

Abstract
INI1 (integrase interactor 1)/hSNF5 is a component of the mammalian SWI/SNF complex and a tumor suppressor mutated in malignant rhabdoid tumors (MRT). We have identified a nuclear export signal (NES) in the highly conserved repeat 2 domain of INI1 that is unmasked upon deletion of a downstream sequence. Mutation of conserved hydrophobic residues within the NES, as well as leptomycin B treatment abrogated the nuclear export. Full-length INI1 specifically associated with hCRM1/exportin1 in vivo and in vitro. A mutant INI1 [INI1(1–319) delG950] found in MRT lacking the 66 C-terminal amino acids mislocalized to the cytoplasm. Full-length INI1 but not the INI1(1–319 delG950) mutant caused flat cell formation and cell cycle arrest in cell lines derived from MRT. Disruption of the NES in the delG950 mutant caused nuclear localization of the protein and restored its ability to cause cell cycle arrest. These observations demonstrate that INI1 has a masked NES that mediates regulated hCRM1/exportin1-dependent nuclear export and we propose that mutations that cause deregulated nuclear export of the protein could lead to tumorigenesis.
Keywords: hSNF5/INI1/NES/nuclear export/tumor suppressor
Introduction
The SWI/SNF complex is one of several evolutionarily conserved multi-protein complexes that remodel chromatin in an ATP-dependent manner (Kingston and Narlikar, 1999). The SNF2/SWI2 component of the complex and its mammalian homologs such as BRG1 and hBRM encode the essential DNA-dependent ATPase activity (Khavari et al., 1993; Laurent et al., 1993). INI1/hSNF5/BAF47 is another component of the SWI/SNF complex and one of the few subunits common to all mammalian SWI/SNF complexes identified thus far (Wang et al., 1996). The exact role of INI1 in chromatin remodeling is not understood.
INI1 was initially isolated by the yeast two-hybrid system through its interaction with HIV-1 integrase (IN; Kalpana et al., 1994) and was shown to co-immunoprecipitate with BRM1 (Muchardt et al., 1995). Recently, it has been implicated as a tumor suppressor in atypical teratoid and malignant rhabdoid tumors (ATRT and MRT, respectively; Versteege et al., 1998; Biegel et al., 1999). The majority of these tumors are homozygous null for INI1 with deletions spanning part of or the entire gene. Several tumors also contain point mutations in the gene that would apparently lead to the production of either a truncated protein or a full-length protein with substitutions. Mutations in the INI1 gene predispose individuals to various types of cancer including MRT and tumors of the central nervous system (Sevenet et al., 1999). Gene-targeted disruption of the INI1 gene in mice demonstrated that while the homozygous deletions are embryonic lethal, the heterozygous mice develop a high frequency of tumors (Klochendler-Yeivin et al., 2000; Roberts et al., 2000; Guidi et al., 2001). The tumors exhibited reduction or complete absence of INI1 expression, confirming that it is a tumor suppressor.
The mechanism by which INI1 exerts its tumor suppressor function remains to be elucidated. Several proteins, such as c-MYC, ALL1/hTRX/MLL and GADD35, have been shown to bind to INI1 (Rozenblatt-Rosen et al., 1998; Adler et al., 1999; Cheng et al., 1999). Structure–function analysis indicated that the first of the two highly conserved repeats (Rpt) of INI1 is involved in these interactions. For example, HIV-1 IN and c-MYC specifically interact with Rpt1 but not Rpt2 of INI1 (Morozov et al., 1998; Cheng et al., 1999). However, the function of Rpt2 is unknown. Apart from these two repeats, INI1 has a C-terminal coiled-coil domain (amino acids 334–376), which is moderately conserved in all homologs (homology region 3 or HR3), and a putative N-terminal leucine zipper domain (amino acids 36–64), both of whose functions remain to be determined (Morozov et al., 1998).
It is crucial to characterize the exact function of each of the conserved domains of INI1 in order to understand its function. In this report, we provide evidence for the existence of a hitherto unsuspected masked nuclear export signal (NES) within the Rpt2 domain of INI1 that mediates hCRM1/exportin-dependent nuclear export. Furthermore, our findings, for the first time, provide insight into one of several possible mechanisms of inactivating INI1 in MRT. We report here that unmasking or deregulating the nuclear export property of INI1 leads to mislocalization of the protein and affects its function in regulating the cell cycle, which in turn contributes to oncogenesis in certain MRTs.
Results
INI1 has an NES within the highly conserved Rpt2 region
Although INI1/hSNF5 is a nuclear protein, during our analysis of inhibitors of HIV-1, we discovered that a dominant-negative mutant of INI1 was mislocalized to the cytoplasm (Yung et al., 2001). We used affinity-purified polyclonal antibodies raised against the N-terminal 130 amino acids of the protein [α-N-INI1(PB3)] to confirm that INI1 is strongly nuclear (Figure 1A). We found that INI1/hSNF5 staining was diffuse in the nucleus, exclusive of the nucleoli. Many nuclei exhibited 2–9 distinct and bright punctate spots. To identify the domain(s) responsible for its subcellular localization, we constructed N- and C-terminal fusions of INI1 to green fluorescent protein (GFP) (GFP–INI1 and INI1–GFP, respectively; Figure 1B) and expressed them in HeLa-TetOn cells. Confocal microscope analysis revealed that both GFP–INI1 and INI1–GFP produced a strong nuclear signal consistent with the immunostaining results of endogenous INI1, suggesting that these GFP fusion proteins could be used for further studies (Figure 1C, rows 2 and 3, respectively). We then constructed a panel of INI1 truncations (Morozov et al., 1998) as N-terminal fusions of GFP (Figure 1C). The constructs GFP–N-INI1(1–130) or GFP–ΔINI1(1–245) were localized to the nucleus, indicating that the first 130 amino acids at the N-terminus of INI1 harbor determinants responsible for its nuclear localization (Figure 1C, rows 4 and 5, respectively).

Fig. 1. Subcellular localization of INI1 and its truncations. (A) Indirect immunofluorescence of endogenous INI1 using affinity-purified α-N-INI1(PB3) antibodies. Left panels indicate the fluorescence due to antibodies, while the right panels indicate the DAPI staining of the nuclei. (B) Cartoon illustrating GFP fusions of INI1 truncations. GFP is indicated in green. Rectangular boxes indicate the INI1 region that is retained in the fusion and the thin line indicates the region that is deleted. Within INI1, boxes with arrows indicate repeat domains. Rpt1, repeat 1; Rpt2, repeat2; HR3, homology region 3. Rpt1 is indicated with a gray box, Rpt2 with a white box and HR3 with a black box. Numbers on top of each line indicate the amino acid positions. (C) Confocal microscope analysis of the localization of GFP–INI1 truncations. In each row, the left panel illustrates the GFP fluorescence, the middle panel illustrates nuclear staining by propidium iodide (PI) and the right panel illustrates the overlay of the two. Note the stark cytoplasmic localization of GFP–C19 and GFP–ΔINI1(1–294). The images of HeLa-TetOn cells transfected with GFP–ΔINI1(189–385) were captured on a cooled CCD camera.
Interestingly, we found that GFP–INI1 truncations containing either amino acids 1–276 (GFP–C19) or amino acids 1–294 [GFP–ΔINI1(1–294)] demonstrated dramatic cytoplasmic accumulation (Figure 1C, rows 6 and 7, respectively). This could have been a result of the absence of a potential C-terminal nuclear localization signal (NLS). However, this alone appears to be unlikely, as the INI1 truncation GFP–ΔINI1(1–245), which also lacked the C-terminal domain, was still nuclear. Furthermore, expression of fragments from regions within the C-terminus such as Rpt1 or Rpt2 (as described below) or a fragment containing the HR3 region (amino acids 259–385, data not shown) did not confer nuclear localization to GFP, suggesting that there are no NLS sequences in the C-terminal fragment. These data suggested that the sequence between amino acids 246 and 276 of INI1 is responsible for the cytoplasmic accumulation of truncations lacking C-terminal sequences. To determine whether this small region of INI1 is sufficient to confer cytoplasmic accumulation to a heterologous proteins, we expressed GFP–ΔINI1(247–277) in HeLa-TetOn cells (Figure 2A and B). This GFP fusion protein was predominantly localized to the cytoplasm (Figure 2A, lower panel, row 1). Since amino acids 247–277 span the highly conserved Rpt2 domain, we expressed GFP fusions of Rpt2 (either amino acids 259–319 or 247–319) and found them to localize to the cytoplasm as well. These data indicated that the sequence at 259–277 is sufficient for this process (Figure 2A, lower panel, rows 2 and 3). We then fused the above three fragments to GFP–N-INI1 (Figure 2B, upper panel), which we had shown above to localize to the nucleus of transfected cells. All three fragments were able to dramatically change the nuclear localization of GFP–N-INI1 into a mainly cytoplasmic distribution (Figure 2B, lower panel, rows 1–3).


Fig. 2. Nuclear export signal (NES) in INI1. (A and B) Top panels, cartoon illustrating fusions of INI1 to GFP and GFP–N-INI1. Labeling is as in Figure 1. Bottom panels, confocal microscope analysis of the indicated deletions. Note the stark cytoplasmic localization of all the fusions except GFP–ΔINI1(183–245) and GFP–N-INI1(183–245). (C) Alignment of various NES with the postulated NES of INI1.The symbol Ψ in the consensus sequence designates any of the hydrophobic amino acids leucine, isoleucine or valine and X designates any amino acid. Invariant L266 residue in INI1 is indicated in bold. (D) Alignment of amino acids 259–276 within the Rpt2 region of INI1 with its homologs. The alignment was performed by the PILEUP program in GCG and displayed using the PRETTY program. The proteins aligned are INI1 and its homologs from mouse (mSNF5), the fish Tetradon flaviatus (tfSNF5), Drosophila (SNR1), Caenorhabditis elegans (ceSNF5), the fission yeast Schizosaccharomyces pombe (spSNF5), the budding yeast Saccharomyces cerevisiae (SNF5 and SFH1) and the plant Arabidopsis thaliana (atSNF5). Invariant L266 residue of INI1 is in bold and all the residues that are >70% conserved are in upper case. The gray area indicates the region of INI1 that has homology to NES found in other proteins. Numbers above the sequence indicate the amino acid residues of INI1.
The observed cytoplasmic distribution of INI1 truncations could be due either to a strong cytoplasmic retention activity of amino acids 259–277 or to the presence of an NES within the sequence, suggesting shuttling of the proteins between nucleus and cytoplasm. Amino acids 259–277 of INI1 are rich in highly conserved hydrophobic residues similar to the NES found in proteins that are exported in hCRM1/exportin-dependent pathways (Mattaj and Englmeier, 1998). When amino acids 259–276 of INI1 were aligned with the NES of other proteins, a good match was observed between the proposed ΨXnΨX2ΨXΨ consensus sequence (where Ψ indicates a hydrophobic residue and X indicates any amino acid) and residues 266-LNIHVGNISLV-276 of INI1 (Figure 2C). Additionally, the L266 residue of this sequence is invariant among all INI1 homologs across the phyla, implying a functional significance (Figure 2D). These analyses suggested that this region of INI1 could be acting as an NES.
Disruption of NES abrogates nuclear export
To determine whether the postulated sequence of INI1 is an NES, dependent on hCRM1/exportin1, we carried out the following experiments. First, we carried out an alanine scanning mutagenesis of the hydrophobic residues within region 259–276 of GFP–C19. Introduction of alanine at the invariant L266 residue caused a strong nuclear accumulation of GFP–C19 (Figure 3A, bottom panel, row 2). Furthermore, cytoplasmic localization of GFP– C19 was disrupted when residues aligning with the consensus sequence (L266, V270, I273 and L275) were replaced with alanine (Figure 3B and data not shown). In addition, mutation of I263 to alanine also disrupted the cytoplasmic localization (Figure 3B). Interestingly, this residue may distinguish the nuclear export property of Rpt2 from that of Rpt1, as described below.


Fig. 3. Disruption of nuclear export of INI1 truncation. (A) Top, cartoon illustrating the position of substitutions in GFP–C19(1–276). Bottom, confocal microscope analysis of GFP–C19 containing the substitutions indicated. Labeling is similar to that in Figure 1. (B) Alanine scanning mutagenesis of hydrophobic residues in the NES to determine their effect on nuclear export. Substitution mutations generated in GFP–C19 are designated on the left side of the diagram and the localization of the mutant protein is indicated on the right side. The position of the alanine substitution is indicated. The gray bar indicates those alanine substitutions that affect the localization. (C) Alignment of Rpt1 and Rpt2 regions of INI1 and its homologs. Alignment was performed using the PILEUP program of GCG as above and displayed using the PRETTY program. The invariant leucine and proline residues of Rpt1 and Rpt2 are indicated in bold. The numbers above the sequence indicate the positions of amino acids in Rpt1 and Rpt2 of INI1. Assignment of abbreviations for INI1 homologs is as in Figure 2D. (D) Confocal microscope analysis of the HeLa-TetOn cells transfected with GFP–C19, when treated with LMB for 6 h. (E) GFP–C19 is shuttling between the nucleus and the cytoplasm. The panels indicate the time-lapse video micrography of GFP fluorescence from HeLa-TetOn cells transfected with GFP–C19 in the presence of LMB and cycloheximide. The pictures of the cell were taken at 4 min intervals. Note the movement of fluorescence from the cytoplasm into the nucleus.
Although the two conserved imperfect repeats of INI1 (Rpt1 and Rpt2) have extensive homology to each other, Rpt1 is not sufficient for nuclear export (Figure 1B and C). Furthermore, fusion of Rpt1 itself to GFP or to GFP–N-INI1 (amino acids 1–130) did not result in cytoplasmic accumulation (Figure 2A, lower panel, row 4 and B, lower panel, row 4, respectively). Alignment of Rpt2 with Rpt1 from all INI1 homologs reveals that while Rpt2 of INI1 contains an isoleucine residue at position 263, Rpt1 contains an invariant proline in the corresponding position (Figure 3C). We mutated I263 of Rpt2 to proline in GFP–C19 to more closely mimic the homologous region of Rpt1 (Figure 3A). The results indicated that GFP–C19(I263P) no longer localized to the cytoplasm exclusively but was seen throughout the cell (Figure 3A, bottom panel, row 4). These results suggested that in addition to the hydrophobic residues that match the NES consensus, residue I263 of Rpt2 is important for NES function.
Treatment with leptomycin B abrogates nuclear export
We next examined the effect of leptomycin B (LMB) on the cytoplasmic accumulation of GFP–C19. LMB is a fungal compound that inhibits nuclear export by covalently modifying and thus inactivating hCRM1/exportin1 (Kudo et al., 1998, 1999). We transfected HeLa-TetOn cells with GFP–C19 and incubated them with 10 ng/ml LMB. Within 2 h, GFP–C19 that was exclusively located in the cytoplasm before the addition of LMB began to appear in the nucleus. After 6 h, GFP–C19 exhibited a robust nuclear signal (Figure 3D). To further confirm these results, we carried out time-lapse video micrography of the LMB-treated HeLa-TetOn cells transfected with GFP–C19 in the presence of the protein synthesis inhibitor cycloheximide at 4 min intervals (Figure 3E). Within 12 min, GFP–C19 began to appear in the nucleus. After ∼44 min, GFP–C19 exhibited a robust nuclear signal. Accumulation of the protein in the nucleus upon the addition of LMB indicates that GFP–C19 probably undergoes nucleo-cytoplasmic shuttling. Our observed time course of inhibition is similar to those of reported studies with other NES-containing proteins (Wolff et al., 1997; Kudo et al., 1998; Toyoshima et al., 1998).
INI1 is able to interact with hCRM1/exportin1 in vivo and in vitro
hCRM1/exportin1 interacts with proteins bearing an NES (Fornerod et al., 1997a,b; Fukuda et al., 1997; Ossareh-Nazari et al., 1997; Stade et al., 1997). Since LMB abrogates the nuclear export of INI1 truncations, it is likely that the observed export is mediated by the interaction of its NES with hCRM1/exportin1. Therefore, we performed immunoprecipitations of cells transiently transfected with either GFP–INI1 or GFP alone (as a control) and analyzed the immunoprecipitates using α-hCRM1 antibodies (Fornerod et al., 1997b). The results indicated that GFP antiserum was able to pull down the nuclear export receptor hCRM1/exportin1 only in the presence of GFP–INI1 (Figure 4A, lane 2), but not GFP (Figure 4A, lane 6).

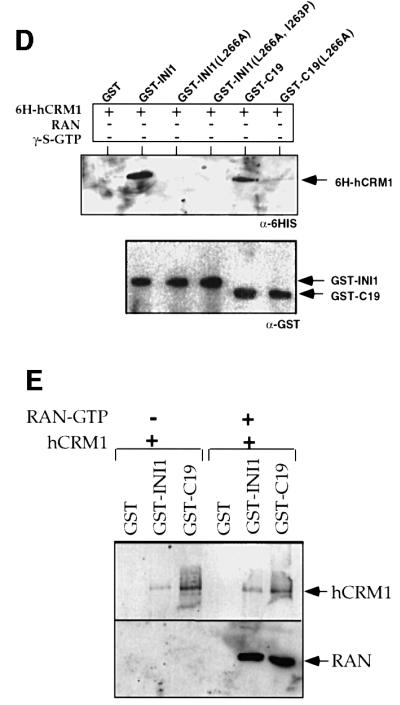
Fig. 4. INI1 interacts with hCRM1/exportin1 in vivo and in vitro. (A) Co-immunoprecipitation of endogenous hCRM1/exportin1 with transiently transfected GFP–INI1. The total proteins isolated from HeLa-TetOn cells transfected either with GFP or with GFP–INI1 were immunoprecipitated using α-GFP antibodies, α-HA antibodies or no antibodies. The resulting immunoprecipitates were separated by SDS–PAGE, blotted onto nitrocellulose and probed with α-hCRM1 antibodies to determine the ability of GFP–INI1 to immunoprecipitate hCRM1/exportin1. IP indicates the antibodies used for immunoprecipitation. Lysate is a fraction (1/20) of input. (B) Immunoblot analysis of total proteins isolated from the cell line EE, stably expressing tetracycline-inducible GFP–INI1. (C) Immunoprecipitation of GFP–INI1 with hCRM1/exportin1 in EE stable cell line. IP indicates the antibodies used for immunoprecipitation. The top blot illustrates the results of immunoblotting with α-hCRM1 antibodies and the lower blot illustrates the immunoblotting results of the same samples with α-GFP antibodies. The lysate indicates the fraction of (1/100) of the input lysate. Note that in the lower blot, lane 4 (Lysate) shows a band corresponding to GFP–INI1, which is only seen upon longer exposure (not shown). (D) In vitro interaction of INI1 with hCRM1. The diagram illustrates the immunoblot analysis of the binding reaction using the indicated GST–proteins and His6-hCRM1 expressed in bacteria. The top blot illustrates the immunoblot analysis of the reaction using α-His6 antibodies; the bottom blot illustrates the immunoblot analysis of the same blot after stripping, using α-GST antibodies. The band from the GST lane is not illustrated in the bottom blot due to excessive signal. Note: His6-hCRM1 is shown as 6H-hCRM1 in the figure. (E) Interaction of INI1 and C19 with Ran–GTP. The binding reaction between GST–INI1, GST–C19 with His6-hCRM1 was carried out in the presence and absence of Ran–GTP. The top blot illustrates the immunoblot analysis of the reactions using α-His6 antibodies. The bottom blot illustrates the immunoblot analysis of the same blot using α-Ran antibodies.
We next determined whether hCRM1/exportin1 associates with GFP–INI1 when expressed at physiological levels. We isolated a HeLa-TetOn cell line (termed EE) carrying stably integrated GFP–INI1 under the control of a tetracycline-inducible promoter (Gossen and Bujard, 1992). When EE cells were treated at 1 µg/ml doxycycline, GFP–INI1 was induced at levels that were similar to or less than that of endogenous INI1, as determined by immunoblot analysis using α-N-INI1(PB3) antibodies (Figure 4B). Total proteins isolated from induced cells were co-immunoprecipitated using α-GFP antibodies and probed with α-hCRM1 antibodies as before. The results clearly indicated that α-GFP antiserum was able to co-precipitate hCRM1/exportin1 with GFP–INI1 in this stable cell line, whereas α-HA antiserum or the no antiserum control was not (Figure 4C, lanes 1–3). Thus, we concluded that hCRM1/exportin1 could interact with physiological levels of INI1.
To determine whether the interaction between full-length INI1 and hCRM1/exportin1 is direct and whether the interaction of INI1 with hCRM1 is abrogated by the substitution of NES residues by alanine, we tested the ability of glutathione S-transferase (GST) fusions of INI1, INI1(L266A), INI1(L266A, I263P), C19 and C19(L266A) to interact with His6-tagged hCRM1, expressed in bacteria. After the binding reaction, the bound proteins were analyzed using α-His6 antibodies to determine the level of bound His6–hCRM1. The results indicate that GST–INI1 and GST–C19 proteins were able to interact with hCRM1 (Figure 4D, top panel). However, GST and the GST fusions harboring mutant NES did not, despite the presence of similar levels of proteins in the reaction (Figure 4D, top and bottom). These results are consistent with the subcellular localization studies, where both L266A and I263P mutants disrupted the cytoplasmic localization, suggesting that we have identified a bona fide NES in INI1.
The in vitro reactions described above were not quantitative and were performed using an excess of proteins in the reaction. To determine the relative strengths of interaction of INI1 and C19 and to determine whether INI1 and C19 can form a ternary complex with hCRM1 and Ran–GTP, we carried out in vitro reactions as above in the presence and absence of purified Ran–GTP. The proteins bound to GST, GST–INI1 or GST–C19 were analyzed by sequential immunoblot analysis using α-His6 and α-Ran antibodies. The results indicated that GST–C19 protein exhibited relatively higher interaction levels with hCRM1 compared with that of GST–INI1 (Figure 4E). Interestingly, when Ran–GTP was present in the reaction, GST–INI1 and GST–C19 proteins were able to pull down both hCRM1 and Ran–GTP, indicating that a ternary complex is formed between the three proteins (Figure 4E). However, the presence of Ran–GTP did not enhance the ability of C19 to bind to hCRM1. Thus, it appears that INI1 and C19 have the unusual ability to bind directly to hCRM1 in the absence of Ran–GTP.
Altered subcellular localization of mutant INI1 found in MRT
Several rhabdoid tumors contain INI1 mutations in various parts of the gene which potentially result in truncated proteins (Versteege et al., 1998; Biegel et al., 1999; Grand et al., 1999; Rousseau-Merck et al., 1999; Sevenet et al., 1999). We noticed that one rhabdoid tumor (WT) had been determined to have a deletion of 1 bp at nucleotide 950 of INI1 (Versteege et al., 1998). This mutation is predicted to cause a premature stop codon at amino acid position 319, resulting in a truncated protein with deletion of HR3, but still retaining the other two highly conserved regions Rpt1 and 2 (Figure 5A). Based on our analysis, deletion of the region downstream of amino acids 276 and 294 results in the constitutive nuclear export of INI1. We surmised that the mutation in WT would also result in the removal of the inhibitory region at the C-terminus, which may lead to the cytoplasmic accumulation of the truncated protein. To test this hypothesis, we introduced a 1 bp deletion identical to that found in WT at nucleotide 950 of GFP–INI1 and monitored the subcellular localization of this mutant protein in HeLa-TetOn cells. As expected, GFP– INI1(1–319 delG950) exhibited dramatic cytoplasmic accumulation (Figure 5B). When cells transfected with GFP–INI1(1–319 delG950) were treated with LMB, it resulted in abrogation of the cytoplasmic accumulation (Figure 5B). To further substantiate these results, we performed immunohistochemistry on the cell line STA-WT1, which is derived from the WT1 tumor using affinity-purified [α-N-INI1(PB3)] antibodies. Unlike the nuclear staining observed for HeLa-TetOn cells, STA-WT1 cells exhibited distinct cytoplasmic staining (Figure 5C). All these data are consistent with our hypothesis that removal of the C-terminal domain results in constitutive nuclear export of the truncated protein.


Fig. 5. Altered localization of INI1 mutants as a mechanism for tumorigenesis in MRT. (A) Schematic representation of INI1 mutation found in one of the MRT, WT. Deletion of a nucleotide (delG950) downstream of Rpt2 of INI1 introduces a stop codon at position 319, deleting the postulated inhibitory region that masks the NES. (B) Confocal microscope analysis of a HeLa-TetOn cell transfected with mutant GFP–ΔINI1 (1–319 delG950) in the presence and absence of LMB. Panels are as indicated in Figure 1. (C) Immunostain analysis of endogenous ΔINI1 protein in STA-WT1 cells with affinity-purified N-INI (PB3) antibody or pre-immune serum. (D) Introduction of L266A mutation into delG950 mutant causes a shift in the subcellular localization of the mutant to the nucleus. The top three panels illustrate the GFP fluorescence and the bottom three panels illustrate PI staining. (E and F) Nuclear localization is necessary for flat cell formation by INI1 in MON cells. The bar chart illustrates the percentage of flat cells formed when INI1–/– MON cells were transfected with GFP–INI1, GFP–INI1(1–319delG950) or GFP–INI1(1–319delG950)L266A. (F) Representative images of the cells from the above transfection are included below the graph, illustrating the flat cells.
Full-length but not the constitutive export mutant of INI1 causes flat cell formation in MRT-derived cell lines
To demonstrate that the constitutive nuclear export results in inactivation of crucial INI1 function, we carried out an INI1-mediated cell cycle arrest assay that we have developed in the laboratory (Z.Zhang and G.V.Kalpana, submitted). We have observed that when INI1 is introduced into MON and other cell lines derived from MRT or ATRT harboring biallelic deletions of the INI1 gene (but not into INI1+/+ cell lines), it causes cell cycle arrest and flat cell formation. These properties are consistent with the tumor suppressor function of INI1. Similar flat cells have been observed when the retinoblastoma protein (Rb) is reintroduced into the Rb-negative SAOS-2 cell line (Zhang et al., 2000) and when BRG1 is reintroduced into the BRG1-deficient SW13 cell line (Dunaief et al., 1994). We utilized this ability of INI1 to determine the significance of its nuclear export property in tumorigenesis.
We surmised that the cytoplasmic accumulation might make the delG950 mutant protein unavailable for its function of cell cycle arrest. We tested the ability of GFP–INI1(1–319 delG950) to cause flat cell formation when introduced into MON cells. After transfection, the MON cells were selected for G418 resistance and at ∼12–15 days post-transfection the percentage of flat cells formed was determined. The results indicated that expression of full-length GFP–INI1 in MON cells caused nearly 90% of the transfected cells to turn flat (Figure 5E). This also indicated that the GFP fusion protein we have utilized in our studies is functional. Interestingly, unlike full-length GFP–INI1, GFP–INI1(1–319 delG950) exhibited a drastic reduction in flat cell formation (Figure 5E and F), suggesting that cytoplasmic accumulation abrogates the ability to cause flat cell formation.
However, it is possible that the inability of the mutant protein to cause flat cell formation may simply be due to the lack of the 66 C-terminal amino acids and not to its cytoplasmic accumulation. To resolve this, we forced the delG950 mutant into the nucleus by introducing the L266A mutation which disrupted the NES function [INI1 (1–319 delG950 L266A)] and tested its ability to cause flat cell formation. Microscope analysis indicated that the mutant was localized to the nucleus, as opposed to the parent protein, which localized to the cytoplasm (Figure 5D, third panel). Western analysis of the transfected cells suggested that GFP–INI1(1–319 delG950) and GFP–INI1 (1–319 delG950)L266A are both expressed at similar levels in MON cells (data not shown). The results of the flat cell assay suggested that unlike GFP–INI1(1–319 delG950), NES-defective GFP–INI1 (1–319 delG950)L266A exhibited a significant degree of flat cell formation similar to that of the wild-type GFP–INI1 (Figure 5E and F). These results indicate that the mutant INI1 found in WT tumors is not able to cause cell cycle arrest due to its cytoplasmic localization and that redirecting the mutant protein to the nucleus by disrupting its NES is sufficient to revert this phenotype.
Discussion
We have discovered the presence of an NES within the Rpt2 region of INI1 that has the ability to confer cytoplasmic localization to a reporter protein. The following evidence strongly supports this view. First, the NES of INI1 closely conformed to the consensus of typical nuclear export signals, ΨXnΨX2ΨXΨ, where Ψ is any hydrophobic residue (leucine, isoleucine or valine) and X is any amino acid (for NES review see Nigg, 1997; Ullman et al., 1997; Izaurralde and Adam, 1998; Mattaj and Englmeier, 1998). In particular, the L266 residue, which is present within the NES, was invariant among all INI1 homologs (Morozov et al., 1998). Secondly, mutagenesis of the hydrophobic residues that match the consensus of other NES sequences caused a radical redistribution of the protein from the cytoplasm to predominantly the nucleus, consistent with other NES mutagenesis studies (Wen et al., 1995; Wada et al., 1998). Thirdly, GFP–INI1 and hCRM1/exportin1, a recently discovered receptor mediating the export of proteins with an NES in a Ran–GTP-dependent fashion (Fornerod et al., 1997a,b; Fukuda et al., 1997; Ossareh-Nazari et al., 1997; Stade et al., 1997), was shown to interact by co-immunoprecipitation and in vitro interaction studies. Mutations in the NES that disrupted nuclear export reduced the interaction of full-length INI1 as well as C19 with hCRM1, in vitro. Fourthly, LMB, a specific inhibitor of nuclear export mediated by hCRM1/exportin1, inhibited nuclear export of INI1 lacking the C-terminal domain, indicating shuttling of this protein. Interestingly, Rpt1 alone appeared not to be sufficient for nuclear export, probably due to the presence of an invariant proline residue. This observation provides additional evidence for differential function of the two repeats despite their similarity (Morozov et al., 1998).
Models of nuclear export of INI1
The presence of an NES in INI1 is unprecedented, as it is a nuclear protein involved in chromatin remodeling and transcription. The data that we have presented above indicate that removing the C-terminal domain results in constitutive hCRM1/exportin1-dependent nuclear export of the truncated protein. One model that explains our data is as follows (Figure 6). INI1 has an N-terminal NLS and a latent NES within Rpt2. The NES is either not active or shows a lower rate of export when the downstream C-terminal 319–385 fragment is present. When present, the C-terminal domain somehow hinders the NES either by slowing the rate of export or by inhibiting it. It may achieve this task either via steric hindrance or by binding to a cellular protein in such a way that either only the N-terminal NLS is active in full-length INI1 or the rate of import is greater than the rate of export. It is possible that at the appropriate time and by proper stimuli, either a change in conformation or altered binding of a cellular protein to INI1 could relieve the block on the NES, leading to active nuclear export. This model accounts for why the full-length protein is primarily nuclear and explains why the NES is unmasked by deletion of the C-terminus, as seen in the mutants found in the tumors. A heterokaryon assay to determine whether full-length GFP–INI1 protein shuttles was negative (data not shown), consistent with the idea that the C-terminus inhibits the export.

Fig. 6. Model for regulated nuclear export of INI1. (1) INI1 is usually nuclear as its NES is masked either by the binding of a cellular protein that blocks the NES or by steric hindrance caused by the folding of a C-terminal inhibitory sequence. (2) Physiological signal(s) could reveal the NES either due to the removal of a protein that masks the NES or by relieving the steric hindrance caused by the inhibitory sequence by altering the protein conformation. (3) Unmasking of the NES mediates the active nuclear export. (4) Deletion of the downstream inhibitory sequence unmasks the NES and results in constitutive nuclear export, as exemplified by the INI1/hSNF5 mutant found in the MRT cell line, WT. NLS, nuclear localization signal; NES, nuclear export signal; IS, inhibitory sequence that blocks the NES; X, hypothetical cellular protein; Rpt1, repeat 1; Rpt2, repeat 2; HR3, homology region 3.
Nature of the sequence that masks the NES
There are several precedents for how an NES can be regulated by other sequences within a protein. For example, p53 was found to have an NES within its tetramerization domain and its subcellular localization was found to be influenced by its oligomerization state (Stommel et al., 1999). The influenza NS1 protein contains charged residues upstream of its NES (Li et al., 1998), which, when deleted or mutated, relieve the inhibition of NES activity. E4 34 kDa is responsible for up-regulating the export of adenoviral mRNA and contains a dominant arginine-rich C-terminal retention domain upstream of its NES, thus preventing cytoplasmic localization in the context of the full-length protein (Dobbelstein et al., 1997). In the case of INI1, the C-terminal inhibitory sequence also contains an arginine-rich sequence ∼100 amino acids downstream of the NES (363- KKIRDQDRNTRRMRRL-378) and this sequence falls within the highly conserved coiled-coil domain of INI1. All our mutants that localized to the cytoplasm lacked this arginine-rich sequence. Further experiments are needed to determine whether this motif acts as an inhibitory domain. Alternatively, the C-terminal domain may have a second NLS that is dominant over the NES and its removal would cause the NES to be unmasked. However, our deletion analysis suggests that fragments containing Rpt1, Rpt2 or HR3 did not show exclusive nuclear localization (data not shown), thus excluding the possibility that removal of an NLS is the cause of unmasking of the NES.
Stimuli that unmask the NES of INI1
At present, it is not clear what normal physiological stimuli may unmask the NES to result in nuclear export of INI1. Since INI1 is present as part of the SWI/SNF complex, it is possible that its association with another protein of the complex will mask the NES, and when that association is disrupted, the NES of INI1 could be activated. By co-immunoprecipitation studies, it was demonstrated that hBRM1, the ATPase component of the SWI/SNF complex, associates with INI1 (Muchardt et al., 1995). However, it is likely that the association of INI1 and hBRM1 or BRG1 is indirect, as indicated by the reconstitution studies (Phelan et al., 1999). Interestingly, it appears that infection of cells by HIV-1 may stimulate nuclear export of INI1. Turelli et al. (2001) have described the induced export of INI1 during the early stages of HIV-1 infection, demonstrating the co-localization of INI1 and another nuclear protein, promyelocytic leukemia protein (PML), with the incoming viral DNA. This export appears to be inhibited by LMB, consistent with our model of export of INI1 in an hCRM1/exportin1-dependent pathway. It is yet to be determined whether disruption of the NES abrogates the export of INI1 in HIV-1-infected cells, to support the hypothesis that the NES of INI1 is important for its HIV-1-induced export. Nevertheless, these observations suggest that full-length INI1 could be induced to be exported. These studies also suggest new lines of research to address the molecular nature of the stimulus and the exact nature of the interaction between viral and cellular proteins.
INI1 nuclear export and cancer
Introduction of INI1 into cells derived from MRT and ATRT results in the formation of flat cells and cell cycle arrest, consistent with INI1 being a tumor suppressor (Z.Zhang and G.V.Kalpana, submitted). Mutations that either disrupt the NES or result in constitutive nuclear export may mislocalize INI1, blocking its normal function. Consistent with this idea, a few truncating mutations that result in removal of the downstream inhibitory sequence of the postulated NES of INI1 have been identified in MRTs and we have demonstrated that mutant INI1 (delG950) found in MRT localizes to cytoplasm. We have also demonstrated that full-length INI1, but not the delG950 mutant, causes cell cycle arrest and that disruption of the NES in delG950 redirects the mutant protein to the nucleus and restores the ability of this protein to cause cell cycle arrest. These data clearly indicate for the first time that nuclear localization is important for the ability of INI1 to cause cell cycle arrest and deletion of the masking domain results in constitutive nuclear export and abrogation of this activity. We suggest that mislocalization and constitutive nuclear export is one of several possible mechanisms of inactivation of INI1 leading to tumorigenesis. In MRT, a wide spectrum of mutations have been found spanning the entire length of the INI1 protein. Since Rpt regions have been demonstrated to exhibit important protein–protein interactions, removal of these domains could make the truncated protein non-functional. However, it was not apparent why truncating the region downstream of Rpt2 could result in tumorigenesis. Our studies indicate that this could be due to the constitutive nuclear export of the truncated protein. Interestingly, p53 also appears to have a masked NES, which is unmasked in certain cancer cells, leading to hyperactive nuclear export and cytoplasmic accumulation of mutant protein (Moll et al., 1992; Schlamp et al., 1997).
In summary, we report a nuclear export sequence in the highly conserved Rpt2 domain of INI1. The fact that NES is conserved among all INI1 homologs and is masked in the full-length protein strongly suggests that INI1 nuclear export is an important and highly regulated process. It is likely that there is a dynamic equilibrium between the exposed and masked form, which would allow the nucleo-cytoplasmic trafficking by INI1. Elucidating the factors responsible or conditions that favor and promote the unmasking of this signal will be a crucial step in understanding fully the role of INI1 export in cellular and viral processes.
Materials and methods
Plasmids
All cloning was performed in the DH5α strain of Escherichia coli unless noted otherwise. Plasmid pGFP-INI1 (GFP fused to the N-terminus of INI1) was generated by inserting a 1.3 kb EcoRI fragment of pSH2-INI1 (Cheng et al., 1999) into the EcoRI site of pEGFP-C2 (Clontech). Plasmid pINI1-GFP (C-terminal fusion of GFP to INI1) was generated by inserting the PCR-amplified BamHI and EcoRI INI1 fragment into pEGFP-N1 (Clontech). A panel of plasmids expressing INI1 truncations as a fusion to GFP, such as GFP–N-INI1 (amino acids 1–130), GFP–ΔINI1 (1–245), GFP–C19 (1–276), GFP–ΔINI1 (1–294) and GFP–ΔINI1 (189–385), was constructed by inserting the BamHI and SalI fragment isolated from the corresponding pGADNot clones (Morozov et al., 1998) into pEGFP-C2 digested with BglII and SalI. Fragments of cDNA encoding Rpt1 (INI1 183–245), Rpt2 (INI1 259–319) and amino acids 247–277 of INI1 were amplified using PCR and fused to GFP and GFP–N-INI1. Tetracycline-inducible GFP–INI1 plasmid (pTc-GFP-INI1) was generated by excising an NheI–XbaI fragment encoding GFP–INI1 and cloning into the same sites of pUHD10.3 (Gossen and Bujard, 1992). Both pGFP-INI1 and pUHD10.3 were isolated from the Dam– strain of E.coli GM2163.
Mutagenesis
Site-directed mutagenesis was performed using the QuikChange kit (Stratagene) as described by the manufacturer. All point mutants generated were confirmed by sequence analysis.
Cell culture, transfections and microscopy
HeLa-TetOn (Clontech) cells were propagated in Dubecco’s modified Eagle’s medium with 10% fetal calf serum. Transfections were performed at 30–50% confluency, using 2 µg of DNA in Lipofectin (Gibco) per 100 mm tissue culture dish. After overnight incubation, fresh medium was added and cells were viewed 6–12 h post-transfection. LMB (a kind gift from Dr Yoshida, University of Tokyo) was added to the medium to a final concentration of 10 ng/ml for 6 h. For the purpose of microscopy and immunostaining, cells were cultured and transfected on 12 mm circular coverslips (0.13–0.17 mm thick) that had been autoclaved. Cells were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature. After fixing, the cells were washed in PBS and permeabilized in 0.5% Triton X-100 in PBS for 10 min. Finally, the cells were washed with PBS and then stained with either Hoechst 33258 (1 µg/ml) or treated with 1 µg/ml RNase A for 30 min at 37°C and then stained with propidium iodide (40 µg/ml). Cells were mounted on a glass slide in 90% glycerol in PBS (pH 8.0) with 1 mg/ml p-phenylenediamine as an anti-photobleaching agent. Confocal images were captured on a Bio-Rad MRC6000. An Olympus IX70 was used to capture images digitally with a Photometrics cooled CCD camera or on photographic film.
Indirect immunofluorescence
HeLa-TetOn cells were grown on cover slips and fixed as described previously. Cells were permeabilized in 1% Triton X-100 in PBS, washed in PBS and blocked in 3% bovine serum albumin (BSA) or normal goat serum in PBS. Cells were washed and incubated in either affinity-purified antiserum (see below) or pre-immune serum treated in an identical fashion at a dilution of 1:400–1:100 overnight at 4°C. Cells were then washed three times in PBS and stained with fluorescein-conjugated donkey α-rabbit serum at a concentration of 1:100 (Amersham) and co-stained either with Hoechst 33258 or propidium iodide as described above. The antiserum [α-N-INI1(PB3)] used was raised in rabbits against a GST fusion to the first 130 N-terminal amino acids of INI1 (GST–N-INI1) and was affinity purified.
Immunoprecipitation
Immunoprecipitation was performed as previously described with minor modifications (Cheng et al., 1999). Briefly, HeLa-TetOn cells either transiently transfected with GFP–INI1 or stably expressing GFP–INI1 under inducible conditions were sonicated and polyclonal GFP antiserum (Invitrogen) was added to pre-cleared lysate for 2–6 h at 4°C. Protein A–Sepharose beads (30 µl of a 50% slurry) were then added for 1 h. Beads were washed seven times. The resultant protein was separated by SDS–PAGE and probed with α-hCRM1 antibodies at a dilution of 1:2000 (a kind gift from Dr Gerard Grosveld).
In vitro binding assays
In vitro binding assays were carried out essentially as before using GST–INI1 fusion proteins and His6-hCRM1 expressed in bacteria and using the same buffer conditions as reported (Cheng et al., 1999). To test the formation of the ternary complex between INI1, hCRM1 and Ran–GTP, we took ∼1.25 nmol each of GST, GST–INI1 or GST–C19 bound to G-beads and incubated it with a bacterial lysate containing a similar amount of hCRM1. In one set of binding reactions we also included purified Ran protein (a kind gift from Elias Coutavas, Rockefeller University) and an excess of the non-hydrolysable GTP analog GTP-γ-S to induce the formation of stable Ran–GTP complex (Coutavas et al., 1993). After the reaction, the bound proteins were analyzed using α-His6 antibodies (a kind gift from Dr Peter Davies, Albert Einstein College of Medicine) or α-Ran antibodies (catalogue No. SC-1156, Santa Cruz, CA).
Flat cell assay
MON cells at 30–50% confluency, plated on 3.5 cm or 6-well plates, were transfected with 1.5 µg of plasmids expressing GFP fusions of INI1 or its mutants. After transfection, cells were cultured for 24–48 h prior to drug selection. Cells were maintained under neomycin selection for 12–15 days with several changes of media, prior to observation under the microscope. The percentage flat cell formation was scored by counting the total number of cells as well as the total number of flat cells in 8–12 independent microscope fields totaling several hundred cells. Three independent transfections were carried out to arrive at the values. The average total cells and flat cells per field and per transfection was determined to obtain the percentage flat cell formation.
Acknowledgments
Acknowledgements
We thank Dr Grosveld of St Jude Children’s Research Hospital, Dr Yoshida of the University of Tokyo, Dr Delattre of the Institute Curie, Paris, Dr Ambros of the Children’s Cancer Research Institute, Austria and Dr Coutavas of the Rockefeller University, for reagents. We thank Dr Hope at the University of Illinois, Chicago, and Dr Singer and Dr Meier at the Albert Einstein College of Medicine for advice, Mr Cammer and the staff at the analytical imaging facility for assistance with microscopy, and Drs Emmons, Kielian, Birshtein and Prasad at the Albert Einstein College of Medicine, Dr Goff at Columbia University, and Drs Trono and Turelli at the University of Geneva, Switzerland, for critically reading the manuscript. We also thank E.Yung for help in generating sequence alignments using the PILEUP program and A.Morozov for INI1 clones. This work was supported by NIH grant AM/GM 39951-04A1 and AmFAR grant 02809-30-RGT to G.V.K.
References
- Adler H.T., Chinery,R., Wu,D.Y., Kussick,S.J., Payne,J.M., Fornace,A.J.J. and Tkachuk,D.C. (1999) Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 protein. Mol. Cell. Biol., 19, 7050–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegel J.A., Zhou,J.Y., Rorke,L.B., Stenstrom,C., Wainwright,L.M. and Fogelgren,B. (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res., 59, 74–79. [PubMed] [Google Scholar]
- Cheng S.W., Davies,K.P., Yung,E., Beltran,R.J., Yu,J. and Kalpana,G.V. (1999) c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nature Genet., 22, 102–105. [DOI] [PubMed] [Google Scholar]
- Coutavas E., Ren,M., Oppenheim,J.D., D’Eustachio,P. and Rush,M.G. (1993) Characterization of proteins that interact with the cell-cycle regulatory protein Ran/TC4. Nature, 366, 585–587. [DOI] [PubMed] [Google Scholar]
- Dobbelstein M., Roth,J., Kimberly,W.T., Levine,A.J. and Shenk,T. (1997) Nuclear export of the E1B 55 kDa and E4 34 kDa adenoviral oncoproteins mediated by a rev-like signal sequence. EMBO J., 16, 4276–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief J.L., Strober,B.E., Guha,S., Khavari,P.A., Alin,K., Luban,J., Begemann,M., Crabtree,G.R. and Goff,S.P. (1994) The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell, 79, 119–130. [DOI] [PubMed] [Google Scholar]
- Fornerod M., Ohno,M., Yoshida,M. and Mattaj,I.W. (1997a) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–1060. [DOI] [PubMed] [Google Scholar]
- Fornerod M., van Deursen,J., van Baal,S., Reynolds,A., Davis,D., Murti,K.G., Fransen,J. and Grosveld,G. (1997b) The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J., 16, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Asano,S., Nakamura,T., Adachi,M., Yoshida,M., Yanagida,M. and Nishida,E. (1997) CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature, 390, 308–311. [DOI] [PubMed] [Google Scholar]
- Gossen M. and Bujard,H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand F., Kulkarni,S., Chase,A., Goldman,J.M., Gordon,M. and Cross,N.C. (1999) Frequent deletion of hSNF5/INI1, a component of the SWI/SNF complex, in chronic myeloid leukemia. Cancer Res., 59, 3870–3874. [PubMed] [Google Scholar]
- Guidi C.J., Sands,A.T., Zambrowicz,B.P., Turner,T.K., Demers,D.A., Webster,W., Smith,T.W., Imbalzano,A.N. and Jones,S.N. (2001) Disruption of INI1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol., 21, 3598–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E. and Adam,S. (1998) Transport of macromolecules between the nucleus and the cytoplasm. RNA, 4, 351–364. [PMC free article] [PubMed] [Google Scholar]
- Kalpana G.V., Marmon,S., Wang,W., Crabtree,G.R. and Goff,S.P. (1994) Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science, 266, 2002–2006. [DOI] [PubMed] [Google Scholar]
- Khavari P.A., Peterson,C.L., Tamkun,J.W., Mendel,D.B. and Crabtree,G.R. (1993) BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature, 366, 170–174. [DOI] [PubMed] [Google Scholar]
- Kingston R.E. and Narlikar,G.J. (1999) ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev., 13, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Klochendler-Yeivin A., Fiette,L., Barra,J., Muchardt,C., Babinet,C. and Yaniv,M. (2000) The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep., 1, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N., Wolff,B., Sekimoto,T., Schreiner,E.P., Yoneda,Y., Yanagida,M., Horinouchi,S. and Yoshida,M. (1998) Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res., 242, 540–547. [DOI] [PubMed] [Google Scholar]
- Kudo N., Matsumori,N., Taoka,H., Fujiwara,D., Schreiner,E.P., Wolff,B., Yoshida,M. and Horinouchi,S. (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl Acad. Sci. USA, 96, 9112–9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent B.C., Treich,I. and Carlson,M. (1993) The yeast SNF2/SWI2 protein has DNA-stimulated ATPase activity required for transcriptional activation. Genes Dev., 7, 583–591. [DOI] [PubMed] [Google Scholar]
- Li Y., Yamakita,Y. and Krug,R.M. (1998) Regulation of a nuclear export signal by an adjacent inhibitory sequence: the effector domain of the influenza virus NS1 protein. Proc. Natl Acad. Sci. USA, 95, 4864–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaj I.W. and Englmeier,L. (1998) Nucleocytoplasmic transport: the soluble phase. Annu. Rev. Biochem., 67, 265–306. [DOI] [PubMed] [Google Scholar]
- Moll U.M., Riou,G. and Levine,A.J. (1992) Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc. Natl Acad. Sci. USA, 89, 7262–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A., Yung,E. and Kalpana,G. (1998) Structure–function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc. Natl Acad. Sci. USA, 95, 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C., Sardet,C., Bourachot,B., Onufryk,C. and Yaniv,M. (1995) A human protein with homology to Saccharomyces cerevisiae SNF5 interacts with the potential helicase hbrm. Nucleic Acids Res., 23, 1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E.A. (1997) Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature, 386, 779–787. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B., Bachelerie,F. and Dargemont,C. (1997) Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science, 278, 141–144. [DOI] [PubMed] [Google Scholar]
- Phelan M.L., Sif,S., Narlikar,G.J. and Kingston,R.E. (1999) Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell, 3, 247–253. [DOI] [PubMed] [Google Scholar]
- Roberts C.W., Galusha,S.A., McMenamin,M.E., Fletcher,C.D. and Orkin,S.H. (2000) Haploinsufficiency of Snf5 (integrase inter actor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl Acad. Sci. USA, 97, 13796–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau-Merck M.F., Versteege,I., Legrand,I., Couturier,J., Mairal,A., Delattre,O. and Aurias,A. (1999) hSNF5/INI1 inactivation is mainly associated with homozygous deletions and mitotic recombinations in rhabdoid tumors. Cancer Res., 59, 3152–3156. [PubMed] [Google Scholar]
- Rozenblatt-Rosen O. et al. (1998) The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc. Natl Acad. Sci. USA, 95, 4152–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp C.L., Poulsen,G.L., Nork,T.M. and Nickells,R.W. (1997) Nuclear exclusion of wild-type p53 in immortalized human retinoblastoma cells. J. Natl Cancer Inst., 89, 1530–1536. [DOI] [PubMed] [Google Scholar]
- Sevenet N., Lellouch-Tubiana,A., Schofield,D., Hoang-Xuan,K., Gessler,M., Birnbaum,D., Jeanpierre,C., Jouvet,A. and Delattre,O. (1999) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype–phenotype correlations. Hum. Mol. Genet., 8, 2359–2368. [DOI] [PubMed] [Google Scholar]
- Stade K., Ford,C.S., Guthrie,C. and Weis,K. (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell, 90, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Stommel J.M., Marchenko,N.D., Jimenez,G.S., Moll,U.M., Hope,T.J. and Wahl,G.M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J., 18, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima F., Moriguchi,T., Wada,A., Fukuda,M. and Nishida,E. (1998) Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J., 17, 2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P., Doucas,V., Craig,E., Mangeat,B., Klages,N., Evans,R., Kalpana,G. and Trono,D. (2001) Cytoplasmic recruitment of INI and PML by incoming HIV preintegration complexes: an interference with the immediate early steps of viral replication. Mol. Cell, 7, 1245–1254. [DOI] [PubMed] [Google Scholar]
- Ullman K.S., Powers,M.A. and Forbes,D.J. (1997) Nuclear export receptors: from importin to exportin. Cell, 90, 967–970. [DOI] [PubMed] [Google Scholar]
- Versteege I., Sevenet,N., Lange,J., Rousseau-Merck,M.F., Ambros,P., Handgretinger,R., Aurias,A. and Delattre,O. (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature, 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Wada A., Fukuda,M., Mishima,M. and Nishida,E. (1998) Nuclear export of actin: a novel mechanism regulating the subcellular localization of a major cytoskeletal protein. EMBO J., 17, 1635–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (1996) Purification and biochemical heterogeneity of the mammalian SWI–SNF complex. EMBO J., 15, 5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wen W., Meinkoth,J.L., Tsien,R.Y. and Taylor,S.S. (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell, 82, 463–473. [DOI] [PubMed] [Google Scholar]
- Wolff B., Sanglier,J.-J. and Wang,Y. (1997) Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol., 4, 139–147. [DOI] [PubMed] [Google Scholar]
- Yung E., Sorin,M., Pal,A., Craig,E., Morozov,A., Delattre,O., Kappes,J., Ott,D. and Kalpana,G.V. (2001) Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nature Med., 7, 920–926. [DOI] [PubMed] [Google Scholar]
- Zhang H.S., Gavin,M., Dahiya,A., Postigo,A.A., Ma,D., Luo,R.X., Harbour,J.W. and Dean,D.C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell, 101, 79–89. [DOI] [PubMed] [Google Scholar]