

Abstract
The Escherichia coli melAB promoter is co-dependent upon two transcription activators, MelR and the cyclic AMP receptor protein, CRP. In this study we demonstrate positive co-operativity between the binding of MelR and CRP at the melAB promoter, which provides a simple mechanism for its co-dependence. MelR binds to four sites, centred at positions –42.5, –62.5, –100.5 and –120.5 relative to the melAB transcription start point. When MelR is pre-bound, CRP is able to bind to a target located between MelR at positions –62.5 and –100.5. This increases the occupation of the two downstream sites for MelR, which is essential for transcription activation. We have identified residues within activating region 1 (AR1) of CRP that are important in transcription activation of the melAB promoter. At simple CRP-dependent promoters, the surface of CRP containing these residues is involved in contacting the RNA polymerase α subunit. Our results show that, at the melAB promoter, the surface of CRP containing AR1 contacts MelR rather than RNA polymerase. Thus, MelR and CRP activate transcription by a novel mechanism in which they bind co-operatively to adjacent sites and form a bacterial enhanceosome.
Keywords: co-dependence/CRP/Escherichia coli/protein–protein interactions/transcription activation
Introduction
Regulation of transcription initiation is a key step in the control of gene expression for all organisms, and hundreds of transcription regulators have been studied in prokaryotes and eukaryotes. In prokaryotes, activation of transcription at many promoters is due to the direct interaction of a single activator protein with RNA polymerase (RNAP; Busby and Ebright, 1994). In eukaryotes, transcription activation is more complex, often involving the co-operative binding of multiple transcription activator proteins to an enhancer element within the promoter, forming an enhanceosome (reviewed by Merika and Thanos, 2001).
In bacteria, expression from some promoters is dependent upon two or more activator proteins. Co-dependence of promoter expression provides a simple way of coupling transcription to multiple environmental stimuli. To date, four mechanisms of co-dependence have been identified at bacterial promoters. (i) The two activators make independent contacts with RNAP, e.g. at the Escherichia coli ansB promoter, which is co-dependent on the cyclic AMP receptor protein (CRP) and FNR, CRP and FNR each interact with one of the two RNAP α subunits (Scott et al., 1995). (ii) One activator bends the DNA to facilitate the function of a second activator that can then interact directly with RNAP, e.g. the DNA-bending protein IHF functions in conjunction with a second activator at a number of σ54-dependent promoters (reviewed by Goosen and van de Putte, 1995). (iii) The binding of one activator repositions a second activator from a location where it is unable to activate transcription to a location where it is able to activate transcription, e.g. CRP binding upstream of the E.coli malK promoter repositions three molecules of MalT on the DNA such that they are able to activate transcription (Richet et al., 1991). (iv) The function of one activator is inhibited by the presence of a repressor, which is removed by the binding of a second activator, e.g. FNR can only activate transcription at the E.coli nirB promoter when the FIS and IHF repressors are removed by the binding of NarL or NarP (Browning et al., 2000).
Interestingly in bacteria, no clear example has been found to date for co-dependence that is due to the co-operative binding of two activators, i.e. the presence of one activator is essential for the binding of the second activator. Here we show that MelR, a member of the AraC/XylS family of transcription activators (reviewed by Gallegos et al., 1997), triggered by melibiose, and CRP, a global transcription regulator, triggered by cyclic AMP (cAMP; reviewed by Kolb et al., 1993), bind co-operatively to adjacent DNA sites at the E.coli melAB promoter. This co-operativity results in the recruitment of CRP and increases the occupancy by MelR of the key target site that overlaps the promoter –35 element. Based on this observation, we have constructed artificial promoters where CRP is able to recruit MelR and vice versa. Finally, we have identified a determinant on the surface of CRP that is responsible for interactions with MelR at the melAB promoter. This determinant includes residues located within activating region 1 (AR1), the surface of CRP that contacts the C-terminal domain of the RNAP α subunit (αCTD) at simple CRP-dependent promoters (reviewed by Busby and Ebright, 1999). Our results show that mutation of this determinant alters the ability of CRP to be recruited to the melAB promoter by MelR, and that CRP functions independently of αCTD at the melAB promoter. Thus, MelR and CRP interact directly with one another, and bind co-operatively to the melAB promoter in a manner analogous to that of transcription activators in eukaryotic enhanceosomes.
Results
Co-dependence of the E.coli melAB promoter on MelR and CRP
Transcription initiation at the E.coli melAB promoter is co-dependent upon MelR and CRP. In our previous work (Belyaeva et al., 2000) we showed that MelR binds to four sites at the melAB promoter (sites 1′, 1, 2 and 2′) centred at positions –120.5, –100.5, –62.5 and –42.5, respectively, relative to the transcription start point (Figure 1A and B). In order to activate transcription, MelR must occupy its most downstream site, site 2′. CRP alone does not bind to the melAB promoter. However, in the presence of MelR, CRP is able to bind to the melAB promoter, and increases the occupancy of site 2′ by MelR. Hence, in the absence of MelR, CRP does not activate transcription at the melAB promoter whereas, in the presence of MelR, CRP activates transcription ∼10-fold (Figure 1C).



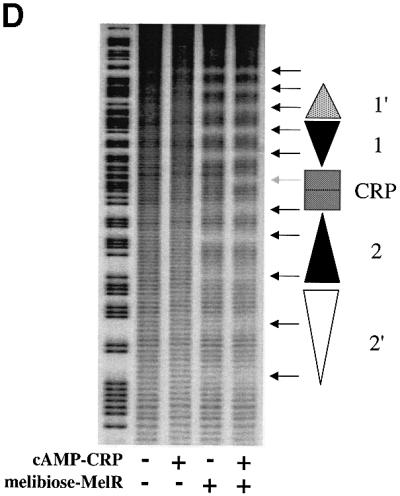
Fig. 1. Role of MelR and CRP at the melAB promoter. (A) Schematic representation of the melAB KK43 fragment carrying the melAB promoter. DNA sequences are numbered with respect to the transcription start (indicated by the horizontal arrow). The KK43 fragment is bounded by an EcoRI site at position –138 and a HindIII site at position +36. The different binding sites for MelR are shown by triangles and the locations of their centres are indicated: filled triangles indicate sites 1 and 2, the lightly shaded triangle indicates site 1′ and the open triangle indicates site 2′. The shaded rectangle indicates the target for CRP, centred at position –81.5. The open box indicates the melAB promoter –10 hexamer element. (B) Base sequence of positions –129 to –34 of the melAB promoter. MelR binding sites 1′, 1, 2 and 2′ are shown in italics. The target for CRP is shown in bold. A consensus 22 bp DNA site for CRP is written above the sequence of the melAB promoter. Position –75 of the melAB promoter is underlined. (C) The activity of the melAB promoter was measured in different backgrounds either with or without CRP and MelR. The figure shows β-galactosidase activities (in Miller units) measured in WAM134 (ΔmelR Δcrp Δlac) cells containing the lac expression vector, pRW50, carrying the KK43 melAB promoter fragment as above. Expression of the melAB promoter::lacZ fusion was measured either in the absence (open bars) or in the presence (shaded bars) of melibiose in cells carrying plamid pDCRP, encoding crp (+CRP), or the control vector pDU9 (–CRP), and pLG314, encoding melR (+MelR), or the control vector pLGΔRS (–MelR). (D) Hydroxyl radical footprint of the KK39 fragment containing the melAB promoter. Footprints were performed in the absence and presence of cAMP-CRP and in the absence and presence of melibiose-MelR as indicated. The position of the different DNA sites for MelR and the target for CRP are indicated by triangles and a rectangle, respectively. Protections due to MelR and CRP are indicated by black and grey arrows, respectively.
We have now used hydroxyl radical footprinting to study the binding of MelR and CRP at the melAB promoter (Figure 1D). MelR alone protects two sets of five consecutive minor grooves, corresponding to two dimers of MelR binding to sites 1′ and 1, and sites 2 and 2′, respectively. CRP alone does not bind to the melAB promoter. However, in the presence of MelR, addition of CRP gives an extra protection at positions –82, –83 and –84 relative to the transcription start point, and protection of MelR binding sites 2 and 2′ is increased. These results, which accord with previous DNase I footprints (Belyaeva et al., 2000), suggest that, in the presence of MelR, CRP binds to a target centred between MelR binding sites 1 and 2, and that this increases the occupancy of sites 2 and 2′ by MelR. Thus, CRP and MelR, present together at the melAB promoter, protect 11 consecutive minor grooves of the DNA, corresponding to a region of >100 bp, suggesting the formation of an ordered nucleoprotein complex. The fact that the locations of the hydroxyl radical footprints created by MelR are the same in both the presence and absence of CRP argues that CRP does not provoke any gross change in the position of MelR binding at the different sites.
The DNA sequence at the melAB promoter between MelR-binding sites 1 and 2 contains a poor match with the consensus DNA site for CRP (Figure 1B), and this accounts for the lack of binding found with CRP alone. We believe that the CRP-dependent protection of this region seen in the presence of MelR is due to the binding of CRP. To confirm this, we mutated position –75 relative to the transcription start point, a position that matches the sequence of a consensus DNA site for CRP. This substitution resulted in a large decrease in transcription activation at the melAB promoter (Figure 2A). It has been shown previously that the effects of substitutions at this position of a DNA site for CRP can be suppressed by the RH180 substitution in the DNA binding domain of CRP (R.H.Ebright, personal communication). Therefore, we compared transcription activation by wild-type CRP and RH180 CRP at the wild-type melAB promoter and at the promoter containing the substitution at position –75. As expected, the RH180 substitution had little effect on CRP-dependent activation of the wild-type melAB promoter. However, the decrease in transcription activation due to the substitution at position –75 was suppressed by the RH180 substitution in CRP (Figure 2A). This confirms that CRP binds directly to the region between sites 1 and 2 for MelR, and permits the exact positioning of the target for CRP, equidistant from sites 1 and 2.


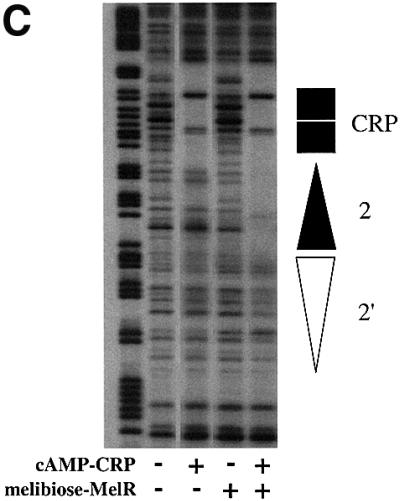
Fig. 2. Interactions of CRP at the melAB promoter. (A) Activity of the wild-type and p75A mutant melAB promoters carried on the KK43 fragment. The fragments with the two promoters are illustrated schematically using the same conventions as in Figure 1A: triangles represent MelR binding sites and the rectangle represents the target for CRP, with the open rectangle indicating the p75A mutation. The promoters were cloned into the lac expression vector, pRW50, and the figure shows β-galactosidase activities (in Miller units) measured in WAM134 (ΔmelR Δcrp Δlac) cells containing the pRW50 derivatives, pLG314 encoding MelR, and pDCRP encoding either wild-type (wt) CRP or His180 CRP. (B) Activity of the JK14 and JK15 melAB promoters. The fragments with the two promoters are illustrated schematically using the same conventions as in Figure 1A: triangles represent MelR binding sites and the rectangle represents the target for CRP, with the dark shading indicating a consensus DNA site for CRP. The promoters were cloned into the lac expression vector, pRW50, and the figure shows β-galactosidase activities (in Miller units) measured in WAM134 (ΔmelR Δcrp Δlac) cells containing the pRW50 derivatives, pLG314 encoding MelR, and either pDCRP encoding wild-type CRP (+CRP) or the vector pDU9 (–CRP). Note that, in the absence of pLG314, the JK15 promoter is inactive. (C) DNase I footprint of complexes at the JK15 fragment. Footprints were performed in the absence and presence of cAMP-CRP and in the absence and presence of melibiose and 150 nM MelR as indicated. The position of MelR binding sites and the target for CRP are indicated by triangles and a rectangle, respectively.
A consensus CRP site short circuits the need for sites 1′ and 1 at the melAB promoter
Previous studies have shown that sites 1′ and 1 for MelR at the melAB promoter are essential for transcription activation by CRP (Belyaeva et al., 2000). One explanation for this may be that CRP is only able to bind to the melAB promoter in the presence of MelR bound both upstream and downstream. To investigate this, we worked with the JK14 promoter, a derivative that lacks sites 1′ and 1 for MelR. First, we confirmed that transcription at the JK14 promoter is not activated by CRP (Figure 2B). We then created the JK15 promoter derivative that contains a correctly positioned consensus DNA site for CRP upstream of site 2. In contrast to the situation with the JK14 promoter, the JK15 promoter can be activated by MelR and CRP (Figure 2B). This activation is dependent on the precise location of the DNA site for CRP, since the insertion of 2 bp just upstream of MelR binding site 2 in the JK15 promoter results in the complete loss of CRP-dependent transcription activation (the JK15+2 promoter; data not shown). These results show that the requirement for MelR-binding sites 1 and 1′ can be short-circuited by a consensus DNA site for CRP and, thus, at the wild-type melAB promoter, MelR binding at sites 1′ and 1 must function to recruit CRP to its poor DNA site.
Co-operative binding of MelR and CRP
We have used DNase I footprinting at the JK15 promoter to investigate the co-operative interactions between MelR and CRP. At this promoter, both MelR and CRP are able to bind independently to their respective DNA sites. However, at a sufficiently low concentration, MelR is only able to bind to sites 2 and 2′ in the presence of CRP (Figure 2C). Thus, whilst MelR recruits CRP at the wild-type melAB promoter, the reciprocal interaction is seen at the JK15 promoter, where CRP can recruit MelR.
We reasoned that the co-operative interactions between MelR and CRP may be sufficient to repress transcription initiation at a promoter where MelR and CRP bind to sites overlapping those for RNAP. In order to investigate this, we performed an experiment with the galP1Δ4 promoter, which is a derivative of the E.coli galP1 promoter. In previous work, we demonstrated factor-independent transcription initiation at galP1 but found that this initiation could be stimulated by the binding of CRP to a site centred at position –41.5 (Bingham et al., 1986). However, when the DNA site for CRP was moved to position –37.5 in the galP1Δ4 promoter, we found that CRP repressed the factor-independent transcription initiation (Bell et al., 1990). In this study we constructed a derivative of the galP1Δ4 promoter that contains a weakened DNA site for CRP such that CRP is less able to repress the promoter (Figure 3). A DNA element carrying MelR-binding sites 1′ and 1 was introduced immediately upstream of the DNA site for CRP, with identical spacing to that found at the melAB promoter. Expression from this promoter, JTW1, was then measured in different backgrounds either with or without CRP and MelR (Figure 3). Our results show that MelR alone hardly affects expression from the JTW1 promoter, whilst in the presence of CRP, MelR increases repression 3-fold, presumably because of co-operative interactions between MelR and CRP.

Fig. 3. Activity of the galP1Δ4 and JTW1 promoters. Fragments with the two promoters are illustrated schematically: galP1Δ4 contains a DNA site for CRP (shown as a rectangle) at a location where CRP represses promoter activity, whilst JTW1 is a derivative carrying a weakened DNA site for CRP (lighter shaded rectangle) and an upstream element carrying MelR binding sites 1′ and 1. The promoters were cloned into the lac expression vector, pRW50, and the figure shows β-galactosidase activities measured in different backgrounds either with or without CRP and MelR. The activities (in arbitrary units) were measured in WAM134 (ΔmelR Δcrp Δlac) cells containing the pRW50 derivatives, and either plasmid pDCRP, encoding crp (+CRP), or the control vector pDU9 (–CRP), together with either pLG314, encoding melR (+MelR), or the control vector pLGΔRS (–MelR).
Substitutions within CRP that affect transcription activation at the melAB promoter
To identify the residues of CRP that are important in transcription activation at the melAB promoter, a library of randomly mutated crp genes was created using error-prone PCR. Plasmids expressing mutated crp genes were transformed into a reporter strain carrying a melAB promoter:: lacZ fusion, and colonies were screened on MacConkey lactose agar for CRP mutants that gave altered levels of expression from the melAB promoter whilst retaining the ability to bind DNA, as judged by repression of the galP1Δ4 promoter. Six independent mutants were isolated, three with increased levels of expression, and three with decreased levels of expression from the melAB promoter. Sequencing of the mutated crp genes identified three substitutions within CRP. Two of these, PS160 and QR164, resulted in decreased levels of transcription activation at the melAB promoter, and the third, TI158, resulted in an increase in transcription activation (Table I). None of the three substitutions affected the ability of CRP to bind DNA as judged by band shift assays in vitro and repression of the galP1Δ4 promoter in vivo (data not shown). Remarkably, all three substitutions identify residues that are located within AR1 of CRP, a surface that contacts αCTD during transcription activation at simple CRP-dependent promoters (i.e. promoters that are activated by a single CRP dimer alone; reviewed by Busby and Ebright, 1999). All three substitutions have previously been shown to reduce CRP-dependent transcription activation at simple CRP-dependent promoters (Zhou et al., 1993, 1994; Niu et al., 1994, 1996), and this was confirmed by measuring activation at the CC(-61.5) promoter, a simple Class I CRP-dependent promoter that is often used as a paradigm (Table I).
Table I. Activation by mutant CRP at different promoters.
| Amino acid substitution | β-galactosidase activity (%)a pmelAB::lac fusion | β-galactosidase activity (%)b CC(-61.5)::lac fusion |
|---|---|---|
| Wild-type CRP | 100 | 100 |
| His159→Leu | 95 | 5 |
| Random mutants | ||
| Thr158→Ile | 225 | 10 |
| Pro160→Ser | 52 | 18 |
| Gln164→Arg | 56 | 18 |
| Alanine scan mutants* | ||
| Lys152→Ala | 95 | 100 |
| Gln153→Ala | 95 | 100 |
| Pro154→Ala | 97 | 100 |
| Asp155→Ala | 92 | 95 |
| Met157→Ala | 105 | 95 |
| Thr158→Ala | 120 | 10 |
| His159→Ala | 80 | 90 |
| Pro160→Ala | 63 | 87 |
| Asp161→Ala | 105 | 95 |
| Gly162→Ala | 90 | 25 |
| Met163→Ala | 105 | 90 |
| Gln164→Ala | 105 | 100 |
| Ile165→Ala | 110 | 100 |
| Lys166→Ala | 61 | 100 |
aβ-galactosidase activities were measured in WAM134 (ΔmelR Δcrp Δlac) cells containing pRW50 carrying the KK43 melAB promoter fragment, pLG314 encoding melR, and pDCRP or pYZCRP (*) encoding different mutant crp derivatives as indicated.
bβ-galactosidase activities were measured in WAM134 (ΔmelR Δcrp Δlac) cells containing pRW50 carrying the CC(-61.5) promoter fragment and pDCRP or pYZCRP (*) encoding different mutant crp derivatives as indicated.
Activities are expressed relative to the activity measured in cells carrying pDCRP or pYZCRP (*) encoding wild-type CRP (taken as 100%). The results listed are the average of at least three independent determinations.
Previous studies by Ebright and colleagues exploited alanine scanning to find the key residues of AR1, and identified Thr158 as providing the most important sidechain for activation at simple CRP-dependent promoters (Niu et al., 1994; Zhou et al., 1994). Thus, expression from the melAB promoter was also measured in the presence of mutant CRP derivatives containing single alanine substitutions at amino acids 152–166 (Table I; note that residue 156 of CRP is Ala). Substantial decreases in transcription activation were seen with alanine substitutions of Pro160 and Lys166 (Table I). Taken together, our results argue that residues Thr158, Pro160, Gln164 and Lys166 of CRP participate in transcription activation at the melAB promoter: these residues identify a surface of CRP that overlaps with AR1 (Figure 4).

Fig. 4. Model of CRP. The figure shows a space-filling model of CRP bound to a 22 bp consensus DNA site (Parkinson et al., 1996). Residues important in transcription activation at the melAB promoter are indicated.
Table I compares the effects of different substitutions in CRP on expression from the melAB promoter with expression from CC(-61.5). The results show that substitutions at certain positions in CRP (e.g. Thr158 and His159) have different effects at the two promoters. This suggests that the role of the AR1-containing surface of CRP at the melAB promoter is different from its role at simple CRP-dependent promoters such as CC(-61.5), where it interacts directly with αCTD. Thus, we have investigated the involvement of αCTD in CRP-dependent activation at the melAB promoter by using a preparation of E.coli RNAP, reconstituted with truncated α subunits that lack the C-terminal 73 residues. To do this, we used in vitro transcription assays with pSR plasmid carrying the KK43 fragment, such that transcripts initiating at the melAB promoter run to an efficient terminator in the pSR vector. In our previous work with purified wild-type RNAP, we showed that synthesis of the transcript initiating at the melAB promoter was dependent on the presence of purified MelR and melibiose (Belyaeva et al., 2000). The transcript could be quantified by reference to the factor-independent synthesis of the RNA-1 transcript, and we showed that initiation of the transcript at the melAB promoter was stimulated nearly 2-fold by the presence of cAMP-CRP in the incubation (the reasons why CRP-dependent activation in vitro is much less than in vivo are not clear). Figure 5 shows the results of an experiment in which the transcript initiating at the melAB promoter was quantified after in vitro assays using preparations of E.coli RNAP reconstituted with wild-type full-length α subunits or truncated α-256 subunits that lack the C-terminal 73 residues, purified MelR and purified CRP. The results show that the truncation of the RNAP α subunit causes a reduction in transcription initiation from the melAB promoter. However, CRP is able to activate transcription from the melAB promoter with RNAP containing both full-length and truncated α subunits. The fact that the same level of CRP-dependent transcription activation is seen in vitro with both RNAP preparations argues that CRP activates transcription at the melAB promoter independently of αCTD. We conclude that the residues within AR1 that are important in transcription activation at the melAB promoter are not involved in a contact with αCTD.
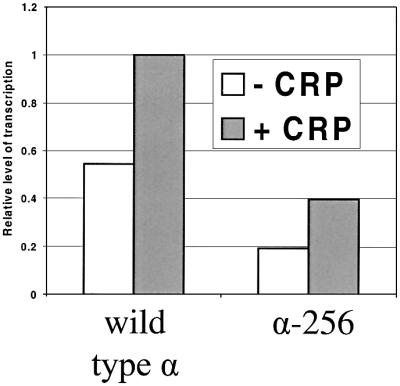
Fig. 5. In vitro transcription from the melAB promoter. The bar chart illustrates amounts of transcripts initiating at the melAB promoter on the KK43 fragment cloned in plasmid pSR. Transcripts were quantified relative to the factor-independent RNA-1 transcript. Assays were performed in the presence of melibiose-MelR and in the absence (open bars) or presence (shaded bars) of cAMP-CRP. Assays were performed with purified RNAP reconstituted with either full-length α (wild-type α) or truncated α lacking the C-terminal 73 residues (α-256).
In order to investigate further the role of residues in and around AR1 in transcription activation at the melAB promoter, we purified CRP carrying the TI158 substitution that improves CRP-dependent activation of the melAB promoter in vivo. We then used DNase I footprinting to study the binding of the wild-type and mutant CRP to the melAB promoter. The results in Figure 6 show that, in the presence of MelR, both wild-type and mutant CRP bind to the CRP target centred at position –81.5 between MelR binding sites 1 and 2. However, mutant Ile158 CRP binds to the promoter with a greater affinity than wild-type CRP and, consequently, the binding of MelR to site 2′ is increased. The simplest explanation for this is that residues in and around AR1 of CRP interact directly with a cognate surface of MelR at the melAB promoter and that substitution of these residues results in altered binding of CRP and MelR to the melAB promoter.
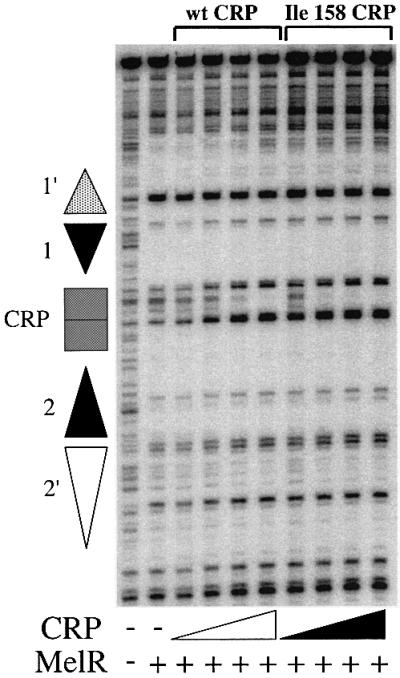
Fig. 6. DNase I footprint of complexes at the melAB promoter. The figure shows footprints obtained in the absence and presence of melibiose-MelR as indicated. Footprints were performed with no CRP, wild-type (wt) CRP (2, 5, 10 or 20 nM) or Ile158 CRP (2, 5, 10 or 20 nM) as shown. The position of MelR binding sites and the target for CRP are indicated by triangles and a rectangle, respectively.
Discussion
MelR and CRP bind together co-operatively at the E.coli melAB promoter to form a large, ordered nucleoprotein complex that covers >100 bp of DNA. The presence of CRP results in an increase in occupancy by MelR of site 2′ that overlaps the promoter –35 element. Our studies have shown that the removal of sites 1′ and 1 upstream of the target for CRP abolishes CRP-dependent transcription activation because upstream-bound MelR is essential for the recruitment of CRP. However, at the promoter (JK14) lacking MelR-binding site 1′ and 1, the introduction of a consensus DNA site for CRP (in the JK15 promoter) restores CRP-dependent transcription activation. This argues that the role of MelR bound at sites 1′ and 1 is to recruit CRP to its target and that upstream-bound MelR is not essential for transcription activation. Working with the JK15 promoter, we were then able to show that pre-bound CRP can recruit MelR to an adjacent binding site. Thus, MelR and CRP bind co-operatively and this provides the mechanism of co-dependence at the melAB promoter. We were able to exploit this co-operative binding to create a promoter where MelR and CRP can function co-operatively together to repress an otherwise factor-independent promoter.
MelR and CRP interact at the melAB promoter to activate transcription, but this interaction can be exploited to repress transcription (Figure 3). In contrast, the anti-activator CytR interacts with CRP to repress transcription at some E.coli promoters (reviewed by Valentin-Hansen et al., 1996). Interestingly, the CytR–CRP interaction can be exploited to activate transcription at an artificial derivative of the lac promoter (Rasmussen et al., 1996). Repression of transcription by CytR is dependent upon a direct protein–protein interaction with CRP involving specific residues within both CytR and CRP (Kallipolitis et al., 1997; Meibom et al., 1999). Similarly, at the melAB promoter, activation by CRP appears to be dependent on direct protein–protein interactions with MelR.
In the second part of this study, we identified substitutions within CRP that alter MelR-dependent transcription activation at the melAB promoter. These substitutions identify the surface of CRP that interacts with MelR (Figure 4). Surprisingly, these substitutions fall within or adjacent to AR1, the surface of CRP that interacts with αCTD at simple CRP-dependent promoters. Since CRP-dependent activation of the melAB promoter is independent of αCTD, we suggest that the role of the CRP surface containing AR1 at the melAB promoter is to contact MelR rather than αCTD. Thus, at this complex promoter, AR1 of CRP has been diverted from its ‘usual’ task of interacting directly with RNAP.
In conclusion, MelR and CRP bind to the melAB promoter co-operatively to adjacent sites in order to activate transcription in a co-dependent manner. This represents a novel mechanism of co-dependence upon two transcription activators at a bacterial promoter. Since this is such a simple mechanism, it is surprising that more examples have not been found at bacterial promoters, especially as this mechanism appears to be widely used in eukaryotes. One of the problems with this mechanism may be that it requires the two activators to be ‘committed’ to each other by carrying complementary contact surfaces, and such ‘commitment’ carries the price of evolutionary inflexibility. With the great increase in our knowledge of bacterial genome sequences, and with many parallel studies on different regulatory regions, it will be especially interesting to see whether the model we have developed for co-dependence at the E.coli melAB promoter turns out to be a ‘one-off’ or a paradigm for many other cases. It will also be interesting to see whether CRP can interact directly with any other member of the large AraC/XylS family of transcription activators.
Materials and methods
Strains, plasmids and promoters
The E.coli K-12 strain used in this work was WAM134, a ΔmelR Δcrp::cm derivative of WAM131 (Belyaeva et al., 2000).
Different EcoRI–HindIII fragments carrying promoters were cloned into the plasmid vectors pRW50 and pSR. pRW50 is a low copy number lac expression vector encoding resistance to 35 µg/ml tetracycline (Lodge et al., 1992), whilst pSR is a pBR322 derivative for cloning promoter fragments upstream of the λ oop transcription terminator (Kolb et al., 1995). By convention, melAB promoter sequences are numbered with respect to the transcription start point with upstream and downstream locations denoted by ‘–’ and ‘+’ prefixes, respectively. The melAB promoter fragments all carry the HindIII site at position +36 and the EcoRI site at position –312 (KK81), –138 (KK43), –188 (KK39) or –94 (JK14), as described by Belyaeva et al. (2000). The Class I CRP-dependent CC(-61.5) promoter was also cloned on an EcoRI–HindIII fragment and used as described by Bell et al. (1990) and Zhou et al. (1994).
Plasmid pLG314, encoding melR expressed from the CRP-independent galP2 promoter, was constructed by subcloning the EcoRI–BamHI fragment from plasmid pCM118-314 (Michán et al., 1995) into the tetR gene of the vector plasmid pLG339, which encodes resistance to 25 µg/ml kanamycin (Stoker et al., 1982). pLG339ΔRS is a derivative of pLG339 carrying a deletion between the EcoRI and SalI sites that results in the loss of tetR (Belyaeva et al., 2000). Wild-type and mutant crp derivatives were cloned in plasmids pDCRP (Bell et al., 1990) and pYZCRP (Zhou et al., 1991), which are pBR322 derivatives encoding resistance to 80 µg/ml ampicillin. As a control plasmid, we used pDU9, a derivative of pDCRP with the crp gene replaced by the EcoRI–HindIII pUC9 polylinker (Bell et al., 1990).
Construction of KK43 p75A
The starting fragment was KK43 (Figure 1A). To construct KK43 p75A, the EcoRI–BglII fragment was replaced with a PCR-amplified fragment generated from KK43 using primer D26440 (5′-AACTCAGATCTTTCGTGAAGCAGC-3′) and a primer upstream of the EcoRI site.
Construction of JK15 and JK15+2
The starting material was the JK14 fragment (Figure 2B). To construct JK15, the JK14 fragment was extended using PCR with primers D27153 (5′-GCAGAATTCAAATGTGATGTACATCACAGGATCTGAGTTTATGG-3′) and D14314 (5′-GCAAAGCTTGGATGCAGGTCGACGGATCTC-3′). To construct JK15+2, the JK14 fragment was extended using PCR with primers D19007 (5′-GCAGAATTCAAATGTGATGTACATCACACAGGATCTGAGTTTATGG-3′) and D14314.
Construction of JTW1
The starting fragments were galP1Δ4 (Bell et al., 1990) and KK43 (Figure 1A). To construct JTW1, an NcoI–HindIII fragment was generated by PCR amplification from galP1Δ4 using primer D29613 (5′-AATAATCCATGGTCACACTGCATCTTTTTTATGCTATGG-3′) and a primer downstream of the HindIII site. An EcoRI–NcoI fragment was generated by PCR amplification from KK43 using primer D29614 (5′-AATAATCCATGGGCAGTAAATCTGAGTTTATGGG-3′) and a primer upstream of the EcoRI site. The EcoRI–NcoI and NcoI–HindIII fragments were ligated together to form the JTW1 promoter.
Screen of library of mutant crp derivatives
Error-prone PCR (Zhou et al., 1991) was used to construct a library of mutant crp derivatives. pDCRP was used as a template with primers D3407 (5′-AGGCGTATCACGAGGCCCT-3′) and D4600 (5′-GTAGTCGGTGTGTTCAC-3′), which anneal upstream of the EcoRI and downstream of the HindIII sites, respectively. EcoRI–HindIII fragments containing mutagenized crp derivatives were subcloned into pDCRP. After eight libraries of pDCRP derivatives had been screened by monitor ing expression of the KK43::lacZ fusion, the base sequence of the entire EcoRI–HindIII insert was determined for all the selected mutants.
Proteins
MelR was overexpressed using the T7 expression plasmid, pCM117-303, as described by Michán et al. (1995). The overexpressed MelR was purified using the method of Caswell et al. (1992), as described by Belyaeva et al. (2000). CRP was purified using the method of Ghosaini et al. (1988), as described by Savery et al. (1998). Holo DNA-dependent RNAP containing either a full-length or a truncated α subunit was a gift from M.Thomas (see Meng et al., 2000).
In vitro transcription assays
These were performed using the system described by Kolb et al. (1995), as described by Savery et al. (1998). Briefly, the KK81 fragment carrying the melAB promoter was cloned into plasmid pSR and the resulting recombinant was purified by caesium chloride gradient centrifugation and used as template for RNA synthesis. After addition of different factors and purified RNAP, a cocktail of labelled nucleoside triphosphate and heparin was added. Transcription initiation at the melAB promoter generates an easily detectable discrete transcript that terminates at the oop terminator, with the control RNA-1 from the ColE1 origin acting as an internal control. The incubation mixture contained 5% glycerol, 20 mM Tris pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 50 µg/ml BSA, 200 µM cAMP, 10 nM melibiose and 10 nM supercoiled plasmid DNA template. MelR (1500 nM) and CRP (50 nM) were added as appropriate, and incubated with 20 nM RNAP for a further 5 min prior to the addition of 200 µM ATP, CTP and GTP, 10 µM α-32P-labelled UTP and 100 µg/ml of heparin. 32P-labelled RNA was analysed on calibrated polyacrylamide sequencing gels precisely as before (Savery et al., 1998). Band intensities were quantified using ImageQuant software (Molecular Dynamics).
Footprinting
DNase I and hydroxyl radical footprinting experiments were performed using protocols described by Savery et al. (1996). In these experiments, incubations contained 4–10 nM of the purified KK81 or KK39 EcoRI–HindIII fragment that had been end-labelled at the HindIII site with [γ-32P]ATP by polynucleotide kinase. Incubations contained 10 mM melibiose, 1000 nM MelR unless otherwise stated, and 200 µM cAMP, 75 nM CRP unless otherwise stated. After DNase I or hydroxyl radical treatment, footprint patterns were analysed on polyacrylamide sequencing gels that were calibrated with Maxam–Gilbert sequence ladders.
Measurement of promoter activities in vivo
DNA fragments containing the promoters were cloned into pRW50, a low copy number lac expression vector, to generate promoter::lac fusions. β-galactosidase levels in cells were measured by the Miller (1972) method: cells were grown in media either with or without melibiose exactly as in our previous work (Webster et al., 1987), and all assays were performed at least three times on independent transformants. Assays were performed in a ΔmelRΔcrp (WAM134) background carrying different plasmids encoding crp and melR.
Acknowledgments
Acknowledgements
We thank Edward Smith, Christine Webster and Rosemary Parslow for excellent technical support, Richard Ebright for providing the pYZCRP alanine scan of AR1 of CRP, and Wenmao Meng and Mark Thomas for supplying purified RNAP with full-length and truncated α subunits. This work was generously supported by the UK BBSRC with a PhD studentship award to J.T.W. and a project grant.
References
- Bell A., Gaston,K., Williams,R., Chapman,K., Kolb,A., Buc,H., Minchin,S., Williams,J. and Busby,S. (1990) Mutations that alter the ability of the Escherichia coli cyclic AMP receptor protein to activate transcription. Nucleic Acids Res., 18, 7243–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva T., Wade,J., Webster,C., Howard,V., Thomas,M., Hyde,E. and Busby,S. (2000) Transcription activation at the Escherichia coli melAB promoter: the role of MelR and the cyclic AMP receptor protein. Mol. Microbiol., 36, 211–222. [DOI] [PubMed] [Google Scholar]
- Bingham A., Ponnambalam,S., Chan,B. and Busby,S. (1986) Mutations that reduce expression from the P2 promoter of the Escherichia coli galactose operon. Gene, 41, 67–74. [DOI] [PubMed] [Google Scholar]
- Browning D., Cole,J. and Busby,S. (2000) Suppression of FNR-dependent transcription activation at the Escherichia coli nir promoter by Fis, IHF and H-NS: modulation of transcription initiation by a complex nucleo-protein assembly. Mol. Microbiol., 37, 1258–1269. [DOI] [PubMed] [Google Scholar]
- Busby S. and Ebright,R. (1994) Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell, 79, 743–746. [DOI] [PubMed] [Google Scholar]
- Busby S. and Ebright,R. (1999) Transcription activation by catabolite activator protein (CAP). J. Mol. Biol., 293, 199–213. [DOI] [PubMed] [Google Scholar]
- Caswell R., Williams,J., Lyddiatt,A. and Busby,S. (1992) Over expression, purification and characterisation of the E. coli MelR transcription activator protein. Biochem. J., 287, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos M.-T., Schleif,R., Bairoch,A., Hofmann,K. and Ramos,J.-L. (1997) AraC/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev., 61, 393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosaini L., Brown,A. and Sturtevant,J. (1988) Scanning calorimetric study of the thermal unfolding of catabolite activator protein from Escherichia coli in the absence and presence of cyclic mono nucleotides. Biochemistry, 27, 5257–5261. [DOI] [PubMed] [Google Scholar]
- Goosen N. and van de Putte,P. (1995) The regulation of transcription initiation by integration host factor. Mol. Microbiol., 16, 1–7. [DOI] [PubMed] [Google Scholar]
- Kallipolitis B., Norregaard-Madsen,M. and Valentin-Hansen,P. (1997) Protein–protein communication: structural model of the repression complex formed by CytR and the global regulator CRP. Cell, 89, 1101–1109. [DOI] [PubMed] [Google Scholar]
- Kolb A., Busby,S., Buc,H., Garges,S. and Adhya,S. (1993) Transcrip tional regulation by cAMP and its receptor protein. Annu. Rev. Biochem., 62, 749–795. [DOI] [PubMed] [Google Scholar]
- Kolb A., Kotlarz,D., Kusano,S. and Ishihama,A. (1995) Selectivity of the Escherichia coli RNA polymerase Eσ38 for overlapping promoters and ability to support CRP activation. Nucleic Acids Res., 23, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge J., Fear,J., Busby,S., Gunasekaran,P. and Kamini,N. (1992) Broad host-range plasmids carrying the E.coli lactose and galactose operons. FEMS Microbiol. Lett., 74, 271–276. [DOI] [PubMed] [Google Scholar]
- Meibom K., Søgaard-Andersen,L., Mironov,A. and Valentin-Hansen,P. (1999) Dissection of a surface-exposed portion of the cAMP–CRP complex that mediates transcription activation and repression. Mol. Microbiol., 32, 497–504. [DOI] [PubMed] [Google Scholar]
- Meng W., Savery,N., Busby,S. and Thomas,M. (2000) The Escherichia coli RNA polymerase α subunit linker: length requirements for transcription activation at CRP-dependent promoters. EMBO J., 19, 1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M. and Thanos,D. (2001) Enhanceosomes. Curr. Opin. Genet. Dev., 11, 205–208. [DOI] [PubMed] [Google Scholar]
- Michán C., Busby,S. and Hyde,E. (1995) The Escherichia coli MelR transcription activator: production of a stable fragment containing the DNA-binding domain. Nucleic Acids Res., 23, 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Niu W., Zhou,Y., Dong,Q., Ebright,Y. and Ebright,R. (1994) Characterization of the activating region of Escherichia coli catabolite gene activator protein (CAP). I. Saturation and alanine-scanning mutagenesis. J. Mol. Biol., 243, 595–602. [DOI] [PubMed] [Google Scholar]
- Niu W., Kim,Y., Tau,G., Heyduk,T. and Ebright,R. (1996) Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell, 87, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson G., Wilson,C., Gunasekara,A., Ebright,Y., Ebright,R. and Berman,H. (1996) Structure of the CAP–DNA complex at 2.5 Å resolution: a complete picture of the protein–DNA interface. J. Mol. Biol., 260, 395–408. [DOI] [PubMed] [Google Scholar]
- Rasmussen P., Holst,B. and Valentin-Hansen,P. (1996) Dual-function regulators: the cyclic AMP receptor protein and CytR can act either to repress or to activate transcription depending on the context. Proc. Natl Acad. Sci. USA, 93, 10151–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet E., Vidal-Ingigliardi,V. and Raibaud,O. (1991) A new mechanism for coactivation of transcription: repositioning of an activator triggered by the binding of a second activator. Cell, 66, 1185–1195. [DOI] [PubMed] [Google Scholar]
- Savery N., Belyaeva,T. and Busby,S. (1996) Protein:DNA interactions. In Docherty,K. (ed.), Gene Transcription: DNA Binding Proteins, Essential Techniques. Wiley/BIOS Press, Chichester, UK, pp. 1–5 and 21–33.
- Savery N., Lloyd,G., Kainz,M., Gaal,T., Ross,W., Ebright,R., Gourse,R. and Busby,S. (1998) Transcription activation at class II CRP-dependent promoters: identification of determinants in the C-terminal domain of the RNA polymerase α subunit. EMBO J., 17, 3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S., Busby,S. and Beacham,I. (1995) Transcriptional co-activation at the ansB promoters: involvement of the activating regions of CRP and FNR when bound in tandem. Mol. Microbiol., 18, 521–531. [DOI] [PubMed] [Google Scholar]
- Stoker N., Fairweather,N. and Spratt,B. (1982) Versatile low-copy number plasmid vectors for cloning in Escherichia coli. Gene, 18, 335–341. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen P., Søgaard-Andersen,L. and Pedersen,H. (1996) A flexible partnership: the CytR anti-activator and the cAMP-CRP activator protein, comrades in transcriptional control. Mol. Microbiol., 20, 461–466. [DOI] [PubMed] [Google Scholar]
- Webster C., Kempsell,K., Booth,I. and Busby,S. (1987) Organisation of the regulatory region of the Escherichia coli melibiose operon. Gene, 59, 253–263. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhang,X. and Ebright,R. (1991) Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res., 19, 6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhang,X. and Ebright,R. (1993) Identification of the activating region of catabolite gene activator protein (CAP): isolation and characterization of mutants of CAP specifically defective in transcription activation. Proc. Natl Acad. Sci. USA, 90, 6081–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Merkel,T. and Ebright,R. (1994) Characterization of the activating region of Escherichia coli catabolite gene activator protein (CAP). II. Role at class I and class II CAP-dependent promoters. J. Mol. Biol., 243, 603–610. [DOI] [PubMed] [Google Scholar]