

Abstract
Cofactor F420 is a 5′-deazaflavin derivative first discovered in methanogenic archaea but later found also to be present in some bacteria. As a coenzyme, it is involved in hydride transfer reactions and as a prosthetic group in the DNA photolyase reaction. We report here for the first time on the crystal structure of an F420-dependent oxidoreductase bound with F420. The structure of F420H2:NADP+ oxidoreductase resolved to 1.65 Å contains two domains: an N-terminal domain characteristic of a dinucleotide-binding Rossmann fold and a smaller C-terminal domain. The nicotinamide and the deazaflavin part of the two coenzymes are bound in the cleft between the domains such that the Si-faces of both face each other at a distance of 3.1 Å, which is optimal for hydride transfer. Comparison of the structures bound with and without substrates reveals that of the two substrates NADP has to bind first, the binding being associated with an induced fit.
Keywords: crystal structure/drug design/F420/hydride transfer/oxidoreductase
Introduction
F420 is a flavin analogue that can exist in an oxidized and a reduced form, oxidoreduction taking place by hydride transfer from and to C5 of the 5′-deazaflavin (Walsh, 1985), as exemplified for the F420H2:NADP+ oxidoreductase reaction in Figure 1 (DiMarco et al., 1990). It is found in methanogenic archaea, sulfate-reducing archaea, haloarchaea, mycobacteria, Streptomyces, cyanobacteria and some eukaryotes, where it functions as a coenzyme of oxidoreductases and/or as a prosthetic group of DNA photolyases (Klein et al., 1996). F420 appears to be absent in most bacteria, plants and animals. In Mycobacterium tuberculosis, F420-dependent enzymes are involved in the metabolic activation of some antitubercular compounds (Stover et al., 2000).

Fig. 1. Reaction catalysed by F420H2:NADP+ oxidoreductase. The Re-faces of the cofactors are shown. The amide group of NADP is shown in the trans conformation.
Eight F420-dependent oxidoreductases are presently known: F420-dependent N5,N10-methylenetetrahydromethanopterin reductase (Kunow et al., 1993a), F420-dependent alcohol dehydrogenase (Klein et al., 1996), F420-dependent glucose-6-phosphate dehydrogenase (Klein et al., 1996), F420-reducing hydrogenase (Teshima et al., 1985; Yamazaki et al., 1985; Fox et al., 1987), F420-dependent formate dehydrogenase (Schauer et al., 1986), F420H2 dehydrogenase complex (Kunow et al., 1993a; Baumer et al., 2000), F420-dependent N5,N10-methylenetetrahydromethanopterin dehydrogenase (Kunow et al., 1993a; Klein and Thauer, 1995) and F420H2:NADP+ oxidoreductase (Yamazaki et al., 1980; Kunow et al., 1993b; Berk and Thauer, 1998; Elias et al., 2000). F420-dependent methylenetetrahydromethanopterin reductase, F420-dependent alcohol dehydrogenase and F420-dependent glucose-6-phosphate dehydrogenase are homologous enzymes, as demonstrated by sequence similarity (Shima et al., 2000). The F420-binding subunits of F420-reducing hydrogenase, F420-dependent formate dehydrogenase and the F420H2 dehydrogenase complex show sequence similarity. The other F420-dependent enzymes do not appear to be phylogenetically related. They show neither sequence similarity nor a common sequence motif for F420 binding. However, all of the F420-dependent enzymes have in common that they are Si-face specific with respect to C5 of the deazaflavin (Klein et al., 1996). The uniform stereospecificity is surprising because flavoproteins are known that catalyse the reduction of synthetic 8-hydroxy-5-deaza-FAD, some Si-face specific and others Re-face specific (Sumner and Matthews, 1992).
However, of the eight F420-dependent enzymes, only the structure of methylenetetrahydromethanopterin reductase has been resolved, yet without F420 bound (Shima et al., 2000). Here, we present the structure of F420H2:NADP+ oxidoreductase in complex with and without its two substrates. The enzyme is a homodimer of a 24 kDa polypeptide and catalyses the reversible reduction of NADP+ by F420H2. In this reaction the proS hydrogen at C5 of F420 is transferred into the proS position at C4 of NADPH (Yamazaki et al., 1980; Kunow et al., 1993b) (Figure 1).
Results and discussion
Overall structure
The structure of F420H2:NADP+ oxidoreductase (Fno) from the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus, heterologously overproduced in Escherichia coli, was initially established at 2.5 Å resolution with the multiple anomalous dispersion (MAD) phasing method (Hendrickson and Ogata, 1997) applied to substrate-free protein crystals (crystal form 1) labelled with selenomethionine (for details see Materials and methods). The structural data presented are based on a substrate-free state (crystal form 2) at 1.8 Å resolution, and on a state complexed with NADP+ and F420 (crystal form 3) at 1.65 Å resolution. Note that the complex is not the Michaelis–Menten complex, since it contains both coenzymes in the oxidized state rather than one in the reduced and the other in the oxidized state. The quality of the electron density map of the latter state is visualized in Figure 2. A superposition of the monomers of the substrate-free and -bound states gives a root mean square deviation (r.m.s.d.) between the Cα atoms of 0.5 Å, which is higher than the r.m.s.d. of 0.41 and 0.39 Å, respectively, between the two monomers in the asymmetric units (GA-FIT; May and Johnson, 1994). Details of the differences between the two enzyme states are discussed below.


Fig. 2. Simulated annealing 2Fo – Fc omit electron density maps of Fno with bound substrates at 1.65 Å resolution contoured at 1.5σ for (A) NADP+ and (B) F420. Contacts are indicated schematically; hydrogen bonds are illustrated as broken lines and hydrophobic contacts as dotted lines. In (A) only solvent molecules conserved in both subunits are shown, which are all bridges to the protein except for O291, O305 and O306. The solvent molecules around F420 are not equivalent in the two subunits. NADP is linked to the polypeptide chain by 12 hydrogen bonds, F420 only by one.
The homodimeric Fno reveals a brick-like shape (Figure 3A), with dimensions of 81 × 44 × 42 Å3. The interface between the monomers is highly hydrophobic and has an area of 1400 Å2, which is 14% of the solvent-accessible surface (Hubbard et al., 1991). The Fno monomer shows a more globular shape composed of two closely attached domains that form an α, β twisted open-sheet structure (Figure 3B). The N-terminal domain includes residues 1–135 and consists of two Rossmann folding units βαβαβ that are arranged as a six-stranded parallel β-sheet (β1–β6) flanked in a characteristic manner by five α-helices. This architecture is found in many other dinucleotide binding proteins (Carugo and Argos, 1997). A novel structural feature is a protruding β-hairpin between sheet β5 and helix α5, which is involved in interface formation. The smaller C-terminal domain (residues 136–212) essentially consists of two parallel β-strands (β7 and β8) and α-helices (α7 and α8). A short third strand (β9) at the C-terminal end only touches the two other strands. Together with helix α8, it is the major constituent of the intersubunit contact area (Figure 3).
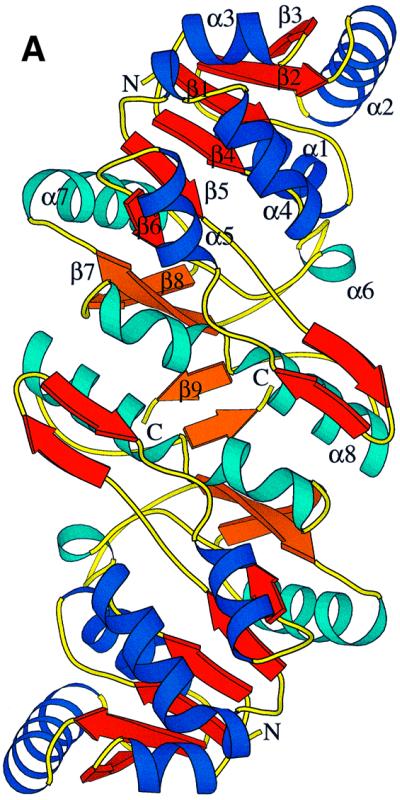

Fig. 3. (A) Ribbon diagram of the homodimer of Fno from A.fulgidus. The interface area between the two monomeric Fnos is 75% non-polar compared with 52% surface for the dimer. Dimer formation might be useful in increasing the stability of the protein but also in providing rigidity to the active site pocket. The fold of Fno can be subdivided into an N-terminal NADP domain consisting of a typical dinucleotide binding fold (coloured in royal blue and red), and a C-terminal F420 domain with an α/β-fold (coloured in sky blue and orange). (B) Stereo diagram of the Cα trace of a monomer of Fno. The colour changes from blue (N-terminus) to red (C-terminus) along the peptide chain.
Both domains together form a common eight-stranded β-sheet with an anti-parallel orientation of the two constituting parallel β-sheets. Between the two domains a deep active-site pocket is built up from the loop regions following strands β4–β6 and from the C-terminal segment (α8–β9). Because of the vicinity of the active site to the dimer interface, dimerization might not only be important for protein stability but also for catalysis.
Fno exhibits a structural relationship to other proteins of the dinucleotide binding family despite the fact that there is no significant sequence similarity (<14%) between them. The most similar structures, according to a search using the DALI server (Holm and Sander, 1993), are shown by the NADP-dependent dehydrogenases: human l-3-hydroxyacyl CoA dehydrogenase (Barycki et al., 1999), Arthrobacter N-(1-d-carboxylethyl)-l-norvaline dehydrogenase (Britton et al., 1998), sheep 6-phosphogluconate dehydrogenase (Adams et al., 1991) and Spinacia oleracea acetohydroxy-acid isomeroreductase (Biou et al., 1997), with r.m.s.d. values of 2.7, 3.1, 2.8 and 2.9 Å, respectively, using in each case >70% of the Fno-Cα atoms for alignment. Substantial differences between the NADP domains are only observed in connecting segments between the secondary structures (i.e. the additional β-hairpin of ∼15 residues) and in the C-terminal segment, which exhibits a fold totally different from the enzymes discussed.
The F420 binding site
Coenzyme F420 is associated mainly with the C-terminal domain. The catalytically relevant deazaflavin moiety dips deeply into the active site pocket with atoms C6 and C7 of the hydroxybenzyl ring (see Figure 1) directed towards the bottom of the pocket and with the more polar 2,4-pyrimidinedione ring towards the solvent. Whereas the Si-face of the deazaflavin faces the nicotinamide ring, the Re-face looks towards a hydrophobic wall formed by the side chains of Val98, Phe107, Thr192, Ile195, Leu196 and Met199. The only direct protein–F420 contact is provided by the hydrogen bond between the benzyl hydroxyl group of F420 and the carbonyl oxygen of Leu207 (Figure 2B). Accordingly, the enzyme does not catalyse the reduction of F420 derivatives lacking the benzyl hydroxyl group (Yamazaki et al., 1982; Eker et al., 1989). Moreover, a methyl group linked to atom C7, as in flavins, would interfere with the backbone at Thr134 of Fno. Thus, these specific interactions seem to be crucial for the orientation of the ring and the exclusion of a flavin from binding within the pocket. In agreement with these structural considerations, the enzyme does not catalyse the oxidation of reduced FMN or FAD with NADP+ (Kunow et al., 1993b). The hydroxybenzyl side of F420 is in van der Waals contact with the backbone (i.e. with the loops after strand β6 and before β9). This is markedly different from the respective environment of the dimethylbenzyl moiety of flavin in several NADP-dependent flavoenzymes (Karplus and Schulz, 1989; Ziegler and Schulz, 2000). This difference could be exploited for the design of drugs.
In contrast to the few F420–protein interactions, there are multiple contacts between F420 and NADP, including van der Waals and aromatic interactions between the ring systems as well as polar interactions (Figure 2B). The hydrogen bond between the ribitol hydroxyl O2R of F420 and the nicotinamide oxygen O7 (Figure 4) is apparently strong enough to rotate (by ∼10°) the carboxamide out of the ring plane. This rotation increases the distance between the nicotinamide oxygen and the peptide nitrogen of Ala137 by ∼0.15 Å. The other hydrogen bond between the carbonyl-O4F of F420 and the nicotinamide ribose hydroxyl O2 of NADP (2.7 Å) is also unique in the structurally characterized NAD(P)-flavin systems (see Figures 2 and 4).

Fig. 4. Stereo diagram of the active site of Fno. The nicotinamide ring of NADP+ and the deazaflavin ring of F420 are embedded face on face in the hydrophobic pocket.
The deazaflavin ring is slightly bent, the wings pointing towards the hydrophobic wall. Interestingly, the side chains of Thr192 and Leu196 protrude from helix α8 towards the pyridine ring (Figure 4), and can be considered as a ‘backstop’ for maintaining the butterfly conformation. The flexibility is low throughout the deazaflavin ring, the lowest temperature factor being found for the atoms deep in the pocket. In contrast, the temperature factor increases continuously for the ribitol and the phosphate groups, which are arranged parallel to the short helical region at the N-terminus of the C-terminal domain without touching them. This high flexibility and the fact that beyond the phosphate group no electron density is visible lead to the conclusion that the binding of F420 in Fno is based mainly on the binding of the deazaflavin.
The NADP binding site
The binding of NADP to the N-terminal domain is analogous to those observed in other members of the dinucleotide binding protein family (Carugo and Argos, 1997). The ADP moiety of NADP fits into a shallow crevice formed at the C-terminus of the β-sheet and the nicotinamide mononucleotide moiety is embedded into the deeper active site pocket described above. The interactions between NADP and the protein matrix are illustrated in Figure 2A.
The nicotinamide ring is flanked at its Si-side by the deazaflavin ring of F420 (see above for the interactions) and on its Re-side by several hydrophobic side chains including Leu12. The only direct hydrogen bond between the nicotinamide and the protein matrix is formed by the amide oxygen and the peptide nitrogen of Ala137 (Figure 2A). It is assumed that this interaction determines the trans conformation of the amide group, which is the only one found in enzymatic systems (Torres et al., 1999). As expected, the pyridine moiety of the oxidized NADP is present in a planar conformation. Interestingly, the short distance of 3.1 Å between the carbonyl oxygen of Ile135 and atom C4 of NADP might be of catalytic relevance (Figure 4).
A central role in the binding of the ADP moiety is played by the pyrophosphate and the ribose phosphate groups. The solvent-exposed pyrophosphate moiety interacts in typical fashion with residues of the glycine-rich loop, and its negative charge is primarily compensated for by a positive dipole formed at the N-terminal end of helix α1 (Figure 3B). The negatively charged ribose phosphate group is linked to the side chains of Thr9, Ser31, Arg32 and Lys36 (Figure 2A). These residues are conserved in the Fno from the three organisms (A.fulgidus, Methanothermobacter marburgensis and Methanococcus jannaschii) with known primary structures. This large number of interactions appears to reflect the physiological necessity to discriminate between NADP(H) and NAD(H).
A comparison of the NADP binding site with and without substrates indicates a substrate-induced conformational change (induced fit) of the enzyme: the more open and softened NADP binding site (Figure 5A) becomes closed and rigid upon NADP binding (Figure 5B), as has been observed in principle for other NADP-dependent dehydrogenases (Korkhin et al., 1998; Cobessi et al., 1999). Especially large positional and temperature factor changes are observed in the glycine-rich loop and in regions involved in fixing the adenosine moiety. For example, the loop between strand β4 and helix α4, partly distorted in the substrate-free enzyme, undergoes a temperature factor decrease of ∼30 Å2 after NADP binding and moves by ∼3 Å towards the crevice, accompanied by the formation of an additional turn at the N-terminus of helix α4. Moreover, the flexible side chains of Arg32 and Arg33 are rearranged with atoms displaced by >5 Å towards the binding crevice upon NADP binding. Substrate binding also leads to a contraction of the active site pocket by ∼0.5–1.6 Å, which strengthens the hydrophobic interactions between non-polar side chains and the nicotinamide and deazaflavin ring systems.


Fig. 5. The NADP binding site of Fno in the states (A) without substrates, (B) with bound substrates and for comparison the superimposed structure without substrates in grey. The models are coloured according to their temperature factors (blue for B <14 Å2, red for B >25 Å2). The NADP binding site is highly flexible prior to NADP binding and becomes rigid with NADP bound.
The catalytic reaction
Kinetic measurements revealed a ternary complex catalytic mechanism for Fno (Berk and Thauer, 1997), which is now confirmed by the structural data. The Km of Fno from A.fulgidus for F420H2 was 20 µM and for NADP it was 40 µM at 65°C and pH 8.0, the optimum pH for NADP reduction with F420H2; the Km for F420 was 10 µM and for NADPH it was 40 µM at 65°C and pH 5.5, the optimum pH for F420 reduction with NADPH (Kunow et al., 1993b). From the kinetic data it is not possible to deduce whether binding of the two substrates is random or sequential and, if sequential, which substrate binds first (Koshland, 1958).
The crystal structure of Fno with both substrates bound revealed that interaction of NADP with the enzyme is much more extensive than that of F420 (see Figure 2), and that F420 in part binds to the enzyme via NADP. This finding suggests that substrate binding is sequential, with NADP having to bind first. In order to find out experimentally the binding sequence of the substrates, equimolar amounts of Fno and NADP+, NADPH and/or F420 were mixed, subsequently ultrafiltrated and the concentration of the substrate(s) determined in the ultrafiltrate. Both at pH 7.5 and 6.0, >80% of NADP+ or NADPH but <5% of F420 was bound in the binary enzyme–substrate complexes. In the presence of NADP+ or NADPH, the binding of F420 to Fno increased to ∼20% at pH 6.0. The increase was less pronounced at pH 7.5. These data clearly demonstrate that NADP+ or NADPH is required for F420 binding. Moreover, the observed substantial positional and conformational changes upon substrate binding suggest that NADPH binding is associated with an induced fit, which preforms the F420 binding site.
Fno is the first F420-dependent enzyme that provides the structural basis for a discussion of the hydride transfer. Basically, the active site of Fno formed at the interface between the NADP and F420 domains is designed as a hydrophobic pocket where the nicotinamide and deazaflavin rings are packed together in a roughly parallel arrangement. Both ring planes are flanked by non-polar side chains, which lock them in a suitable relative position and provide the exclusion of bulk solvent necessary for the hydride transfer. A similar molecular design was observed for NADP-dependent oxidoreductases with FAD as a prosthetic group in, for example, glutathione reductase (Karplus and Schulz, 1989) and adrenodoxin reductase (Ziegler and Schulz, 2000), where the hydride transfer occurs between isoalloxazine and nicotinamide ring systems. In contrast, the deazaflavin- and FAD-dependent DNA photolyase (Tamada et al., 1997), the only other known structure with a deazaflavin bound, shows no structural and mechanistic relationships with Fno.
Concentrating on the region directly involved in hydride transfer reveals that the rings are not laid one upon another, but are laterally shifted such that the amide group of the nicotinamide is positioned above the deazaflavin moiety and the C4 atom of NADP exactly above the C5 atom of F420 (Figure 2B). Their distance of 3.1 Å is the shortest one between the two substrates. This optimal distance for the hydride transfer (Almarsson and Bruice, 1993) might have been adjusted by the bending of the deazaflavin ring and by the deviation from the parallel arrangement of the ring systems. Theoretical papers on the hydride transfer (Almarsson and Bruice, 1993; Young and Post, 1996; Torres et al., 1999) propose a transition state with a boat conformation for the pyridine part, which would further reduce the distance between the interacting carbon atoms. Quite recently, a distorted boat conformation for NADH was found in the atomic resolution structure of its complex with horse liver alcohol dehydrogenase (Meijers et al., 2001). The present structure corroborates the model of a transient boat conformation. First, the observation that the C4 atom shows the highest B-values within NADP (mean values for the two molecules). Secondly, the slight rotation of the carboxamide group of NADP due to a hydrogen bond with F420 weakens the nicotinamide aromatic system and in turn reduces the activation energy for the boat conformation. Thirdly, the boat conformation would again make the C4 atom roughly coplanar with NADP atoms C3, C7 and O7 (see Figures 1 and 4), which stabilizes this transition state. Fourthly, the unfavourably short distance between C4 and the carbonyl oxygen of Ile135 C4 [d(C–O) = 3.1 Å] would be increased. On the other hand, the close vicinity of this carbonyl oxygen might ease the hydride transfer by ‘guiding’ the proR-H to its new position (Young and Post, 1996). However, it has to be considered that the structural data are based on oxidized states of both substrates and that the actual redox reaction might imply minor structural rearrangements.
The stereochemical analysis of the hydride transfer leads to the conclusion that the observed orientation of the Si-face of F420 towards the Si-face of NADP allows only the transfer of the proS hydrogen at C5 to the proS position at C4 and vice versa (Figure 6). The structural data are in agreement with the observed Si-face stereospecificity of Fno with respect to both F420 and NADP (Yamazaki et al., 1980; Kunow et al., 1993b). A general explanation for the finding that hydride transfer involving F420 always proceeds Si-face stereospecifically cannot be inferred from the Fno structure. Nevertheless, the conserved nicotinamide orientation within the dinucleotide binding family and the preferred orientation of the deazaflavin ring with its more non-polar hydroxy-benzyl group pointing to the bottom of the pocket provide a comprehensible structural explanation for the observed stereochemistry in the case of Fno.

Fig. 6. Hydride transfer. The hydride is transferred from the Si-face of F420 towards the Si-face of NADP. The distance between C5 of F420 and C4 of NADP is 3.1 Å. The proR and proS hydrogen atoms are modelled using the conformation of NADP+ as found in Fno complexed with F420 and NADP+, and assuming identical conformations in the oxidized and reduced state. Also shown in dark red is the carbonyl O of Ile135.
Materials and methods
Preparation of the enzyme
The product of gene AF0892 in the A.fulgidus genome (Klenk et al., 1997) has significant sequence similarity (36% identity) to Fno from M.marburgensis (Berk and Thauer, 1998). The fno gene was amplified by PCR, cloned into the expression vector pET24b (Novagen) (Studier et al., 1990) and overexpressed in E.coli BL21 (DE3) under the control of the T7lac promoter of the expression plasmid following the protocol given by the manufacturer. The recombinant E.coli strain grew at 37°C in mineral salts medium (Sambrook et al., 1989). For selenomethionine labelling experiments, the expression vector containing the fno gene was introduced into a methionine auxotrophic strain, E.coli B834 (DE3)(pLysS). The recombinant E.coli strain grew in the medium supplemented with 0.2 mM selenomethionine. When the OD600 of the culture reached 0.6, fno gene expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside. The cells were harvested after 16 h induction.
The overproduced enzyme in the soluble fraction was purified by heat precipitation of E.coli proteins at 90°C in 1.5 M potassium phosphate pH 8.0 for 30 min. After centrifugation at 4°C, the supernatant was applied to Source 15 Phe (1.6 × 14 cm) (Pharmacia-Amersham Biotech), which was equilibrated at 4°C with 2 M ammonium sulfate in 50 mM MOPS–KOH pH 7.0. Fno eluted from the column at 1.0–0.6 M ammonium sulfate in a linear decreasing gradient of the salt (145 ml, 2–0 M). The fractions containing Fno were combined and concentrated by filtration (30 kDa cut-off) (Millipore), and diluted with 10 mM potassium phosphate pH 7.0. The solution was applied to a ceramic hydroxyapatite column (1.3 × 10 cm) (Bio-Rad), which was equilibrated at 4°C with 10 mM potassium phosphate pH 7.0. Fno eluted at 50–125 mM potassium phosphate pH 7 in a linear increasing gradient of the salt (130 ml, 10–500 mM). The fractions containing Fno were combined and concentrated by filtration (30 kDa cut-off), and diluted with 10 mM MOPS–KOH pH 7.0. The purified enzyme exhibited a specific activity of 280 U/mg at 65°C under standard assay conditions (Kunow et al., 1993b).
Substrate affinity measurement in solution
Binding of NADP+, NADPH and/or F420 to the enzyme was determined by measuring the decrease in the free substrate concentration upon addition of equimolar amounts of Fno: 25 µl of Fno solution (0.5 mM, 11.4 mg/ml) was mixed with 25 µl of 200 mM HEPES/NaOH pH 7.5 or 200 mM Mes/NaOH pH 6.0 containing the substrate(s) at 0.5 mM. The mixture was incubated for 10 min at 4°C. The solution was then ultrafiltrated at 4°C by centrifugation at 13 000 g through Microcon 10 (10 kDa cut-off, Millipore) and the concentration of the substrate(s) in the 30 µl ultrafiltrate was determined by UV/visible spectroscopy using a Zeiss Specord S10 diode array spectrophotometer [light path, 0.3 cm; extinction coefficients, 18.0 mM–1 cm–1 (NADP+) at 260 nm, 6.2 mM–1 cm–1 (NADPH) at 339 nm and 25.9 mM–1 cm–1 (F420) at 401 nm (Dawson et al., 1986; DiMarco et al., 1990)]. From the substrate concentration in the ultrafiltrate, the amount of substrate bound to the enzyme was calculated assuming that the free substrate concentration in the 20 µl concentrate was the same as in the 30 µl ultrafiltrate. The results obtained were corrected for unspecific binding of the substrates to the ultrafiltration membrane and to proteins, which was determined in blanks containing bovine serum albumin (11.4 mg/ml) and the substrate(s).
Crystallization and data collection
Fno was crystallized in three different crystal forms at a temperature of 4°C using the hanging drop vapour diffusion method. Crystals grew in a drop consisting of 1 µl of enzyme solution (12 mg/ml) and 1 µl of reservoir solution. Initial screenings were performed using a sparse matrix crystallization kit (Hampton Research) (Jancarik and Kim, 1991). Crystals of form 1 were obtained using a reservoir solution composed of 29% methyl-2,4-pentanediol (MPD), 0.1 M acetate/NaOH pH 4.6 and 0.24 M CaCl2. Microseeding was required to produce large-size crystals at this condition. Their space group was P6222 and the lattice parameters were a = 87.4 Å and c = 93.6 Å, which is most compatible with one monomer per asymmetric unit. Native data to 1.8 Å resolution were collected at the Max-Planck beamline BW6 at the Deutsches Elektronen Synchrotron in Hamburg (DESY) using flash-frozen crystals. Data processing and scaling were performed with the HKL suite (Otwinowski and Minor, 1996) (see Table I). Crystals of form 2 were obtained using a reservoir solution with 30% MPD, 0.1 M acetate/NaOH pH 4.6 and 0.4 M MgCl2. The space group and the lattice parameters were P21 and a = 38.64 Å, b = 72.14 Å, c = 70.40 Å and β = 90.12°, respectively, indicating the presence of one dimer in the asymmetric unit. Diffraction data were collected at –180°C at the ESRF beamline ID 14-4 in Grenoble, France (see Table I). For preparation of form 3 crystals, the enzyme solution (12 mg/ml) was supplemented with 1 mM F420 and 1 mM NADP, then combined with a reservoir solution containing 0.1 M HEPES/NaOH pH 7.5 and 1.4 M sodium citrate. The space group was P212121 and the lattice parameters were a = 54.9 Å, b = 69.8 Å and c = 145.5 Å, most compatible with one dimer per asymmetric unit. Diffraction data up to a resolution of 1.65 Å were collected at the microfocus beamline ID 13 at ESRF, Grenoble, under cryogenic conditions (Table I).
Table I. Crystallographic and refinement data of Fno from A.fulgidus.
| Crystal form |
|||
|---|---|---|---|
| 1 | 2 | 3 | |
| Space group | P6222 | P21 | P212121 |
| a (Å) | 87.4 | 38.64 | 54.9 |
| b (Å) | 72.14 | 69.8 | |
| c (Å) | 93.6 | 70.4 | 145.5 |
| β (°) | 90 | 90.12 | 90 |
| No. of molecules/ asymmetric unit | 1 | 2 | 2 |
| Solvent content (%) | 48 | 35 | 54 |
| Resolution (Å) | 1.8 (1.9–1.8) | 1.8 (1.9–1.8) | 1.65 (1.8–1.65) |
| Rsym (%) | 2.7 (25.3) | 5.7 (7.1) | 8.3 (28.5) |
| I/σ | 33.0 (5.9) | 17.2 (13.3) | 12.3 (2.2) |
| Completeness (Å) | 95.5 (87.2) | 98.5 (99.2) | 91.1 (73.2) |
| Number of reflections | 18 624 | 35 340 | 58 152 |
| Multiplicity | 3.9 | 3.7 | 4.7 |
| Solution method | MAD (Se) | MR | MR |
| Rcryst (%) | 32 | 18.5 | 18.2 |
| Rfree (%) | 36 | 22.1 | 20.6 |
| R.m.s.d. bonds (Å) | 0.006 | 0.010 | 0.013 |
| R.m.s.d. angles (°) | 1.36 | 1.48 | 1.59 |
| Residues per monomer | 212 | 212 | 212 |
| Ligands per monomer | – | – | 1 NADP, 1 F420 |
| No. of water molecules | 24 | 270 | 324 |
| Ions | 1 K+ | 2 Na+, 1 Mg2+ | 2 Na+ |
All data were measured under cryogenic conditions. Rsym, I/σ and completeness for the last resolution shell are given in parentheses.
Phase determination and model building
Phase determination was performed using the MAD method by collecting a three-wavelengths data set (λ = 0.9789, 0.9792 and 0.9395 Å) of the Se-methionine-substituted protein at the ESRF beamline ID14-4. The calculations were carried out within the program SOLVE (Terwilliger and Berendzen, 1999). After solvent flattening using DM (Cowtan, 1994), the quality of the electron density was sufficient to build a model, except for the poorly resolved regions around residues 85 and 105 using the program O (Jones et al., 1991) and the density skeletonization option of program MAPMAN (Kleywegt and Jones, 1996). The phases for crystal forms 2 and 3 were determined by the molecular replacement method with AMoRe (Navaza, 1994) using the coordinates of crystal forms 1 and 2, respectively, as the search model.
Model refinement and quality of the model
For unknown reasons, the model of crystal form 1 could not be refined to an Rfree-factor sizeably below 40% (1.8–20 Å), both for the native and the Se-methionine data (CNS; Brünger et al., 1998). Tests for twinning were negative. The refinement worked successfully with crystal forms 2 and 3 by performing alternately computer-assisted molecular dynamics and conjugate gradient minimization rounds and manual correction rounds using program O (Jones et al., 1991). A bulk solvent correction at low resolution up to 20 Å was achieved and water molecules were introduced if the 2Fo – Fc electron density was >1σ, the Fo – Fc electron density >2σ, and the shape of the electron density approximately spherical. The molecules NADP and F420 were modelled into the electron density of crystal form 3. Three electron density peaks were assigned to cations on the basis of their coordination and interatomic distances. No non-crystallographic symmetry restraints were applied during refinement. For crystal form 2, the Rcryst and Rfree factors converged to 18.5 and 22.1% (1.8–20.0 Å) and for crystal form 3 to 18.2 and 20.6% (1.65–30.0 Å), respectively (Table I). The quality of the final model is summarized in Table I. The final model of crystal form 2 comprises 2 × 212 residues, 270 water molecules and three putative cations (two sodium and one magnesium), and that of crystal form 3, 2 × 212 residues, two NADP and 0.9 and 0.8 F420, 324 water molecules and two sodium ions (at positions equivalent to those in crystal form 2). The occupancy of the deazaflavin of F420 was fitted by several cycles of alternate occupancy and temperature factor refinement. The reduced and vanished electron density of the residual part of F420 was attributed to increasing disorder and modelled by further reduced occupation. Alternative conformations were observed for 15 and 10 residues and the average temperature factor was 21.7 and 22.6 Å2, respectively. Model errors were assessed with the programs CNS and PROCHECK (Laskowski et al., 1993). In both structures, all non-glycine and non-proline residues were either in most favoured (94% for crystal form 2 and 95% for crystal form 3) or in additionally allowed regions (6% for crystal form 2 and 5% for crystal form 3) of the Ramachandran plot.
Preparation of figures
Figures 2–6 were generated using the programs MOLSCRIPT (Kraulis, 1991), BOBSCRIPT (Esnouf, 1997) and RASTER3D (Merritt and Murphy, 1994).
Coordinates
The coordinates have been deposited at the Protein Data Bank under accession codes 1JAX and 1JAY.
Acknowledgments
Acknowledgements
We thank Hartmut Michel for support, the staff of the EMBL and Max-Planck outstations at DESY and of ID-13 at ESRF for help during data collection, and Wim Burmeister of ID14-4 at ESRF for MAD data collection. This work was supported by the Max-Planck-Gesellschaft and by the Fonds der Chemischen Industrie.
References
- Adams M.J., Gover,S., Leaback,R., Phillips,C. and Somers,D.O. (1991) The structure of 6-phosphogluconate dehydrogenase refined at 2.5 Å resolution. Acta Crystallogr. B, 47, 817–820. [DOI] [PubMed] [Google Scholar]
- Almarsson Ö. and Bruice,T.C. (1993) Evaluation of the factors influencing reactivity and stereospecificity in NAD(P)H dependent dehydrogenase enzymes. J. Am. Chem. Soc., 115, 2125–2138. [Google Scholar]
- Barycki J.J., O’Brien,L.K., Bratt,J.M., Zhang,R., Sanishvili,R., Strauss,A.W. and Banaszak,L.J. (1999) Biochemical characterization and crystal structure determination of human heart short chain l-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry, 38, 5786–5798. [DOI] [PubMed] [Google Scholar]
- Baumer S., Ide,T., Jacobi,C., Johann,A., Gottschalk,G. and Deppenmeier,U. (2000) The F420H2 dehydrogenase from Methanosarcina mazei is a redox-driven proton pump closely related to NADH dehydrogenases. J. Biol. Chem., 275, 17968–17973. [DOI] [PubMed] [Google Scholar]
- Berk H. and Thauer,R.K. (1997) Function of coenzyme F420-dependent NADP reductase in methanogenic archaea containing an NADP-dependent alcohol dehydrogenase. Arch. Microbiol., 168, 396–402. [DOI] [PubMed] [Google Scholar]
- Berk H. and Thauer,R.K. (1998) F420H2:NADP oxidoreductase from Methanobacterium thermoautotrophicum: identification of the encoding gene via functional overexpression in Escherichia coli. FEBS Lett., 438, 124–126. [DOI] [PubMed] [Google Scholar]
- Biou V., Dumas,R., Cohen-Addad,C., Douce,R., Job,D. and Pebay-Peyroula,E. (1997) The crystal structure of plant acetohydroxy acid isomeroreductase complexed with NADPH, two magnesium ions and a herbicidal transition state analog determined at 1.65 Å resolution. EMBO J., 16, 3405–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton K.L., Asano,Y. and Rice,D.W. (1998) Crystal structure and active site location of N-(1-d-carboxylethyl)-l-norvaline dehydro genase. Nature Struct. Biol., 5, 593–601. [DOI] [PubMed] [Google Scholar]
- Brünger A. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determinations. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carugo O. and Argos,P. (1997) NADP-dependent enzymes. II: Evolution of the mono- and dinucleotide binding domains. Proteins, 28, 29–40. [DOI] [PubMed] [Google Scholar]
- Cobessi D., Tete-Favier,F., Marchal,S., Azza,S., Branlant,G. and Aubry,A. (1999) Apo and holo crystal structures of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol., 290, 161–173. [DOI] [PubMed] [Google Scholar]
- Cowtan K.D. (1994) ‘DM’: an automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newsl. Protein Crystallogr., 31, 83–91. [Google Scholar]
- Dawson R.M.C., Elliott,D.C., Elliott,W.H. and Jones,K.M. (1986) Data for Biochemical Research, 3rd edn. Oxford University Press, New York, NY.
- DiMarco A.A., Bobik,T.A. and Wolfe,R.S. (1990) Unusual coenzymes of methanogenesis. Annu. Rev. Biochem., 59, 355–394. [DOI] [PubMed] [Google Scholar]
- Eker A.P., Hessels,J.K. and Meerwaldt,R. (1989) Characterization of an 8-hydroxy-5-deazaflavin:NADPH oxidoreductase from Streptomyces griseus. Biochim. Biophys. Acta, 990, 80–86. [DOI] [PubMed] [Google Scholar]
- Elias D.A., Juck,D.F., Berry,K.A. and Sparling,R. (2000) Purification of the NADP+:F420 oxidoreductase of Methanosphaera stadtmanae. Can. J. Microbiol., 46, 998–1003. [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 133–138. [DOI] [PubMed] [Google Scholar]
- Fox J.A., Livingston,D.J., Orme-Johnson,W.H. and Walsh,C.T. (1987) 8-Hydroxy-5-deazaflavin-reducing hydrogenase from Methano bacterium thermoautotrophicum: 1. Purification and characterization. Biochemistry, 26, 4219–4227. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. and Ogata,C.M. (1997) Phase determination for multiwavelength anomalous diffraction measurements. Methods Enzymol., 276, 307–315. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Secondary structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Hubbard S.J., Campbell,S.F. and Thornton,J.M. (1991) Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol., 220, 507–530. [DOI] [PubMed] [Google Scholar]
- Jancarik J. and Kim,S.H. (1991) Sparse-matrix sampling—a screening method for crystallization of proteins. J. Appl. Crystallogr., 24, 409–411. [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Karplus P.A. and Schulz,G.E. (1989) Substrate binding and catalysis by glutathione reductase as derived from refined enzyme: substrate crystal structures at 2 Å resolution. J. Mol. Biol., 210, 163–180. [DOI] [PubMed] [Google Scholar]
- Klein A.R. and Thauer,R.K. (1995) Re-face specificity at C14a of methylenetetrahydromethanopterin and Si-face specificity at C5 of coenzyme F420 for coenzyme F420-dependent methylenetetrahydro methanopterin dehydrogenase from methanogenic Archaea. Eur. J. Biochem., 227, 169–174. [DOI] [PubMed] [Google Scholar]
- Klein A.R., Berk,H., Purwantini,E., Daniels,L. and Thauer,R.K. (1996) Si-face stereospecificity at C5 of coenzyme F420 for F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis and F420-dependent alcohol dehydrogenase from Methanoculleus thermophilicus. Eur. J. Biochem., 239, 93–97. [DOI] [PubMed] [Google Scholar]
- Klenk H.P. et al. (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature, 390, 364–370. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Jones,T.A. (1996) xdIMAPMAN and xdIDATAMAN—programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr. D, 52, 826–828. [DOI] [PubMed] [Google Scholar]
- Korkhin Y., Kalb,A.J., Peretz,M., Bogin,O., Burstein,Y. and Frolow,F. (1998) NADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol., 278, 967–981. [DOI] [PubMed] [Google Scholar]
- Koshland D.E. Jr, (1958) Application of a theory of enzyme specificity to protein synthesis. Proc. Natl Acad. Sci. USA, 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kunow J., Schwörer,B., Setzke,E. and Thauer,R.K. (1993a) Si-face stereospecificity at C5 of coenzyme F420 for F420-dependent N5,N10-methylenetetrahydromethanopterin dehydrogenase, F420-dependent N5,N10-methylenetetrahydromethanopterin reductase and F420H2: dimethylnaphthoquinone oxidoreductase. Eur. J. Biochem., 214, 641–646. [DOI] [PubMed] [Google Scholar]
- Kunow J., Schwörer,B., Stetter,K.O. and Thauer,R.K. (1993b) A F420-dependent NADP reductase in the extremely thermophilic sulfate reducing Archaeoglobus fulgidus. Arch. Microbiol., 160, 199–205. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- May A.C.W. and Johnson,M.S. (1994) Protein structure comparisons using a combination of a genetic algorithm, dynamic programming and least-squares minimization. Protein Eng., 7, 475–485. [DOI] [PubMed] [Google Scholar]
- Meijers R., Morris,R.J., Adolph,H.W., Merli,A., Lamzin,V.S. and Cedergren-Zeppezauer,E.S. (2001) On the enzymatic activation of NADH. J. Biol. Chem., 276, 9316–9321. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Murphy,M.E.P. (1994) Raster3D Version 2.0—A program for photorealistic molecular graphics. Acta Crystallogr. A, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Navaza J. (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr. A, 50, 157–163. [Google Scholar]
- Otwinowski Z. and Minor,W. (1996) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schauer N.L., Ferry,J.G., Honek,J.F., Orme-Johnson,W.H. and Walsh,C. (1986) Mechanistic studies of the coenzyme F420 reducing formate dehydrogenase from Methanobacterium formicicum. Biochemistry, 25, 7163–7168. [DOI] [PubMed] [Google Scholar]
- Shima S., Warkentin,E., Grabarse,W., Sordel,M., Wicke,M., Thauer,R.K. and Ermler,U. (2000) Structure of coenzyme F-420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J. Mol. Biol., 300, 935–950. [DOI] [PubMed] [Google Scholar]
- Stover C.K. et al. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature, 405, 962–966. [DOI] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- Sumner J.S. and Matthews,R.G. (1992) Stereochemistry and mechanism of hydrogen transfer between NADPH and methylenetetrahydrofolate in the reaction catalyzed by methylenetetrahydrofolate reductase from pig liver. J. Am. Chem. Soc., 114, 6949–6959. [Google Scholar]
- Tamada T., Kitadokoro,K., Higuchi,Y., Inaka,K., Yasui,A., de Ruiter, P.E., Eker,A.P. and Miki,K. (1997) Crystal structure of DNA photolyase from Anacystis nidulans. Nature Struct. Biol., 4, 887–891. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima T., Nakaji,A., Shiba,T., Tsai,L. and Yamazaki,S. (1985) Elucidation of stereospecificity of a selenium-containing hydrogenase from Methanococcus vannielii—Syntheses of (R)- and (S)-[4-2H1]-3,4-dihydro-7-hydroxy-1-hydroxyethylquinolinone. Tetrahedron Lett., 26, 351–354. [Google Scholar]
- Torres R.A., Schiøtt,B. and Bruice,T.C. (1999) Molecular dynamics simulations of ground and transition states for the hydride transfer from formate to NAD+ in the active site of formate dehydrogenase. J. Am. Chem. Soc., 121, 8164–8173. [Google Scholar]
- Walsh C. (1985) Naturally occurring 5-deazaflavin coenzymes: biological redox roles. Acc. Chem. Res., 19, 216–221. [Google Scholar]
- Yamazaki S., Tsai,L., Stadtman,T.C., Jacobson,F.S. and Walsh,C. (1980) Stereochemical studies of 8-hydroxy-5-deazaflavin-dependent NADP+ reductase from Methanococcus vannielii. J. Biol. Chem., 255, 9025–9027. [PubMed] [Google Scholar]
- Yamazaki S., Tsai,L. and Stadtman,T.C. (1982) Analogues of 8-hydroxy-5-deazaflavin cofactor: relative activity as substrates for 8-hydroxy-5-deazaflavin-dependent NADP+ reductase from Methano coccus vannielii. Biochemistry, 21, 934–939. [DOI] [PubMed] [Google Scholar]
- Yamazaki S., Tsai,L., Stadtman,T.C., Teshima,T., Nakaji,A. and Shiba,T. (1985) Stereochemical studies of a selenium-containing hydrogenase from Methanococcus vannielii: determination of the absolute configuration of C-5 chirally labeled dihydro-8-hydroxy-5-deazaflavin cofactor. Proc. Natl Acad. Sci. USA, 82, 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. and Post,C.B. (1996) Catalysis by entropic guidance from enzymes. Biochemistry, 35, 15129–15133. [DOI] [PubMed] [Google Scholar]
- Ziegler G.A. and Schulz,G.E. (2000) Crystal structures of adrenodoxin reductase in complex with NADP+ and NADPH suggesting a mechanism for the electron transfer of an enzyme family. Biochemistry, 39, 10986–10995. [DOI] [PubMed] [Google Scholar]