMed] [Google Scholar]
- 4.Greenwell, P. W., Kronmal, S. L., Porter, S. E., Gassenhuber, J., Obermaier, B. & Petes, T. D. (1995) Cell 82, 823-829. [DOI] [PubMed] [Google Scholar]
- 5.Metcalfe, J. A., Parkhill, J., Campbell, L., Stacey, M., Biggs, P., Byrd, P. J. & Taylor, A. M. (1996) Nat. Genet. 13, 350-353. [DOI] [PubMed] [Google Scholar]
- 6.Qi, L., Strong, M. A., Karim, B. O., Armanios, M., Huso, D. L. & Greider, C. W. (2003) Cancer Res. 63, 8188-8196. [PubMed] [Google Scholar]
- 7.Bi, X., Wei, S. D. & Rong, Y. S. (2004) Curr. Biol. 14, 1348-1353. [DOI] [PubMed] [Google Scholar]
- 8.Oikemus, S. R., McGinnis, N., Queiroz-Machado, J., Tukachinsky, H., Takada, S., Sunkel, C. E. & Brodsky, M. H. (2004) Genes Dev. 18, 1850-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva, E., Tiong, S., Pedersen, M., Homola, E., Royou, A., Fasulo, B., Siriaco, G. & Campbell, S. D. (2004) Curr. Biol. 14, 1341-1347. [DOI] [PubMed] [Google Scholar]
- 10.Song, Y.-H., Mirey, G., Betson, M., Haber, D. A. & Settleman, J. (2004) Curr. Biol. 14, 1354-1359. [DOI] [PubMed] [Google Scholar]
- 11.Ritchie, K. B., Mallory, J. C. & Petes, T. D. (1999) Mol. Cell. Biol. 19, 6065-6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craven, R. J., Greenwell, P. W., Dominska, M. & Petes, T. D. (2002) Genetics 161, 493-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mieczkowski, P. A., Mieczkowska, J. O., Dominska, M. & Petes, T. D. (2003) Proc. Natl. Acad. Sci. USA 100, 10854-10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naito, T., Matsuura, A. & Ishikawa, F. (1998) Nat. Genet. 20, 203-206. [DOI] [PubMed] [Google Scholar]
- 15.Matsuura, A., Naito, T. & Ishikawa, F. (1999) Genetics 152, 1501-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura, T. M., Moser, B. A. & Russell, P. (2002) Genetics 161, 1437-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Queiroz-Machado, J., Perdigao, J., Simoes-Carvalho, P., Herrmann, S. & Sunkel, C. E. (2001) Chromosoma 110, 10-23. [DOI] [PubMed] [Google Scholar]
- 18.Ciapponi, L., Cenci, G., Ducau, J., Flores, C., Johnson-Schlitz, D., Gorski, M. M., Engels, W. R. & Gatti, M. (2004) Curr. Biol. 14, 1360-1366. [DOI] [PubMed] [Google Scholar]
- 19.Gorski, M. M., Romeijn, R. J., Eeken, J. C., de Jong, W., van Veen, B. L., Szuhai, K., Mullenders, L. H., Ferro, W. & Pastink, A. (2004) DNA Repair 3, 603-615. [DOI] [PubMed] [Google Scholar]
- 20.Pardue, M. L. & DeBaryshe, P. G. (2003) Annu. Rev. Genet. 37, 485-511. [DOI] [PubMed] [Google Scholar]
- 21.Abad, J. P., De Pablos, B., Osoegawa, K., De Jong, P. J., Martin-Gallardo, A. & Villasante, A. (2004) Mol. Biol. Evol. 21, 1620-1624. [DOI] [PubMed] [Google Scholar]
- 22.Biessmann, H., Carter, S. B. & Mason, J. M. (1990) Proc. Natl. Acad. Sci. USA 87, 1758-1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levis, R. W., Ganesan, R., Houtchens, K., Tolar, L. A. & Sheen, F. M. (1993) Cell 75, 1083-1093. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad, K. & Golic, K. G. (1998) Genetics 148, 775-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gatti, M., Bonaccorsi, S. & Pimpinelli, S. (1999) Methods Cell. Biol. 44, 371-391. [DOI] [PubMed] [Google Scholar]
- 26.Cenci, G., Siriaco, G., Raffa, G. D., Kellum, R. & Gatti, M. (2003) Nat. Cell Biol. 5, 82-84. [DOI] [PubMed] [Google Scholar]
- 27.Agudo, M., Losada, A., Abad, J. P., Pimpinelli, S., Ripoll, P. & Villasante, A. (1999) Nucleic Acids Res. 27, 3318-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shareef, M. M., King, C., Damaj, M., Badagu, R., Huang, D. W. & Kellum, R. (2001) Mol. Biol. Cell. 12, 1671-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hari, K. L., Santerre, A., Sekelsky, J. J., McKim, K. S., Boyd, J. B. & Hawley, R. S. (1995) Cell 82, 815-821. [DOI] [PubMed] [Google Scholar]
- 30.Laurencon, A., Purdy, A., Sekelsky, J., Hawley, R. S. & Su, T. T. (2003) Genetics 164, 589-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gatti, M. (1979) Proc. Natl. Acad. Sci. USA 76, 1377-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mason, J. M., Green, M. M., Shaw, K. E. & Boyd, J. B. (1981) Mutat. Res. 81, 329-343. [DOI] [PubMed] [Google Scholar]
- 33.Brodsky, M. H., Sekelsky, J. J., Tsang, G., Hawley, R. S. & Rubin, G. M. (2000) Genes Dev. 14, 666-678. [PMC free article] [PubMed] [Google Scholar]
- 34.Bi, X., Gong, M., Srikanta, D. & Rong, Y. (2005) Genetics, in press. [DOI] [PMC free article] [PubMed]
- 35.Ritchie, K. B. & Petes, T. D. (2000) Genetics 155, 475-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Digweed, M., Reis, A. & Sperling, K. (1999) BioEssays 21, 649-656. [DOI] [PubMed] [Google Scholar]
- 37.Leicht, B. G. & Bonner, J. J. (1988) Genetics 119, 579-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGowan, C. H. (2002) BioEssays 24, 502-511. [DOI] [PubMed] [Google Scholar]
- 39.Chen, Y. & Sanchez, Y. (2004) DNA Repair 3, 1025-1032. [DOI] [PubMed] [Google Scholar]
- 40.Fogarty, P., Campbell, S. D., Abu-Shumays, R., Phalle, B. S., Yu, K. R., Uy, G. L., Goldberg, M. L. & Sullivan, W. (1997) Curr. Biol. 7, 418-426. [DOI] [PubMed] [Google Scholar]
- 41.Sibon, O. C., Stevenson, V. A. & Theurkauf, W. E. (1997) Nature 388, 93-97. [DOI] [PubMed] [Google Scholar]
- 42.Brodsky, M. H., Weinert, B. T., Tsang, G., Rong, Y. S., McGinnis, N. M., Golic, K. G., Rio, D. C. & Rubin, G. M. (2004) Mol. Cell. Biol. 24, 1219-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cortez, D., Guntuku, S., Qin, J. & Elledge, S. J. (2001) Science 294, 1713-1716. [DOI] [PubMed] [Google Scholar]
- 44.Fanti, L., Giovinazzo, G., Berloco, M. & Pimpinelli, S. (1998) Mol. Cell 2, 527-538. [DOI] [PubMed] [Google Scholar]
- 45.Craven, R. J. & Petes, T. D. (2000) Mol. Cell. Biol. 20, 2378-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cryderman, D. E., Morris, E. J., Biessmann, H., Elgin, S. C. & Wallrath, L. L. (1999) EMBO J. 18, 3724-3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrini, B., Piacentini, L., Fanti, L., Altieri, F., Chichiarelli, S., Berloco, M., Turano, C., Ferraro, A. & Pimpinelli, S. (2004) Mol. Cell 15, 467-476. [DOI] [PubMed] [Google Scholar]
- 48.Savitsky, M., Kravchuk, O., Melnikova, L. & Georgiev, P. (2002) Mol. Cell. Biol. 22, 3204-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takata, H., Kanoh, Y., Gunge, N., Shirahige, K. & Matsuura, A. (2004) Mol. Cell 14, 515-522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}