

Abstract
HIV has evolved many strategies to avoid neutralizing antibody responses, particularly to conserved regions on the external glycoprotein spikes of the virus. Nevertheless, a small number of antibodies have been evolved by the human immune system to recognize conserved parts of the glycoproteins, and therefore, have broadly neutralizing cross-strain activities. These antibodies constitute important tools in the quest to design immunogens that can elicit broadly neutralizing antibodies in humans and hence contribute to an effective HIV vaccine. Crystallographic analyses of the antibodies, in many cases in an antigen-complexed form, have revealed novel and, in some instances, remarkable structural adaptations to attain virus recognition. Antibodies, like HIV, can evolve relatively rapidly through mutation and selection. It seems that the structures of these broadly neutralizing antibodies bear witness to a heroic struggle between two titans of rapid evolution.
Despite emerging in the human population only relatively recently (1), HIV has unleashed a pandemic that has killed ≈20 million people. Currently, ≈40 million individuals are infected (2), most in resource-poor countries, and many or most of these are expected to die of AIDS. The virus has been so successful in part because it has evolved many mechanisms of immune evasion (3–8). The means by which HIV evades the antiviral effects of antibodies are mostly attributable to the characteristics of the envelope spike decorating the viral surface, which is the target for antiviral (neutralizing) antibodies.
The functional envelope spike of HIV is thought to consist of a trimer of heterodimers formed of two glycoproteins, gp120 and gp41. gp120 is a highly glycosylated protein, with approximately half of its mass being N-linked carbohydrates (9). Sequence analyses from different HIV isolates reveal that gp120 can be organized into variable (V1–V5) regions and conserved (C1–C5) regions. The glycoprotein has a receptor site for the CD4 molecule, which defines the tropism of HIV for CD4 T cells, and a second site for binding to chemokine receptors, usually CCR5 or CXCR4. The crystal structure of the conserved core of gp120, lacking most of the variable domains or loops and the C and N termini, and with trimmed carbohydrate chains, revealed that the protein has two domains linked by a bridging sheet (10, 11). This structure provided clues as to some mechanisms of viral evasion from neutralizing antibodies (3). Much of the surface of the core of gp120 is covered by carbohydrate, and most of the rest is expected to be involved in interaction with gp41 or other gp120 units in the trimeric envelope spike. The relatively conserved CD4 binding site is recessed and arguably difficult for antibody to access. The conserved coreceptor site is largely inaccessible on monomeric gp120 unless CD4 binds and triggers conformational changes to expose the site to antibody. Further, a recent structure of the core of simian immunodeficiency virus (SIV) gp120 suggests that the coreceptor site may not be formed in the absence of CD4 (12). The coreceptor site also appears to be hidden on trimeric gp120 in Env spikes. However, CD4 binding in the context of viral infection does not appear to expose the coreceptor site sufficiently for antibody binding. It appears that the binding of CD4 arrayed on a target cell membrane reveals the coreceptor site in a more sterically restricted environment than on monomeric gp120, and this environment is associated with limited antibody access (see below).
The role of gp120 is primarily to attach HIV to its target cells and to bring the virus close to the membrane of these target cells. Once that is achieved, the transmembrane protein gp41 occupies center stage because it now mediates the fusion of viral and target cell membranes to enable the genetic information of the virus to flow in to the target cell (13). gp41 is relatively well conserved, and most of its surface appears to be hidden from antibody recognition in Env spikes before attachment and fusion (14).
The enormous variability of HIV is an effective mechanism for evading neutralizing antibody. The sequence variation in one isolate from a single HIV-infected individual sampled a few years after infection is greater than the global variation of an influenza epidemic strain during a flu season (15). In HIV Env, sequence variability is concentrated in the variable loops (V1–V5), which appear to be a major target for neutralizing antibody responses. Escape from these responses is readily achieved by mutations in the loops that have only minor consequences for viral fitness. Longitudinal studies in humans show that a neutralizing antibody response to the dominant virus does develop but, once a threshold is reached, an escape variant is selected (16, 17). In turn, an antibody response to this variant develops over time that results in the selection of a new escape variant and further repetition of the process. Antibodies that can recognize many different variants of HIV, so-called broadly neutralizing antibodies, are thought to evolve slowly and only in some individuals. It is precisely these type of antibodies that one would like to elicit by vaccination, and so they have attracted special interest.
In fact, a small panel of broadly neutralizing mAbs has been identified (18). These mAbs are important in vaccine design but, unexpectedly, elucidation of their 3D structures has provided remarkable insight into the adaptability of antibodies when challenged by a virus that has developed an extensive armament of anti-antibody features. Here, we review the structures of these broadly neutralizing antibodies that are, importantly, most often in complex with an Env antigen. The approximate locations of the epitopes recognized by the mAbs are mapped onto a model of the Env spike in Fig. 1.
Fig. 1.

The trimeric Env spike of HIV-1/SIV. (a) Electron micrographs of SIV particles showing trimeric Env spikes on the surface (63). This micrograph (courtesy of Ken Roux, Florida State University, Tallahassee) shows trimers on the surface of an SIV particle expressing high levels of Env. HIV-1 Env appears to be less stable than Env of SIV, and there is likely heterogeneity in the number of Env spikes per virion. (b) Model of the Env spike based on the structure of core gp120 (11, 64), with three gp120 monomers shown in gray, pale green, and pale blue. gp41 is shown schematically as three pink tubes. Carbohydrate chains are shown in yellow, and the oligomannose cluster proposed to interact with mAb 2G12 is shown in cyan. The approximate locations of the epitopes for broadly neutralizing mAbs are indicated.
b12: An Antibody That Recognizes the CD4 Binding Site of gp120
The crystal structure of b12 was originally solved as an intact human IgG1 molecule (19). The most prominent feature of the antibody combining site is a heavy-chain complementarity-determining region 3 (CDRH3) that extends directly out from the surface of the antibody like a finger (Fig. 2). The finger structures are seen on two independent Fabs in the crystal asymmetric unit and in different crystal forms, suggesting that the extended finger is not an artifact of crystallization. Computerdocking and mutagenesis studies on gp120 and b12 have been used to argue that the finger probes the recessed CD4 binding site of gp120 (19–21). The CDRH3 is 18 aa long, which is exceptionally long, compared with those typically found in well studied mouse mAbs to protein antigens (average CDRH3 length of about 10 residues). It also is relatively long, compared with the average in anti-protein human antibody reported to be about 13 residues (22). However, human antibodies to viral pathogens seem to have longer-thanaverage CDRH3 regions (22). Indeed, it is an attractive notion that recessed receptor sites on pathogens could be recognized by extended CDR fingers on human antibodies. Clearly, more antibodies in other systems need to be studied before definitive conclusions can be made.
Fig. 2.
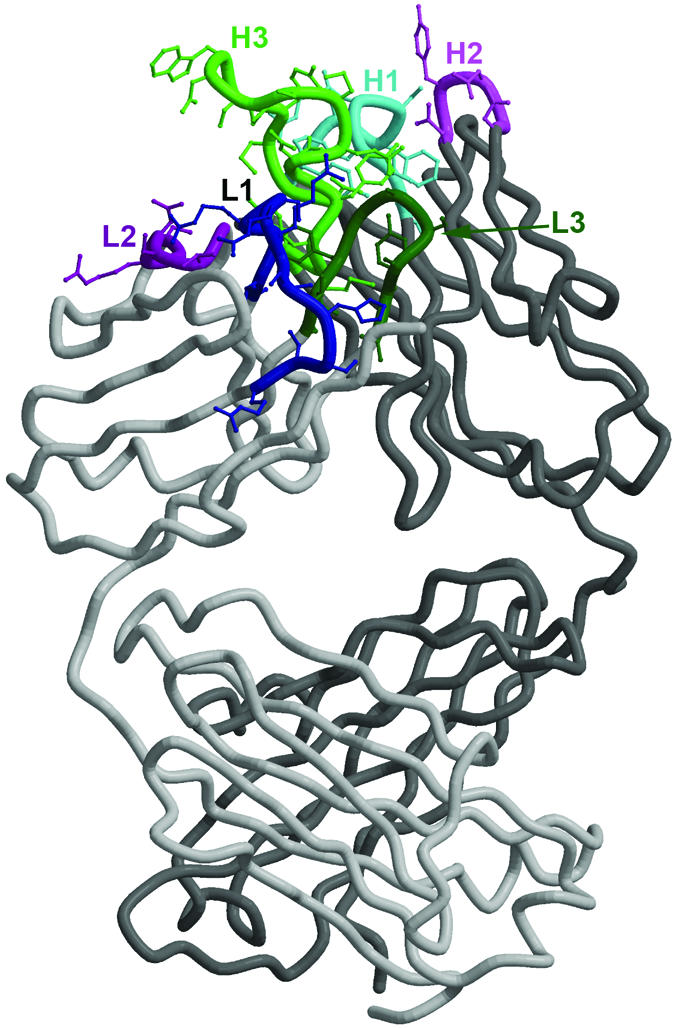
The structure of mAb b12. The structure of Fab b12 as described in ref. 19 is shown with the CDRs presented in colors and labeled as belonging to the heavy (H) or light (L) chain. The long HCDR3 (H3) is clearly visible as an extended finger-like structure. Critical aromatic residues on the CDRs for binding to gp120 (19–21) are prominent.
The CD4 binding site of gp120 is recognized by many other human mAbs that do not broadly neutralize HIV. Current evidence suggests that, although these mAbs are able to recognize monomeric g120, they cannot do so in the context of gp120 organized in Env spikes on the viral surface (23–25). It appears that only b12 of these antibodies described to date is able to gain access to the CD4 binding site on the Env spike, indicating a strong selection pressures on HIV to conceal this crucial and conserved site from antibody recognition. Interestingly, a number of the nonneutralizing mAbs also have long CDRH3 regions, but their structures have not yet been determined (26). Finally, it should be noted that b12 was originally derived from an immune phage library in which heavy and light chains are shuffled. However, the heavy chain, at the very least, is believed to be the one used in vivo according to well rehearsed arguments (26, 27).
Antibodies That Recognize the Coreceptor Binding Site on gp120 [CD4-Induced (CD4i) Antibodies]
During infection, the interaction of Env spikes on the virus with CD4 on target cells makes available the conserved coreceptor binding site on gp120 for interaction with the chemokine receptors CCR5 or CXCR4. Similarly, the coreceptor site on monomeric gp120 is revealed by CD4 ligation, when it is also then recognized by a set of CD4i antibodies. CCR5 is a seven transmembrane-domain protein that interacts with gp120 in part through its extracellular N-terminal peptide (28). This peptide is rich in tyrosines, many of which are sulfated, thereby providing negative charge for interaction with the largely basic coreceptor binding site (29). In a stunning demonstration of convergent evolution, a number of CD4i antibodies have acquired posttranslational modifications in the form of sulfated tyrosines on their CDRH3 regions; these tyrosines, in some cases, are important in gp120 binding (30). Thus, for the first time, a chemical modification of an antibody has been correlated with evolution of antibody specificity.
The crystal structures of five CD4i antibodies (31) have been determined, two of them, 17b (10, 11) and X5 (P. Kwong, personal communication), in complex with gp120. The CD4i antibodies fall into two classes, those with long CDRH3s and those with short CDRH3s. Only for a subgroup of the long CDRH3 antibodies does tyrosine sulfation contribute to antigen binding. Otherwise, these antibodies have a high frequency of acidic residues on the CDRH3 loops. Initially, some excitement arose from studies of the Fab fragment of X5 isolated from a phage library because it showed neutralizing activity against a range of HIV-1 primary isolates (32). However, the intact Ig, IgG1X5, lacked such activity (33). In fact, it seems that a number of CD4i antibodies have neutralizing activity as antibody fragments with an inverse relationship between molecular size and neutralization, i.e., single-chain Fv > Fab > IgG in neutralizing potency. Thus, it appears that during infection, the virus exposes the coreceptor site sufficiently to allow access by CCR5 and antibody fragments but not by the physiologically relevant IgG molecule. These observations are consistent with the expected close proximity of the coreceptor site to the target cell membrane and the orientation and mode of access from the target cell membrane side that would be required for antibody recognition of the coreceptor site (Fig. 3).
Fig. 3.

A model that illustrates accessibility of the coreceptor site region to CD4i antibody fragments after the engagement of the HIV-1 Env spike by CD4 (33). CD4 (yellow) on the target cell membrane engages the CD4bs on gp120 molecules to assemble and expose the coreceptor binding site. In the context of virus–target cell interaction, the site now appears accessible to single-chain Fv (scFv) and Fab fragments of CD4i antibodies but not to intact antibody molecules. In contrast for free monomeric gp120, soluble CD4 binding triggers the binding of intact CD4i antibodies.
447–52D: An Antibody to the V3 Loop of gp120
The V3 loop of gp120, as its name implies, varies in sequence between different isolates of HIV. However, the crown of the loop has a relatively conserved sequence motif GPGR or GPGQ that may be important in binding to the coreceptor (34). The antibody 447–52D (hereafter shortened to 447) neutralizes a range of isolates bearing the GPGR motif (35–37). The crystal structure of 447 complexed with a V3-loop peptide reveals how an antibody can evolve to recognize a motif with a conserved core but with a good deal of flanking variation (38). The antibody interacts specifically with the GPGR crown of the V3 loop, but the flanking sequence is bound by interaction with main-chain atoms (Fig. 4). A parallel is with MHC recognition of peptides (39), where the MHC class I and class II interact with the main-chain atoms of a variable peptide sequence in combination with conserved anchor residues (40). Perhaps in response to selection pressure by antibodies akin to 447, many primary viruses appear to have reduced accessibility of the V3 loop to the point where they are no longer recognized by such antibodies (37, 41).
Fig. 4.

The structure of a V3-loop peptide in the binding site of the antibody 447 (38). The CDRH3 loop (pink, numbered) forms a mixed β-sheet with the V3 loop (blue). GPGR forms the turn in the peptide structure and interacts with the base of the CDRH3 loop. Main-chain interactions dominate the interaction of the peptide with the CDRH3 loop.
2G12: An Antibody That Recognizes Glycans on the Silent Face of gp120
gp120 is ≈50% carbohydrate by weight, with the majority of the glycosylation being N-linked glycans. The structure of the core of gp120 reveals that the N-glycans are arranged on one face of the protein (3, 42). This structural feature was termed the silent face, denoting immunosilence, because glycans are attached by the host glycosylation machinery and are, therefore, expected to be “self” and nonimmunogenic. In essence, the dense carbohydrate shield protects the protein from antibody recognition. Therefore, initial reports suggesting that the neutralizing mAb 2G12 recognized glycans were carefully qualified to include likely polypeptide involvement in binding (43). However, it was subsequently shown that 2G12 does indeed recognize glycans exclusively, and a number of oligomannose candidates were identified (44, 45). This discovery led to a number of key questions. How did 2G12 apparently break tolerance to recognize what is expected to be a self-antigen? How does 2G12 achieve nanomolar binding affinity when typical antibody affinities for glycans are in the micromolar range? How can the oligomannose chains predicted to be involved in 2G12 binding be accommodated within a typical antibody footprint when they are quite spatially separated on the gp120 surface?
These questions were resolved when the crystal structures of 2G12 free and complexed with the disaccharide Manα1–2Man and with the oligosaccharide Man9GlcNAc2 were determined (Fig. 5) (46). A previously uncharacterized structure in which the two Fabs of the IgG assemble into an interlocked VH domain-swapped dimer was revealed. Biochemical, biophysical, and mutagenesis data strongly supported an Fab-dimerized antibody as the predominant form that recognizes gp120. The extraordinary configuration of this antibody provides an extended surface consisting of two classical binding sites (VL–VH) and one previously uncharacterized dimer interface region (VH–VH) for multivalent interaction with a conserved cluster of oligomannoses on gp120. Similar clusters do not appear to be present on any other proteins, because 2G12 is uniquely reactive with high affinity for gp120. Therefore, although individual oligomannose chains are self, the cluster of oligomannoses can be regarded as nonself. The unusually high affinity of 2G12 can be understood in terms of the recognition of a multivalent array of oligomannose residues by an equivalent array of antibody combining sites that match the spacing of the individual sugars within the oligomannose cluster on gp120. Finally, the dimeric configuration of 2G12 is consistent with the relatively large footprint required to cover multiple oligomannose moieties on gp120 as described above.
Fig. 5.

A model of mAb 2G12 Fab2 bound to the HIV-1 Env spike. The heavy chains of 2G12 are shown in dark blue and light red, and the light chains are shown in azure. The domain-swapped structure of 2G12 uncomplexed and complexed with Manα1–2Man and with Man9GlcNAc2 is described in ref. 46. The gp120 oligomannose residues important in 2G12 binding were assigned based on data from a number of different approaches (44, 45). Docking of the structure of 2G12, complexed in the conventional VH–VL combining sites with Man9GlcNAc2, onto gp120 places the GlcNac2 groups very close to N332 and N392 (outer dark red moieties). The Man9GlcNAc2 group attached to N339 (middle dark red) can be readily modeled to interact with the nonconventional VH–VH interface region.
The unusual architecture of 2G12 is achieved by a number of changes to the typical residues found in antibody V domains. These changes include the acquisition of a proline in the bridge between VH and CH1 domains, mutations that destabilize the usual VH–VL interface, and mutations that stabilize the new VH–VH interface and, hence, dimer formation. The changes are achieved by extensive somatic mutation from the closest germ-line antibody VH and VL genes and suggest that 2G12 is the product of many cycles of mutation and selection. At the present time, the frequency of occurrence of domainexchanged antibodies in human responses is unknown, although it is likely to be relatively low. Nevertheless, this experiment of nature provides a powerful insight into how antibodies could be engineered for high-affinity recognition of repeating epitopes, such as those that might be found on cell or microbial surfaces.
2F5 and 4E10: Antibodies That Recognize the Membrane-Proximal External Region (MPER) of gp41
Although most of the surface of gp41 appears to be occluded from antibody binding on Env spikes, a region close to the viral membrane, the MPER (47), has some accessibility to the neutralizing human mAbs 2F5 and 4E10 (47–53). The description of broadly neutralizing mAbs that recognize contiguous epitopes on HIV-1 suggests that the MPER could be a highly promising vaccine target (47). Some evidence has emerged that the epitopes of these two mAbs are accessible, and possibly more so, after CD4 binding to gp120 (54). Both mAbs appear to recognize linear gp41 epitopes in that they bind with relatively high affinity to short peptides corresponding to cognate gp41 sequences, and their neutralizing activity can be effectively inhibited by such peptides. However, all attempts to date to generate neutralizing activity by immunization with sequences from these epitopes, either as peptides or incorporated into proteins, have been unsuccessful. These results have suggested that the antibodies may have evolved to recognize the gp41 peptide sequences in a specialized context, such as their proximity to the viral membrane. The MPER is very rich in tryptophans, which may aid interaction of this region of gp41 with the viral membrane.
The structure of 2F5 was originally described in complex with its short core epitope (55). A recent structure in complex with a longer 17-mer peptide confirms that the core epitope adopts a β-turn and reveals the peptide otherwise to be in a relatively extended conformation (56). Notably, the peptide makes contact with the base of the 22-aa-long CDRH3 region but not with much else of this loop. However, mutagenesis studies have shown that changes in the apex of the loop can reduce antibody binding to peptide and have an even more pronounced adverse effect on neutralization by 2F5 (57). These results suggest not only that the long loop is required for the creation of the peptide binding site, but also that the tip of the loop may be involved in further interactions. A favored current hypothesis is that the loop may interact with the viral membrane, and some support for this view is provided by observations of enhanced 2F5 binding to MPER peptides when attached to a membrane (56, 58).
In contrast to 2F5, a 13-residue peptide containing the core epitope for 4E10 adopts a helical conformation in complex with antibody (59). The key contact residues map to one face of the helix and bind in a highly hydrophobic pocket in the 4E10 antibody combining site. As for 2F5, the CDRH3 is long and not apparently directly involved in peptide binding. The CDRH3 region contains a number of tryptophan residues. The epitope for 4E10 is even closer to the viral membrane than that for 2F5; a small number of residues separate the putative C terminus of the epitope and the transmembrane domain. Therefore, the possible interaction of the tryptophans on the CDRH3 with the viral membrane is an attractive hypothesis (Fig. 6). Again, this notion is supported by enhanced antibody binding to the MPER in the context of a membrane (56, 58). Very recent studies (60) have highlighted cross-reactivity of 2F5 and 4E10 with cardiolipin; this cross-reactivity may reflect the hydrophobic nature of the combining sites of these antibodies rather than any autoimmune origin. In any case, the two antibodies provide a very interesting paradigm for recognition of protein epitopes close to a membrane and should be further studied.
Fig. 6.

A model of mAbs 2F5 (56) and 4E10 (59) Fabs bound to their epitopes close to the virus membrane. The Fabs are shown as a solvent-accessible surface (gray) with their Cα trace from the heavy (blue) and light (cyan) embedded in the translucent structure. The CDRH3s of the antibodies are shown in purple. The MPER model structure (yellow) is based on connecting the actual structures of peptides observed in the corresponding crystal structures of the Fab–peptide complexes. The potential for simultaneous interaction of the antibody molecules with the viral membrane is illustrated.
Concluding Remarks
The study of broadly neutralizing antibodies has revealed some remarkable adaptations by the antibody molecule to counter the molecular trickery of HIV. The virus can evolve very rapidly because of the combination of the relatively poor fidelity of transcription associated with reverse transcriptase and the high rate of virus production in vivo. Antibodies evolve more slowly, but long-term exposure to antigen during persistent HIV infection provides the opportunity for the selection, in some individuals, of antibody variants able to recognize many different viral isolates. Under these conditions, the titans are most evenly matched. In established infection, the very rapid evolution of the virus will ultimately triumph by the selection of variants that are resistant even to the most broadly neutralizing antibodies (61, 62). However, if the antibodies are present before infection (for example, by vaccination) and before virus diversification, then they are likely to be much more effective.
The adaptations of antibody under HIV challenge could be exploited directly in antibody engineering. For instance, the 2G12 framework may allow the development of antibodies with high affinity for other carbohydrates or even other repeating antigens, such as those found on natural pathogens. The 2F5 and 4E10 antibodies have provided clues as to how one should design antibodies that need to bind close to cell membranes. Our primary interest in these broadly neutralizing antibodies is driven by what they can teach us about HIV vaccine design. The novelty of the antibodies could then work against elicitation of similar antibodies by a putative vaccine. Certainly, some of the antibodies show a high degree of somatic mutation from germ-line genes. However, this observation is not, in itself, a barrier to success. Many of the antibody mutations that have accumulated during a natural response to the virus are probably not required for high-affinity antigen binding. An appropriately designed immunogen should then elicit the requisite antibodies without the need for so many mutations. Nevertheless, there is little doubt that the step from antibody–antigen structures to immunogen design is a huge one that will challenge our understanding of molecular structure and immunology to the limits.
Acknowledgments
This work was supported by funding from the National Institutes of Health, the International AIDS Vaccine Initiative, and the Pendleton Trust.
Author contributions: D.R.B., R.L.S., and I.A.W. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CD4i, CD4-induced; MPER, membrane-proximal external region; SIV, simian immunodeficiency virus; CDRH3, heavy-chain complementarity-determining region 3.
References
- 1.Korber, B., Muldoon, M., Theiler, J., Gao, F., Gupta, R., Lapedes, A., Hahn, B. H., Wolinsky, S. & Bhattacharya, T. (2000) Science 288, 1789–1796. [DOI] [PubMed] [Google Scholar]
- 2.AIDS Epidemic Update, December 2004 (Joint United Nations Programme on HIV/AIDS, Geneva), www.unaids.org/wad2004/report.html.
- 3.Wyatt, R. & Sodroski, J. (1998) Science 280, 1884–1888. [DOI] [PubMed] [Google Scholar]
- 4.Hilleman, M. R. (2004) Proc. Natl. Acad. Sci. USA 101, Suppl. 2, 14560–14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans, D. T. & Desrosiers, R. C. (2001) Immunol. Rev. 183, 141–158. [DOI] [PubMed] [Google Scholar]
- 6.Kwong, P. D., Doyle, M. L., Casper, D. J., Cicala, C., Leavitt, S. A., Majeed, S., Steenbeke, T. D., Venturi, M., Chaiken, I., Fung, M., et al. (2002) Nature 420, 678–682. [DOI] [PubMed] [Google Scholar]
- 7.Johnson, W. E. & Desrosiers, R. C. (2002) Annu. Rev. Med. 53, 499–518. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad, A. & Ahmad, R. (2003) Curr. HIV Res. 1, 295–307. [DOI] [PubMed] [Google Scholar]
- 9.Poignard, P., Saphire, E. O., Parren, P. W. & Burton, D. R. (2001) Annu. Rev. Immunol. 19, 253–274. [DOI] [PubMed] [Google Scholar]
- 10.Kwong, P. D., Wyatt, R., Majeed, S., Robinson, J., Sweet, R. W., Sodroski, J. & Hendrickson, W. A. (2000) Structure Fold. Des. 8, 1329–1339. [DOI] [PubMed] [Google Scholar]
- 11.Kwong, P. D., Wyatt, R., Robinson, J., Sweet, R. W., Sodroski, J. & Hendrickson, W. A. (1998) Nature 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, B., Vogan, E. M., Gong, H., Skehel, J. J., Wiley, D. C. & Harrison, S. C. (2005) Nature 433, 834–841. [DOI] [PubMed] [Google Scholar]
- 13.Weiss, C. D. (2003) AIDS Rev. 5, 214–221. [PubMed] [Google Scholar]
- 14.Zwick, M. B., Saphire, E. O. & Burton, D. R. (2004) Nat. Med. 10, 133–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korber, B., Gaschen, B., Yusim, K., Thakallapally, R., Kesmir, C. & Detours, V. (2001) Br. Med. Bull. 58, 19–42. [DOI] [PubMed] [Google Scholar]
- 16.Richman, D. D., Wrin, T., Little, S. J. & Petropoulos, C. J. (2003) Proc. Natl. Acad. Sci. USA 100, 4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei, X., Decker, J. M., Wang, S., Hui, H., Kappes, J. C., Wu, X., Salazar-Gonzalez, J. F., Salazar, M. G., Kilby, J. M., Saag, M. S., et al. (2003) Nature 422, 307–312. [DOI] [PubMed] [Google Scholar]
- 18.Burton, D. R., Desrosiers, R. C., Doms, R. W., Koff, W. C., Kwong, P. D., Moore, J. P., Nabel, G. J., Sodroski, J., Wilson, I. A. & Wyatt, R. T. (2004) Nat. Immunol. 5, 233–236. [DOI] [PubMed] [Google Scholar]
- 19.Saphire, E. O., Parren, P. W., Pantophlet, R., Zwick, M. B., Morris, G. M., Rudd, P. M., Dwek, R. A., Stanfield, R. L., Burton, D. R. & Wilson, I. A. (2001) Science 293, 1155–1159. [DOI] [PubMed] [Google Scholar]
- 20.Pantophlet, R., Ollmann Saphire, E., Poignard, P., Parren, P. W., Wilson, I. A. & Burton, D. R. (2003) J. Virol. 77, 642–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwick, M. B., Parren, P. W., Saphire, E. O., Church, S., Wang, M., Scott, J. K., Dawson, P. E., Wilson, I. A. & Burton, D. R. (2003) J. Virol. 77, 5863–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collis, A. V., Brouwer, A. P. & Martin, A. C. (2003) J. Mol. Biol. 325, 337–354. [DOI] [PubMed] [Google Scholar]
- 23.Roben, P., Moore, J. P., Thali, M., Sodroski, J., Barbas, C. F., III, & Burton, D. R. (1994) J. Virol. 68, 4821–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sattentau, Q. J., Zolla-Pazner, S. & Poignard, P. (1995) Virology 206, 713–717. [DOI] [PubMed] [Google Scholar]
- 25.Fouts, T. R., Binley, J. M., Trkola, A., Robinson, J. E. & Moore, J. P. (1997) J. Virol. 71, 2779–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbas, C. F., III, Collet, T. A., Amberg, W., Roben, P., Binley, J. M., Hoekstra, D., Cababa, D., Jones, T. M., Williamson, R. A., Pilkington, G. R., et al. (1993) J. Mol. Biol. 230, 812–823. [DOI] [PubMed] [Google Scholar]
- 27.Burton, D. R. & Barbas, C. F., III (1994) Adv. Immunol. 57, 191–280. [DOI] [PubMed] [Google Scholar]
- 28.Choe, H., Martin, K. A., Farzan, M., Sodroski, J., Gerard, N. P. & Gerard, C. (1998) Semin. Immunol. 10, 249–257. [DOI] [PubMed] [Google Scholar]
- 29.Farzan, M., Mirzabekov, T., Kolchinsky, P., Wyatt, R., Cayabyab, M., Gerard, N. P., Gerard, C., Sodroski, J. & Choe, H. (1999) Cell 96, 667–676. [DOI] [PubMed] [Google Scholar]
- 30.Choe, H., Li, W., Wright, P. L., Vasilieva, N., Venturi, M., Huang, C. C., Grundner, C., Dorfman, T., Zwick, M. B., Wang, L., et al. (2003) Cell 114, 161–170. [DOI] [PubMed] [Google Scholar]
- 31.Huang, C. C., Venturi, M., Majeed, S., Moore, M. J., Phogat, S., Zhang, M. Y., Dimitrov, D. S., Hendrickson, W. A., Robinson, J., Sodroski, J., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 2706–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moulard, M., Phogat, S. K., Shu, Y., Labrijn, A. F., Xiao, X., Binley, J. M., Zhang, M. Y., Sidorov, I. A., Broder, C. C., Robinson, J., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 6913–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labrijn, A. F., Poignard, P., Raja, A., Zwick, M. B., Delgado, K., Franti, M., Binley, J., Vivona, V., Grundner, C., Huang, C. C., et al. (2003) J. Virol. 77, 10557–10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartley, O., Klasse, P. J., Sattentau, Q. & Moore, J. P. (2005) AIDS Res. Hum. Retroviruses 21, 171–189. [DOI] [PubMed] [Google Scholar]
- 35.Gorny, M. K., Conley, A. J., Karwowska, S., Buchbinder, A., Xu, J. Y., Emini, E. A., Koenig, S. & Zolla-Pazner, S. (1992) J. Virol. 66, 7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zolla-Pazner, S., Zhong, P., Revesz, K., Volsky, B., Williams, C., Nyambi, P. & Gorny, M. K. (2004) AIDS Res. Hum. Retroviruses 20, 1254–1258. [DOI] [PubMed] [Google Scholar]
- 37.Binley, J. M., Wrin, T., Korber, B., Zwick, M. B., Wang, M., Chappey, C., Stiegler, G., Kunert, R., Zolla-Pazner, S., Katinger, H., et al. (2004) J. Virol. 78, 13232–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanfield, R. L., Gorny, M. K., Williams, C., Zolla-Pazner, S. & Wilson, I. A. (2004) Structure 12, 193–204. [DOI] [PubMed] [Google Scholar]
- 39.Matsumura, M., Fremont, D. H., Peterson, P. A. & Wilson, I. A. (1992) Science 257, 927–934. [DOI] [PubMed] [Google Scholar]
- 40.Kwong, P. D. (2004) Structure (London) 12, 173–174. [DOI] [PubMed] [Google Scholar]
- 41.Bou-Habib, D. C., Roderiquez, G., Oravecz, T., Berman, P. W., Lusso, P. & Norcross, M. A. (1994) J. Virol. 68, 6006–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyatt, R., Kwong, P. D., Desjardins, E., Sweet, R. W., Robinson, J., Hendrickson, W. A. & Sodroski, J. G. (1998) Nature 393, 705–711. [DOI] [PubMed] [Google Scholar]
- 43.Trkola, A., Purtscher, M., Muster, T., Ballaun, C., Buchacher, A., Sullivan, N., Srinivasan, K., Sodroski, J., Moore, J. P. & Katinger, H. (1996) J. Virol. 70, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scanlan, C. N., Pantophlet, R., Wormald, M. R., Ollmann Saphire, E., Stanfield, R., Wilson, I. A., Katinger, H., Dwek, R. A., Rudd, P. M. & Burton, D. R. (2002) J. Virol. 76, 7306–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders, R. W., Venturi, M., Schiffner, L., Kalyanaraman, R., Katinger, H., Lloyd, K. O., Kwong, P. D. & Moore, J. P. (2002) J. Virol. 76, 7293–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calarese, D. A., Scanlan, C. N., Zwick, M. B., Deechongkit, S., Mimura, Y., Kunert, R., Zhu, P., Wormald, M. R., Stanfield, R. L., Roux, K. H., et al. (2003) Science 300, 2065–2071. [DOI] [PubMed] [Google Scholar]
- 47.Zwick, M. B., Labrijn, A. F., Wang, M., Spenlehauer, C., Saphire, E. O., Binley, J. M., Moore, J. P., Stiegler, G., Katinger, H., Burton, D. R., et al. (2001) J. Virol. 75, 10892–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buchacher, A., Predl, R., Tauer, C., Purtscher, M., Gruber, G., Heider, R., Steindl, F., Trkola, A., Jungbauer, A. & Katinger, H. (1992) in Vaccines '92: Modern Approaches to New Vaccines Including Prevention of AIDS, eds. Brown, F., Chanock, R., Ginsberg, H. S. & Lerner, R. (Cold Spring Harbor Lab. Press, Woodbury, NY), pp. 191–194.
- 49.Muster, T., Steindl, F., Purtscher, M., Trkola, A., Klima, A., Himmler, G., Ruker, F. & Katinger, H. (1993) J. Virol. 67, 6642–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muster, T., Guinea, R., Trkola, A., Purtscher, M., Klima, A., Steindl, F., Palese, P. & Katinger, H. (1994) J. Virol. 68, 4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purtscher, M., Trkola, A., Gruber, G., Buchacher, A., Predl, R., Steindl, F., Tauer, C., Berger, R., Barrett, N., Jungbauer, A., et al. (1994) AIDS Res. Hum. Retroviruses 10, 1651–1658. [DOI] [PubMed] [Google Scholar]
- 52.Buchacher, A., Predl, R., Strutzenberger, K., Steinfellner, W., Trkola, A., Purtscher, M., Gruber, G., Tauer, C., Steindl, F., Jungbauer, A., et al. (1994) AIDS Res. Hum. Retroviruses 10, 359–369. [DOI] [PubMed] [Google Scholar]
- 53.Stiegler, G., Kunert, R., Purtscher, M., Wolbank, S., Voglauer, R., Steindl, F. & Katinger, H. (2001) AIDS Res. Hum. Retroviruses 17, 1757–1765. [DOI] [PubMed] [Google Scholar]
- 54.Binley, J. M., Cayanan, C. S., Wiley, C., Schulke, N., Olson, W. C. & Burton, D. R. (2003) J. Virol. 77, 5678–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pai, E. F., Klein, M. H., Chong, P. & Pedyczak, A. (2000) World Intellectual Property Organization Patent WO0061618.
- 56.Ofek, G., Tang, M., Sambor, A., Katinger, H., Mascola, J. R., Wyatt, R. & Kwong, P. D. (2004) J. Virol. 78, 10724–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zwick, M. B., Komori, H. K., Stanfield, R. L., Church, S., Wang, M., Parren, P. W., Kunert, R., Katinger, H., Wilson, I. A. & Burton, D. R. (2004) J. Virol. 78, 3155–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grundner, C., Mirzabekov, T., Sodroski, J. & Wyatt, R. (2002) J. Virol. 76, 3511–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cardoso, R. M., Zwick, M. B., Stanfield, R. L., Kunert, R., Binley, J. M., Katinger, H., Burton, D. R. & Wilson, I. A. (2005) Immunity 22, 163–173. [DOI] [PubMed] [Google Scholar]
- 60.Haynes, B. F., Fleming, J., St. Clair, W. E., Katinger, H., Stiegler, G., Kunert, R., Robinson, J., Scearce, R. M., Plonk, K., Staats, H. F., et al. (2005) Science 308, 1906–1908. [DOI] [PubMed] [Google Scholar]
- 61.Poignard, P., Sabbe, R., Picchio, G. R., Wang, M., Gulizia, R. J., Katinger, H., Parren, P. W., Mosier, D. E. & Burton, D. R. (1999) Immunity 10, 431–438. [DOI] [PubMed] [Google Scholar]
- 62.Trkola, A., Kuster, H., Rusert, P., Joos, B., Fischer, M., Leemann, C., Manrique, A., Huber, M., Rehr, M., Oxenius, A., et al. (2005) Nat. Med. 11, 615–622. [DOI] [PubMed] [Google Scholar]
- 63.Zhu, P., Chertova, E., Bess, J., Jr., Lifson, J. D., Arthur, L. O., Liu, J., Taylor, K. A. & Roux, K. H. (2003) Proc. Natl. Acad. Sci. USA 100, 15812–15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwong, P. D., Wyatt, R., Sattentau, Q. J., Sodroski, J. & Hendrickson, W. A. (2000) J. Virol. 74, 1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]