

Abstract
Ammonium transporters (Amts) are integral membrane proteins found in all kingdoms of life that fulfill an essential function in the uptake of reduced nitrogen for biosynthetic purposes. Amt-1 is one of three Amts encoded in the genome of the hyperthermophilic archaeon Archaeoglobus fulgidus. The crystal structure of Amt-1 shows a compact trimer with 11 transmembrane helices per monomer and a central channel for substrate conduction in each monomer, similar to the known crystal structure of AmtB from Escherichia coli. Xenon derivatization has been used to identify apolar regions of Amt-1, emphasizing not only the hydrophobicity of the substrate channel but also the unexpected presence of extensive internal cavities that should be detrimental for protein stability. The substrates ammonium and methylammonium have been used for cocrystallization experiments with Amt-1, but the identification of binding sites that are distinct from water positions is not unambiguous. The well ordered cytoplasmic C terminus of the protein in the Amt-1 structure has allowed for the construction of a docking model between Amt-1 and a homology model for its physiological interaction partner, the PII protein GlnB-1. In this model, GlnB-1 binds tightly to the cytoplasmic face of the transporter, effectively blocking conduction through the three individual substrate channels.
Keywords: archaebacteria, membrane proteins, nitrogen metabolism
Nitrogen is an essential constituent of all biological macromolecules, and its assimilation by living organisms is a prerequisite for all biosynthetic processes. Uptake of nitrogen almost exclusively occurs in its most reduced form, ammonia (NH3). Although the gaseous and hydrophobic NH3 molecule can cross biological membranes, ammonia concentrations in natural environments are generally low, and the basic NH3 exists as the positively charged ammonium ion (NH4) (1, 2). Under such conditions, its uptake into the cell is mediated by a class of ubiquitous membrane proteins, the ammonium transporters (Amts) or methylammonium permeases (3), whose homologs in humans are the Rhesus blood-type proteins (4). Amt proteins form stable trimers (5), and most recently the crystal structure of the AmtB from Escherichia coli has been determined (6, 7). AmtB contains 11 transmembrane helices, of which helices I–IV and V–X show a pseudotwofold symmetry that has also been observed with atomic force microscopy and cryoelectron microscopy (8). The structure indicates that transport occurs through a channel between these symmetry-related halves within each monomer. It has been suggested that NH3 rather than 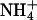 is translocated, and that the binding of substrate does not induce a major conformational change, making AmtB an ammonia channel rather than an ammonium transporter (6). In a second crystal form, however, a conformational change has been observed at the cytoplasmic exit of the transport channel, but it remains inconclusive whether this constitutes a gating mechanism (7).
is translocated, and that the binding of substrate does not induce a major conformational change, making AmtB an ammonia channel rather than an ammonium transporter (6). In a second crystal form, however, a conformational change has been observed at the cytoplasmic exit of the transport channel, but it remains inconclusive whether this constitutes a gating mechanism (7).
In most prokaryotes, Amt proteins are organized in operons paired with GlnK, a protein belonging to the PII family of cytoplasmic regulatory proteins (9). For both the Amt protein of E. coli (10) and Corynebacterium glutamicum (11), evidence for direct interaction with GlnK has recently been presented, and it is presumed that complex formation prevents transport, whereas dissociation of GlnK from the transporter renders it active (11, 12). In most proteobacteria, the association of these two components of the ammonium uptake system is blocked by uridylylation of a conserved tyrosine residue in GlnK (Tyr 51 in E. coli) by the uridylyltransferase GlnD, whereas in cyanobacteria, the same signal is conferred by phosphorylation of a nearby serine (13). Because the function of GlnK is intimately coupled to the regulation of the NifLA as well as the NtrBC systems in nitrogen metabolism (14–17), and AmtB activity was found to be necessary for GlnK deuridylylation (12, 18), Amt proteins have also been described as ammonium sensors (18).
The regulation of nitrogen metabolism in archaea, with the notable exception of the nitrogen-fixing methanogens (19), is less well investigated. Most archaeal genome sequences known to date contain Amt homologs, which in all cases are paired with GlnK-type proteins. Archaea, however, do not use the NtrBC system and neither possess a homolog of GlnD nor do their PII proteins contain a conserved tyrosine or serine residue for covalent modification.
We are studying the ammonium transport system of the hyperthermophilic euryarchaeon Archaeglobus fulgidus. The genome of this organism has been sequenced (20) and found to contain three homologs of Amt proteins, which in the following will be referred to as Amt-1, Amt-2, and Amt-3. All transporters have been purified and crystallized, and we report here the structural analysis of Amt-1 at a resolution of 1.54 Å.
Methods
Crystal Structure Determination. A. fulgidus Amt-1 has been produced and crystallized as described (21). Diffraction data have been collected on beamline BW7A of the European Molecular Biological Laboratory Hamburg outstation at Deutsches Elektronen Synchrotron, Hamburg, Germany. Analysis of the data with xprep (Bruker, Billerica, MA) and cns (22) revealed varying degrees of partial hemihedral twinning. Nevertheless, a molecular replacement solution was found by using molrep (23) with the structure of AmtB from E. coli (Protein Data Bank ID code 1U77) as a search model. This solution contained one monomer per asymmetric unit at a solvent content of 69%, with the crystallographic threefold axis of the R3 cell creating a tightly packed trimer. Interaction of Amt-1 with substrates was examined by cocrystallization with varying concentrations (20–160 mM) of (NH4)2SO4 and 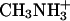 (Table 1).
(Table 1).
Table 1. Data collection and refinement statistics (Protein Data Bank ID codes 2B2F, 2B2H, 2B2I, and 2B2J).
| Dataset | Native | 20 mM (NH4)2SO4 | 80 mM CH3NH3+ | Xenon |
|---|---|---|---|---|
| Unit cell dimensions (hexagonal obverse setting), Å | a = b = 111.31 c = 136.56 | a = b = 111.41 c = 136.64 | a = b = 111.49 c = 136.35 | a = b = 111.46 c = 136.58 |
| Resolution range, Å | 100.00-1.72 (1.75-1.72) | 100.00-1.54 (1.64-1.54) | 100.00-1.85 (1.89-1.85) | 100.00-1.85 (1.95-1.85) |
| Observed reflections | 913,549 | 1,150,467 | 787,055 | 803,954 |
| Independent reflections | 66,323 | 92,775 | 53,457 | 52,810 |
| Completeness, % | 98.4 (90.9) | 98.7 (93.3) | 99.0 (82.0) | 98.6 (91.5) |
| Redundancy | 2.1 (1.3) | 3.9 (2.8) | 2.7 (1.7) | 3.8 (2.5) |
| Mean I/σ (I) | 7.2 (1.5) | 5.8 (1.7) | 13.7 (2.2) | 5.1 (1.6) |
| Rsym | 0.10 (0.52) | 0.09 (0.60) | 0.06 (0.39) | 0.11 (0.58) |
| Twin fraction | 0.113 | 0.238 | 0.441 | 0.087 |
| rms deviation in bond lengths, Å | 0.0064 | 0.0063 | 0.0069 | 0.0069 |
| rms deviation in bond angles, ° | 1.298 | 1.194 | 1.273 | 1.240 |
| Rcryst/Rfree | 0.181/0.200 | 0.154/0.169 | 0.154/0.174 | 0.197/0.221 |
| Mean B factor for protein, Å2 | 24.42 | 28.52 | 23.46 | 36.54 |
| Mean B factor for water, Å2 | 41.34 | 45.06 | 38.62 | 46.45 |
Numbers in brackets represent values for the highest-resolution shells.
Xenon Derivatives. Derivatization of crystals with xenon gas was carried out by using an XCell system (Oxford Cryosystems, Oxford, U.K.). Crystals were pressurized with 15 bar of Xe for 15 min and immediately flash-cooled in liquid nitrogen. Data from these crystals were collected on a Rigaku (Tokyo) rotating anode x-ray generator equipped with a mar345dtb image plate detector (Mar-Research, Hamburg, Germany). At the Cu-Kα x-ray wavelength of 1.54 Å, xenon has a notable anomalous signal equivalent to approximately seven electrons.
Refinement. To account for the observed varying degrees of partial hemihedral twinning, the R3 structures were refined by using cns (22) and shelxl (24, 25), yielding virtually identical results in terms of refinement statistics and quality of the electron density maps. For both programs, an identical set of reflections was chosen for calculation of a free R value. The structures were built by using o (26) and compared and analyzed by using o and pymol (27). For the Xe derivative, an anomalous difference Fourier map was calculated by using programs of the ccp4 suite (23), showing clear maxima at positions where xenon atoms were bound. A xenon atom was assigned only for peaks with an anomalous signal >5 σ. The occupancy of all xenon sites was estimated by matching the B factors of the noble gas atoms to those of the surrounding atoms.
Results
Nitrogen Metabolism in A. fulgidus. Judging from its genome sequence, A. fulgidus neither carries out any of the energy-conserving metabolic pathways from the nitrogen cycle nor can it fix N2 through the nitrogenase system (20). The use of nitrate as an alternative nitrogen source is suggested by the presence of a nitrate reductase (NarGHI, genes AF0176, AF0175, and AF0174) and a siroheme-containing nitrite reductase (NirA, AF0164). With three genes for Amt proteins, however, uptake of ammonia seems to be the preferred route for obtaining nitrogen. The occurrence of several Amts within one genome is frequent, and three copies are also found, for example, in yeast (3), tomato (28), or Methanosarcina acetivorans (29). It is generally assumed that these transporters differ in their substrate affinity (30) and are expressed and regulated according to environmental conditions (31). In the genome of A. fulgidus, the three transporters are found in two separate loci, each one followed directly by a gene for a PII family member, glnB1-3. This is in contrast to the case of E. coli, where the GlnK gene precedes the one for the transporter (9). Amt-1 and GlnB-1 are encoded by genes AF0977 and AF0978, whereas the two other transporters are located in a different region, encoded by AF1746/AF1747 (Amt-2, GlnB-2) and AF1749/AF1750 (Amt-3, GlnB-3) (Fig. 1). The gene AF1748 is transcribed in the same direction and encodes a putative zinc-containing hydrolase. With an amino acid sequence identity of 64.2%, Amt-1 is most closely related to Amt-3, whereas the sequence identity to Amt-2 is only 39.7% for Amt-1 and 40.6% for Amt-3. Amt-1 shows a moderate sequence identity of 41.6% with AmtB from E. coli, which was used as a model for structure solution by molecular replacement (Fig. 1).
Fig. 1.

Amts of A. fulgidus. (Upper) The genome of the archaeon contains three copies of paired genes for an Amt and a protein of the PII family. (Lower) Sequence alignment of A. fulgidus Amt-1–3 and E. coli AmtB. Helices, as derived from the crystal structures, are marked in gray, the numbering corresponds to Amt-1.
Structure of Amt-1. The model of Amt-1 comprises the entire protein sequence from residues 1 through 391, containing 11 transmembrane helices (Fig. 2A). In contrast to both E. coli AmtB and Amt-2 (Fig. 1), the Amt-1 sequence does not contain a cleavable signal peptide and consequently the N terminus of the protein needs to cross the membrane. A common indication for an extracellular N terminus of membrane proteins is the absence of positively charged residues in the sequence before the first helical segment. This is the case for both Amt-1 and -3, such that the topologies of all Amt proteins from A. fulgidus as well as the one of E. coli AmtB are most likely identical. In agreement with this, the electrostatic surface potential of Amt-1 shows a slightly negative charge on the extracellular side of the membrane and a positive charge on the cytoplasmic side, following the positive-inside rule generally observed in membrane protein structures (32). The pseudotwofold symmetry that has been described for E. coli AmtB (6, 7) is retained in Amt-1, with the twofold axis in the plane of the membrane relating helices I–V on one side to helices VI–X on the other. Both halves are then held by the terminal helix XI in a clamp-like fashion (Fig. 2 A). Amt-1 forms a trimer similar to E. coli AmtB (Fig. 2B), with monomer dimensions of ≈40 Å × 47 Å in the plane of the membrane and a height of 57 Å.
Fig. 2.

Structure of A. fulgidus Amt-1. (A) Stereo representation of the Amt-1 monomer. The membrane is indicated in gray, with the extracellular side above and the cytoplasmic side below. The protein chain is colored from blue at the N terminus to red at the C terminus, the 11 transmembrane helices numbered in accordance with Fig. 1. (B) The Amt-1 trimer seen from the extracellular side. (C) B factor representation of Amt-1. The region with the highest B factors is in the loop between helices V and VI, connecting the pseudosymmetry-related halves of the protein.
E. coli AmtB and A. fulgidus Amt-1 superimpose with a rms deviation of 1.38 Å for 307 Cα atoms. Structural conservation is highest within the membrane-spanning helices, although helices II, IX, and XI are significantly shifted (Fig. 3A). The differences are more pronounced in both the extracellular and intracellular loop regions, in particular the one between helices V and VI, which connects the pseudosymmetry-related halves of the protein and shows elevated temperature factors (Fig. 2C).
Fig. 3.

Structural details of Amt-1. (A) Structural comparison of Amt-1 (red) with E. coli AmtB (black). (B) Xenon binding to internal cavities in Amt-1. The surface representation shows the substrate channel and several internal cavities as well as the positions of 9 of a total of 15 Xe atoms located in the structure. The residues depicted as sticks are the same as in Fig. 4.
As in the case of both E. coli structures, Amt-1 has been purified by affinity chromatography with a C-terminally fused hexahistidine linker (21). Although this tag was not cleaved, there is no recognizable electron density visible beyond T391, the terminal residue of the native protein (Fig. 3A). In both E. coli structures, the last 20 aa of the transporter are disordered, and it can be assumed that the addition of the tag caused this disorder in the mesophilic E. coli, whereas the thermostable Amt-1 protein proved more rigid. When examining the interaction with the respective PII protein, these residues are crucial, because they form a significant part of the cytoplasmic face of the transporter and are therefore necessarily involved in any interaction with GlnB-1.
Ammonium-Binding Site. For E. coli AmtB, varying interpretations of data regarding the assignment of ammonia/ammonium electron density peaks have been presented. Only in crystals grown in the presence of ammonium sulfate, Khademi et al. (6) observed three weak electron density peaks in the hydrophobic channel close to H168 and H318 (H157 and H305 in Amt-1, respectively; Fig. 4), that were interpreted to be partially occupied ammonia molecules. Similar electron density peaks were observed by Zheng et al. (7) in crystals grown with, but also without, ammonium and were therefore assigned as water. In all of the substrate cocrystals of Amt-1, no such electron density maxima were found.
Fig. 4.

Ammonium recruitment site and substrate channel. 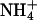 presumably forms a hydrogen bond to the side chain of S208 on the extracellular side of Amt-1 (at the top), stabilized by a π interaction with the side chain of W137. The hydrophobic channel leading to the cytoplasmic side is blocked by F96 and F204, with the latter having significantly elevated B factors, an indication of structural flexibility. The Nδ atoms of two conserved histidine residues, H157 and H305, are in hydrogen-bonding distance and their imidazole rings are almost perfectly coplanar.
presumably forms a hydrogen bond to the side chain of S208 on the extracellular side of Amt-1 (at the top), stabilized by a π interaction with the side chain of W137. The hydrophobic channel leading to the cytoplasmic side is blocked by F96 and F204, with the latter having significantly elevated B factors, an indication of structural flexibility. The Nδ atoms of two conserved histidine residues, H157 and H305, are in hydrogen-bonding distance and their imidazole rings are almost perfectly coplanar.
It has been suggested for E. coli (6, 7) that a recruitment site for ammonium is formed by a hydrogen bond accepting serine residue (S208 in Amt-1) and a nearby tryptophan (W137) that can specifically bind cations through interaction with its π system (Fig. 4). Here, a significant peak was consistently observed in Amt-1, but its exact position and occupancy were not conclusively related to the presence or absence of ammonia in the crystals. Thus, although this site may be crucial for the discrimination from the similarly sized potassium ion, our results indicate that it may as well be occupied by water.
Water and ammonia have an identical number of electrons and are therefore indistinguishable in an x-ray diffraction experiment. In the case of the alternative substrate methylammonium, however, the electron density of an elongated (1.50-Å) 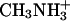 molecule should be distinct from the spherical peak of a water or
molecule should be distinct from the spherical peak of a water or 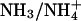 molecule. Nevertheless, although Khademi et al. (6) observed an electron density peak at this position in AmtB that was assigned as methylammonium (Protein Data Bank ID code 1U7C), no equivalent electron density was found in the Amt-1 structure. Even at high resolution, the distinction between a disordered water molecule that can partially occupy two neighboring positions and a diatomic molecule is very difficult, unless the binding of the latter occurs with high affinity in a defined state. Thus, this does not seem to act as a high-affinity substrate-binding site, and it is unlikely that it serves as a selectivity filter in Amt-1.
molecule. Nevertheless, although Khademi et al. (6) observed an electron density peak at this position in AmtB that was assigned as methylammonium (Protein Data Bank ID code 1U7C), no equivalent electron density was found in the Amt-1 structure. Even at high resolution, the distinction between a disordered water molecule that can partially occupy two neighboring positions and a diatomic molecule is very difficult, unless the binding of the latter occurs with high affinity in a defined state. Thus, this does not seem to act as a high-affinity substrate-binding site, and it is unlikely that it serves as a selectivity filter in Amt-1.
Immediately below this site, the transport pathway is blocked by the side chains of F96 and F204, both of which presumably have to change position to allow for the passage of substrate. In Amt-1, F204 has significantly elevated B factors compared with its surroundings, indicative of an increased mobility within the structure. Below these two aromatic residues, a hydrophobic transport channel opens, lined by the side chains of H157 and H305 and the indole moiety of W201. In contrast to the structures of AmtB, the two histidine imidazole rings are almost coplanar, which should increase the stability of the 3.0-Å hydrogen bond between their Nδ atoms (Fig. 4).
Xenon Derivatives. The noble gas xenon shows higher solubility in apolar than in polar solvents. When used for phase determination in x-ray crystallography, it is commonly found to bind to hydrophobic regions of a protein (33) and can thus be used to map such regions in a structure (34). Xenon derivatives of Amt-1 have been prepared, and 15 binding sites per monomer were found at a resolution of 1.85 Å (Fig. 3B). The overall structure of the protein did not show significant changes upon heavy atom binding. Four strong sites were found on the central threefold axis of the Amt-1 trimer, representing xenon atoms that could enter the highly hydrophobic central cavity from the cytoplasmic side and diffuse almost to the extracellular side, where their passage was blocked only by the very N-terminal residues, M1 and S2, of all three monomers. Only a single strong anomalous signal was observed in the entire region facing the membrane. In the crystal, this region is covered by disordered detergent molecules that might hinder access of Xe.
Three strong and one weak xenon site were found directly in the putative transport channel, leading from the constriction at F96 and F204 to the cytoplasmic side. Two of these sites are close to the conserved histidine residues H157 and H305, occupying positions nearly identical to two of the three weak peaks observed in the E. coli AmtB structures and assigned as ammonia (6) or water (7). The other two positions lie in a side arm of the channel that leads to an interface between two monomers of the trimer and is blocked by amino acid side chains in the E. coli structures (Fig. 3B).
Two further Xe sites have been found to occupy a large hydrophobic pocket in the structure, situated on the cytoplasmic side of the membrane-spanning part of the transporter, between helices II, III, IV, and XI. This pocket is present in all Amt-1 structures analyzed and is unoccupied in the absence of Xe. It also exists in the structures of E. coli AmtB at the same position, although a cavity of this extent should be unfavorable for protein stability. Large internal cavities are not commonly observed in typical passive transporters such as the potassium (35) or chloride channels (36), but they are found in active transporters such as the glycerol-3-phosphate transporter GlpT from E. coli (37), where they are needed to accommodate for conformational changes of the protein. In the Amt proteins, this cavity could allow for a major conformational shift of helix V, given that the beginning of this helix, with the highly conserved glycines G152 and G153, serves as a pivot point. Both glycine amide nitrogens are hydrogen-bonded to the side chain of the conserved D149 (D160 in E. coli AmtB), a residue essential for the transport of methylamine (18) but not directly involved in substrate binding at the putative recruitment site. A movement of helix V would cause a shift of its end, made possible by the long flexible loop connecting helices V and VI. This could account for the elevated B factors observed in this loop even in the thermostable A. fulgidus protein. A further remarkable point is the high abundance of glycine residues in the sequence of Amt-1. Glycine accounts for 12.9% of all residues and are known to increase the flexibility of a protein chain; therefore, they should not be favored in a rigid channel.
Transport Mechanism. The precise nature of the substrate of Amt proteins, ammonia vs. ammonium, is still under debate. When the first crystal structure of E. coli AmtB was published, the authors (6) proposed a passive channel rather than an active transporter, based on the finding that no conformational change was observed in the presence or absence of NH3. Upon substrate uptake by AmtB reconstituted into proteoliposomes, the internal pH increased, consistent with an uptake of protons by NH3 after its translocation over the membrane. These results were in agreement with earlier work suggesting AmtB to be the first example of a gas channel (38, 39). Findings conflicting with this hypothesis were obtained in voltage–clamp experiments with the tomato paralog LeAMT1;1, where transport currents increased with voltage and external ammonium concentration, indicating the translocation of a charged species (28), and also in C. glutamicum, where a dependence on the membrane potential was found (40).
Our findings do confirm the hydrophobic nature of the substrate channel by binding of xenon, but in our understanding, the uniport of NH3 poses a fundamental bioenergetic problem: The translocation of NH3 requires deprotonation of 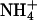 on the extracellular side and reprotonation in the cytoplasm. Effectively, this would result not in passive transport of NH3 but rather in a net antiport of
on the extracellular side and reprotonation in the cytoplasm. Effectively, this would result not in passive transport of NH3 but rather in a net antiport of 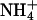 vs. H+. In organisms using a proton gradient for energy conservation, the direction of proton transport would be against this gradient and, as a consequence, uptake of ammonium would lead to the generation of proton motive force. In fact,
vs. H+. In organisms using a proton gradient for energy conservation, the direction of proton transport would be against this gradient and, as a consequence, uptake of ammonium would lead to the generation of proton motive force. In fact, 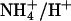 antiport is electroneutral and would therefore not depend on the membrane potential, but it would occur only if the concentration difference of ammonium across the membrane is larger than the one for protons, i.e.,
antiport is electroneutral and would therefore not depend on the membrane potential, but it would occur only if the concentration difference of ammonium across the membrane is larger than the one for protons, i.e., 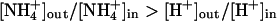 . For a typical bacterial membrane with ΔpH ≈ 1 and an intracellular ammonium concentration of 0.25 mM (41), this means that ammonium could be taken up only if its extracellular concentration was above 2.5 mM. However, the action of Amt proteins seems to be most required at concentrations below 1 mM, when passive diffusion cannot contribute significantly to ammonium uptake (1), and for Klebsiella pneumoniae, it has been shown that its Amt protein becomes derepressed only at
. For a typical bacterial membrane with ΔpH ≈ 1 and an intracellular ammonium concentration of 0.25 mM (41), this means that ammonium could be taken up only if its extracellular concentration was above 2.5 mM. However, the action of Amt proteins seems to be most required at concentrations below 1 mM, when passive diffusion cannot contribute significantly to ammonium uptake (1), and for Klebsiella pneumoniae, it has been shown that its Amt protein becomes derepressed only at 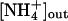 below 2.5 mM (41).
below 2.5 mM (41).
This apparent contradiction might be resolved in a mechanism that does imply a transport of NH3 linked to a parallel transport of protons in the same direction. This would correspond to a net uniport of 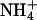 , but it would include a deprotonation of the ammonium ion on the extracellular side and two separate but mechanistically coupled pathways, one for protons and the other for the hydrophobic NH3 molecule. Driven by the proton motive force, this would constitute a secondary active transport and explain the electrogenicity observed in LeAMT1;1 (28) as well as the apparently passive transport of NH3 that Khademi et al. (6) found in the absence of a proton gradient. No clear proton pathway could be discerned within AmtB (6), but the identification of such a structural feature may not be straightforward, also because a conformational change of the protein during the transport cycle cannot presently be ruled out. In a cotransport of NH3 and H+, the membrane potential would favor the import of protons, and the gradients of both substrates would work in the same direction, such that uptake would be possible for [NH3]in/[NH3]out < [H+]out/[H+]in ·exp(–(F/RT)·Δψ). With standard values of ΔpH ≈ 1 and Δψ = –100 mV, this would allow for a nearly 500-fold accumulation of ammonium. At an
, but it would include a deprotonation of the ammonium ion on the extracellular side and two separate but mechanistically coupled pathways, one for protons and the other for the hydrophobic NH3 molecule. Driven by the proton motive force, this would constitute a secondary active transport and explain the electrogenicity observed in LeAMT1;1 (28) as well as the apparently passive transport of NH3 that Khademi et al. (6) found in the absence of a proton gradient. No clear proton pathway could be discerned within AmtB (6), but the identification of such a structural feature may not be straightforward, also because a conformational change of the protein during the transport cycle cannot presently be ruled out. In a cotransport of NH3 and H+, the membrane potential would favor the import of protons, and the gradients of both substrates would work in the same direction, such that uptake would be possible for [NH3]in/[NH3]out < [H+]out/[H+]in ·exp(–(F/RT)·Δψ). With standard values of ΔpH ≈ 1 and Δψ = –100 mV, this would allow for a nearly 500-fold accumulation of ammonium. At an 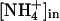 of 0.25 mM (41), Amt proteins could function with
of 0.25 mM (41), Amt proteins could function with 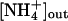 as low as 0.5 mM. This discussion is necessarily prone to the uncertainties in the numbers applied, and it also does not account for passive diffusion of ammonia, but it does highlight the fundamental problem related to the action of an NH3 channel in the presence of a proton gradient. Further functional studies will be required to clarify these questions.
as low as 0.5 mM. This discussion is necessarily prone to the uncertainties in the numbers applied, and it also does not account for passive diffusion of ammonia, but it does highlight the fundamental problem related to the action of an NH3 channel in the presence of a proton gradient. Further functional studies will be required to clarify these questions.
Interaction with the PII Protein GlnB-1. In the A. fulgidus genome, the amt-1 gene is followed by glnB-1, suggesting a direct interaction of the gene products similar to the one postulated for E. coli (10, 18). To further investigate this possibility, a homology model of A. fulgidus GlnB-1 has been constructed, based on the 3D structure of E. coli GlnB (42). GlnB is a compact trimer of 109 residues per monomer, and its most prominent feature is a protruding loop, the T loop. It comprises residues 46–55 and harbors a tyrosine residue that in E. coli can be reversibly uridylylated to exclude association with AmtB when cellular nitrogen levels are low (10, 43) but is absent in all A. fulgidus GlnB proteins. This structure has been chosen rather than the highly similar one of GlnK (44), because in the latter, the crucial T loop is not ordered. E. coli GlnB and GlnB-1 align with a sequence identity of 54% and without gaps or insertions, such that the sequence of GlnB-1 could be modeled onto the GlnB structure, followed by geometry optimization using cns (22).
Because of its cellular localization, GlnB-1 will interact with the cytoplasmic face of Amt-1, a significant part of which is formed by the 20 C-terminal residues that were disordered in E. coli AmtB but are clearly defined in the structure of Amt-1. The proteins were docked manually, followed by geometry optimization with cns. The negative electrostatic surface potential of GlnB-1 matches the slightly positively charged cytoplasmic face of Amt-1, but the most striking aspect of this model is the obvious surface complementarity of both proteins (Fig. 5). Without significant clashes or rearrangements, GlnB-1 and Amt-1 can be docked to form a tight complex with the T loops inserted deeply into the cytoplasmic exit channels of the Amt-1 monomers. This would undoubtedly result in an inactive transporter, simply through sterical blocking of the transport pathway. Dissociation of the complex is presumably regulated through binding of glutamine, ATP and 2-oxoglutarate to the PII protein (45). In the structure of E. coli GlnB, ATP was bound in a large pocket between the GlnB monomers (42), which in the model is freely accessible from the cytoplasm.
Fig. 5.

Docking model for the interaction of Amt-1 with GlnB-1. (A) Side view. GlnB-1 binds tightly to the transporter, with the protruding T loops inserting deeply into the substrate channels. (B) View from the cytoplasmic side. Both GlnB-1 and Amt-1 are stable trimers, and in the docked state, their threefold axes coincide.
Conclusion
Amt proteins are stable trimers with separate transport pathways in each monomer. According to amino acid sequence comparisons, the presumably functional residues and their arrangement are conserved throughout the family. However, substrate affinity and specificity are thought to vary substantially (30) and at present diverging mechanistic models exist.
The first archaeal Amt, Amt-1, adds new aspects to the picture: Of the two conserved phenylalanines that block the transport pathway, F204 consistently shows elevated B factors, whereas F96 does not, indicating that the former has an increased flexibility in the structure. It is situated close to H157, which together with H305 forms the characteristic histidine pair around the twofold pseudosymmetry axis. In contrast to E. coli AmtB, the imidazoles of these residues are practically coplanar in Amt-1, presumably resulting in a more stable hydrogen bond between their Nδ atoms. No clear discrimination between the binding of water, 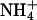 , or methylammonium was possible at the putative substrate recruitment site, but Amt proteins have been found not to conduct water (6). Although this does not speak against this site as a recruitment point for substrate, it does indicate that a further selectivity mechanism must exist.
, or methylammonium was possible at the putative substrate recruitment site, but Amt proteins have been found not to conduct water (6). Although this does not speak against this site as a recruitment point for substrate, it does indicate that a further selectivity mechanism must exist.
Xenon has been used to locate hydrophobic cavities within Amt-1, and its strong binding confirms the hydrophobic nature of the channel leading from the cytoplasmic side up to residue F204. This is consistent with the proposal of a translocation of ammonia rather than the charged species ammonium (6, 7), but because of its high pKa, a unidirectional transport of NH3 would necessarily result in a net antiport of H+ vs. 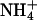 , with protons moving against the their gradient across the cytoplasmic membrane. Further strong xenon sites mark a conserved internal cavity, a possible indication for structural flexibility in Amt-1.
, with protons moving against the their gradient across the cytoplasmic membrane. Further strong xenon sites mark a conserved internal cavity, a possible indication for structural flexibility in Amt-1.
A model has been presented for the interaction of Amt-1 with the PII protein GlnB-1. The proposed docking mode is straight-forward, and the resulting complex is tight and presumably stable, with GlnB-1 effectively blocking ammonium transport by Amt-1.
Acknowledgments
We thank Ruth Schmitz-Streit for stimulating discussions. Diffraction data were collected on beamline BW7A at the European Molecular Biology Laboratory Hamburg outstation at Deutsches Elektronen Synchrotron. This work was supported by a Marie Curie Intra-European Fellowship within the 6th European Community Framework Program (to S.L.A.A.).
Author contributions: S.L.A.A. and O.E. designed research; S.L.A.A., A.D., and O.E. performed research; R.F. contributed new reagents/analytic tools; S.L.A.A. and O.E. analyzed data; and S.L.A.A. and O.E. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: Amt, ammonium transporter.
Data deposition: The structural data have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 2B2F (Native), 2B2H (ammonium sulfate), 2B2I (methylammonium), and 2B2J (xenon)].
References
- 1.Kleiner, D. (1985) FEMS Microbiol. Rev. 32, 87–100. [Google Scholar]
- 2.Lide, D. R. (2001) Handbook of Chemistry and Physics (Chemical Rubber Publishing, Cleveland).
- 3.Marini, A. M., Vissers, S., Urrestarazu, A. & Andre, B. (1994) EMBO J. 13, 3456–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludewig, U., von Wirén, N., Rentsch, D. & Frommer, W. B. (2001) Genome Biol. 2, 1010.1–1010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blakey, D., Leech, A., Thomas, G. H., Coutts, G., Findlay, K. & Merrick, M. (2002) Biochem. J. 364, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khademi, S., O'Connell, J., Remis, J., Robles-Colmenares, Y., Miericke, L. J. W. & Stroud, R. M. (2004) Science 305, 1587–1594. [DOI] [PubMed] [Google Scholar]
- 7.Zheng, L., Kostrewa, D., Berneche, S., Winkler, F. K. & Li, X. D. (2004) Proc. Natl. Acad. Sci. USA 101, 17090–17095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conroy, M. J., Jamieson, S. J., Blakey, D., Kaufmann, T., Engel, A., Fotiadis, D., Merrick, M. & Bullough, P. A. (2004) EMBO Rep. 5, 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas, G., Coutts, G. & Merrick, M. (2000) Trends Genet. 16, 11–14. [DOI] [PubMed] [Google Scholar]
- 10.Javelle, A. & Merrick, M. (2005) Biochem. Soc. Trans. 33, 170–172. [DOI] [PubMed] [Google Scholar]
- 11.Strosser, J., Lüdke, A., Schaffer, S., Kramer, R. & Burkovski, A. (2004) Mol. Microbiol. 54, 132–147. [DOI] [PubMed] [Google Scholar]
- 12.Coutts, G., Thomas, G., Blakey, D. & Merrick, M. (2002) EMBO J. 21, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forchhammer, K. (2003) Symbiosis 35, 101–115. [Google Scholar]
- 14.Blauwkamp, T. A. & Ninfa, A. J. (2003) Mol. Microbiol. 48, 1017–1028. [DOI] [PubMed] [Google Scholar]
- 15.Jack, R., De Zamaroczy, M. & Merrick, M. (1999) J. Bacteriol. 181, 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stips, J., Thummer, R., Neumann, M. & Schmitz, R. A. (2004) Eur. J. Biochem. 271, 3379–3388. [DOI] [PubMed] [Google Scholar]
- 17.Zhang, Y. P., Pohlmann, E. L. & Roberts, G. P. (2005) J. Bacteriol. 187, 1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Javelle, A., Severi, E., Thornton, J. & Merrick, M. (2004) J. Biol. Chem. 279, 8530–8538. [DOI] [PubMed] [Google Scholar]
- 19.Ehlers, C., Grabbe, R., Veit, K. & Schmitz, R. A. (2002) J. Bacteriol. 184, 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klenk, H. P., Clayton, R. A., Tomb, J. F., White, O., Nelson, K. E., Ketchum, K. A., Dodson, R. J., Gwinn, M., Hickey, E. K., Peterson, J. D., et al. (1997) Nature 390, 364–370. [DOI] [PubMed] [Google Scholar]
- 21.Andrade, S. L. A., Dickmanns, A., Ficner, R. & Einsle, O. (2005) Acta Crystallogr. F61, 861–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brünger, A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grosse Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 24.Herbst-Irmer, R. & Sheldrick, G. M. (1998) Acta Crystallogr. B 54, 443–449. [Google Scholar]
- 25.Sheldrick, G. M. & Schneider, T. R. (1997) Methods Enzymol. 277, 319–343. [PubMed] [Google Scholar]
- 26.Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjelgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 27.DeLano, W. L. (2002) The PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA).
- 28.Ludewig, U., von Wirén, N. & Frommer, W. B. (2002) J. Biol. Chem. 277, 13548–13555. [DOI] [PubMed] [Google Scholar]
- 29.Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., Calvo, S., Engels, R., Smirnov, S., Atnoor, D., et al. (2002) Genome Res. 12, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montesinos, M. L., Muro-Pastor, A. M., Herrero, A. & Flores, E. (1998) J. Biol. Chem. 273, 31463–31470. [DOI] [PubMed] [Google Scholar]
- 31.Lauter, F. R., Ninnemann, O., Bucher, M., Riesmeier, J. W. & Frommer, W. B. (1996) Proc. Natl. Acad. Sci. USA 93, 8139–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Heijne, G. & Gavel, Y. (1988) Eur. J. Biochem. 174, 671–678. [DOI] [PubMed] [Google Scholar]
- 33.Soltis, S. M., Stowell, M. H., Wiener, M. C., Phillips, G. N. & Rees, D. C. (1997) J. Appl. Crystallogr. 30, 190–194. [Google Scholar]
- 34.Prangé, T., Schiltz, M., Pernot, L., Colloc'h, N., Longhi, S., Bourguet, W. & Fourme, R. (1998) Proteins Struct. Funct. Genet. 30, 61–73. [PubMed] [Google Scholar]
- 35.Doyle, D. A., Cabral, J. M., Pfuetzner, R. A., Kuo, A. L., Gulbis, J. M., Cohen, S. L., Chait, B. T. & MacKinnon, R. (1998) Science 280, 69–77. [DOI] [PubMed] [Google Scholar]
- 36.Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 415, 287–294. [DOI] [PubMed] [Google Scholar]
- 37.Huang, C. H. & Liu, P. Z. (2001) Blood Cells Mol. Dis. 27, 90–101. [DOI] [PubMed] [Google Scholar]
- 38.Soupene, E., Chu, T., Corbin, R. W., Hunt, D. F. & Kustu, S. (2002) J. Bacteriol. 184, 3396–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soupene, E., Lee, H. & Kustu, S. (2002) Proc. Natl. Acad. Sci. USA 99, 3926–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siewe, R. M., Weil, B., Burkovski, A., Eikmanns, B. J., Eikmanns, M. & Krämer, R. (1996) J. Biol. Chem. 271, 5398–5403. [DOI] [PubMed] [Google Scholar]
- 41.Kleiner, D. (1985) FEBS Lett. 187, 237–239. [DOI] [PubMed] [Google Scholar]
- 42.Xu, Y. B., Carr, P. D., Huber, T., Vasudevan, S. G. & Ollis, D. L. (2001) Eur. J. Biochem. 268, 2028–2037. [DOI] [PubMed] [Google Scholar]
- 43.Son, H. S. & Rhee, S. G. (1987) J. Biol. Chem. 262, 8690–8695. [PubMed] [Google Scholar]
- 44.Xu, Y. B., Cheah, E., Carr, P. D., van Heeswijk, W. C., Westerhoff, H. V., Vasudevan, S. G. & Ollis, D. L. (1998) J. Mol. Biol. 282, 149–165. [DOI] [PubMed] [Google Scholar]
- 45.Arcondéguy, T., Jack, R. & Merrick, M. (2001) Microbiol. Mol. Biol. Rev. 65, 80–105. [DOI] [PMC free article] [PubMed] [Google Scholar]