

Abstract
The crystal structure of the catalytic core of murine terminal deoxynucleotidyltransferase (TdT) at 2.35 Å resolution reveals a typical DNA polymerase β-like fold locked in a closed form. In addition, the structures of two different binary complexes, one with an oligonucleotide primer and the other with an incoming ddATP-Co2+ complex, show that the substrates and the two divalent ions in the catalytic site are positioned in TdT in a manner similar to that described for the human DNA polymerase β ternary complex, suggesting a common two metal ions mechanism of nucleotidyl transfer in these two proteins. The inability of TdT to accommodate a template strand can be explained by steric hindrance at the catalytic site caused by a long lariat-like loop, which is absent in DNA polymerase β. However, displacement of this discriminating loop would be sufficient to unmask a number of evolutionarily conserved residues, which could then interact with a template DNA strand. The present structure can be used to model the recently discovered human polymerase µ, with which it shares 43% sequence identity.
Keywords: crystal structure/incoming nucleotide/nucleotidyltransferase/polymerase/primer strand
Introduction
Terminal deoxynucleotidyltransferase (TdT; EC 2.7.7.31) is the only known DNA polymerase that elongates DNA strands in a template-independent manner (Bollum, 1974). Unlike any other polymerase, TdT can incorporate both ribo- and deoxyribonucleotides in vitro (Roychoudhury, 1972; Boulé et al., 2001) as well as a large array of unnatural nucleoside triphosphates (Semizarov et al., 1997; Krayevsky et al., 2000). TdT has only been found in vertebrates, where it is highly conserved (reviewed in Boulé et al., 2000). Its appearance seems to coincide with the emergence of an adaptive immune system, which relies in part on the combinatorial diversity generated by recombination of gene segments in lymphocytes (Lewis, 1994). TdT brings additional diversity in the immune repertoire by adding nucleotides, called N regions, to the V(D)J recombination junction sites of immunoglobulin and T-cell receptor genes (Kallenbach et al., 1990; Gilfillan et al., 1993; Komori et al., 1993).
TdT belongs to the family of polymerases called pol X (Ito and Braithwaite, 1991), a subclass of an ancient nucleotidyltransferase (NT) superfamily (Holm and Sander, 1995). The NT superfamily includes nucleic acid polymerases such as DNA polymerase β (pol β), DNA polymerase λ and DNA polymerase µ (Aoufouchi et al., 2000; Dominguez et al., 2000; Garcia-Diaz et al., 2000), as well as CCA-adding enzymes (Yue et al., 1996) and poly(A) polymerases (Martin and Keller, 1996). Numerous other NTs transferring a nucleotide to non-nucleic acid molecules and involved in various metabolic pathways also belong to the NT superfamily (Aravind and Koonin, 1999). Sequence similarities among members of the NT superfamily are low and essentially confined to short motifs involved in nucleotide binding and catalysis.
The sequence alignment between TdT and pol β encompasses the whole catalytic domain, but TdT has an additional 13 kDa N-terminal region, which contains a nuclear localization sequence (Peterson et al., 1985) and a protein–protein interaction BRCT-like domain, as shown in Figure 1A (Bork et al., 1997; Callebaut and Mornon, 1997). The X-ray structure of the XRCC1 BRCT domain has been reported (Zhang et al., 1998). In TdT, this domain is thought to interact with Ku70/86 (Mahajan et al., 1999), a protein heterodimer involved in the processing of double strand breaks during V(D)J recombination (Zhu et al., 1996).


Fig. 1. General structure and domain organization of TdT. (A) Linear arrangement of the different domains of C-TdT (blue), compared with pol β. The two motifs C and A and the two HhH consensus sequences are indicated in red and green, respectively. The disordered part of C-TdT in the crystal is in cyan and the N-terminal extension of C-TdT compared with pol β is in magenta. The part of the molecule absent from the crystallized construct is in white. (B) General architecture of the TdT structure drawn with Molscript (Kraulis, 1991). The three catalytic aspartate residues are shown in ball-and-stick representation. The N- and C-termini are in dark blue, while the two strictly conserved stretches of sequences implicated in the binding of the incoming dNTP are in cyan and magenta. Loop1 is shown in yellow. An intrinsic magnesium ion (grey) as well as the two putative sodium ions bound to the HhH motifs are represented in CPK mode (blue); only the Na+ ion with ligands in an octahedral geometry is labelled.
The structures of only three NTs have as yet been solved: kanamycin nucleotidyltransferase (Sakon et al., 1993; Pedersen et al., 1995), pol β (Davis et al., 1994; Sawaya et al., 1994) and poly(A) polymerase (Bard et al., 2000; Martin et al., 2000). At the structural level, the best known NT is pol β, solved at atomic resolution alone and as various binary and ternary complexes (Pelletier et al., 1994, 1996a,b; Sawaya et al., 1997). All structurally known proteins of the NT family contain a catalytic domain made of a five-stranded mixed β-sheet and two α-helices, which is topologically different from the structures of other DNA polymerases from the pol I (Ollis et al., 1985; Doublié et al., 1998) and pol α families. (Delarue et al., 1990; Wang et al., 1997). Despite these topological differences, the local structure of the catalytic site of both families is made of three carboxylate side chains and two divalent cations in the same spatial arrangement. Furthermore, the hand metaphor first introduced for Escherichia coli DNA polymerase I Klenow fragment (Ollis et al., 1985) holds true for pol β, with a palm domain containing the catalytic carboxylate triad and a finger and a thumb domain involved in the binding of the nucleic acid and nucleotide substrates (Pelletier et al., 1994). Finally, pol β as well as several polymerases from both pol I and pol α families have been shown to adopt two different conformations, the open and closed forms (reviewed in Doublié et al., 1999).
The natural abundance of TdT in calf thymus has made it one of the earliest eukaryotic DNA polymerases to be isolated (Bollum, 1960), but so far it has eluded structural characterization. Here we present the crystal structure of the catalytic core of murine TdT at 2.35 Å resolution. In addition, X-ray data at 3.0 Å resolution including anomalous scattering measurements have been obtained for two binary complexes, one with a brominated DNA primer and the other with an incoming nucleotide–Co2+ complex. Both complexes have been refined, allowing a model for the ternary complex to be proposed.
Results and discussion
Overall structure
The crystal structure reported here includes only the catalytic core of TdT, hereafter referred to as C-TdT, which encompasses residues 130–510 (see Figure 1A). The N-terminal domain of TdT, which is a typical BRCT-like fold (Bork et al., 1997; Callebaut and Mornon, 1997), is absent from the construct that could be crystallized. C-TdT retains catalytic activity in the crystal (Sukumar et al., 2000). The structure was solved initially with MIRAS phases at 3.0 Å resolution (Table I) and refined up to 2.35 Å resolution using standard protocols (Table II). The first 18 residues were found to be disordered in the crystal state. All atoms of residues 148–510 are included in the final model except for three presumably mobile residues (amino acids 421–423) situated at the tip of an external loop, called loop2 (see Figure 1B), which is distant from the active site.
Table I. Data collection and phasing statistics.
| Nat1 | EtHgCl | SmCl3 | IrCl6 | ddATP-Co2+ | (5Br-dU)9 | |
|---|---|---|---|---|---|---|
| Concentration (mM) | – | 0.1 | 10 | 1 | 1 | 2 |
| Soaking time (h) | – | 3 | 72 | 24 | 24 (×3) | 24 (×3) |
| X-ray source | ESRF, ID14-3 | ESRF, ID14-3 | ESRF, ID14-3 | ESRF, ID14-2 | ESRF, ID14-3 | ESRF, ID14-4 |
| Wavelength in Å | 0.940 | 0.933 | 0.933 | 0.933 | 0.940 | 0.960 |
| Detector | MARCCD | MARCCD | MARCCD | ADSC | MARCCD | ADSC |
| a, b, c (Å) P212121 | 47.1, 85.2, 111.7 | 47.1, 85.4, 111.1 | 47.2, 85.0, 111.1 | 47.0, 84.7, 112.2 | 46.8, 85.2, 108.3 | 46.8, 85.0, 111.4 |
| Resolution (Å)a | 20–2.35 (2.4–2.35) | 20–2.60 (2.7–2.6) | 20–2.9 (2.98–2.92) | 20–2.6 (2.7–2.6) | 20–3.0 (3.11–3.0) | 20–3.0 (3.11–3.0) |
| No. of observed reflections | 99 454 | 50 119 | 34 353 | 50 449 | 30 995 | 25 666 |
| No. of unique reflectionsa | 19 351 (1224) | 14 338 (1795) | 9906 (1323) | 13 913 (1954) | 8995 (894) | 8998 (897) |
| Multiplicitya | 5.2 (4.1) | 3.5 (2.6) | 3.5 (2.2) | 3.6 (2.5) | 3.4 (2.1) | 2.8 (1.8) |
| Completeness (%)a | 99.8 (98.4) | 97.5 (99.5) | 97.6 (91.9) | 96.1 (92.7) | 97.9 (99.4) | 89.0 (87.4) |
| Rmergea,b | 0.059 (0.340) | 0.060 (0.136) | 0.078 (0.360) | 0.061 (0.257) | 0.074 (0.480) | 0.100 (0.608) |
| I/σ(I)a | 25.3 (4.1) | 8.5 (4.4) | 7.1 (1.6) | 8.3 (2.8) | 8.4 (1.7) | 8.5 (2.5) |
| Risoc (no. of sites) | – | 0.387 (6) | 0.180 (3) | 0.231 (5) | 0.360 | 0.300 |
| Phasing power (res. Å) | – | 1.10 (15–3.0) | 1.30 (15–4.2) | 1.02 (15–3.0) | – | – |
| RCullis (anom.) | – | 0.84 (0.90) | 0.77 | 0.86 |
aThe number in parentheses refers to the last (highest) resolution shell.
bRmerge = ΣhΣi(Ihi – <Ih>)/Σh,iIhi, where Ihi is the ith observation of the reflection h, while <Ih> is its mean intensity.
cRiso = Σ|FPH – FP|/ΣFP, where FPH and FP are the derivative and the native structure-factor amplitudes, respectively.
Table II. Refinement statistics.
| Native | Co2+-ddATP | (Br-dU)9 | |
|---|---|---|---|
| Resolution range (Å) | 18.0–2.35 | 27.0–3.0 | 27.0–3.0 |
| Intensity cut-off [F/σ(F)] | 0 | 1 | 0 |
| No. of reflections | |||
| completeness (%) | 98.1 | 94.7 | 91.5 |
| used for refinement | 17 984 | 7138 | 8174 |
| used for Rfree calculation | 973 | 770 | 405 |
| No. of non-hydrogen atoms | |||
| protein (missing residues in loop1 and 2) | 2910 (0, 3) | 2829 (3, 3) | 2849 (4, 3) |
| Na+ | 1 | 1 | 1 |
| Mg2+ (or Co2+) | 1 | (2) | 1 |
| water (ligand) | 214 | 18 (29) | 1 (81) |
| R-factora (%) | 21.0 | 26.7 | 24.1 |
| Rfreeb (%) | 25.8 | 31.9 | 29.8 |
| R.m.s. deviations from ideality | |||
| bond lengths (Å) | 0.0064 | 0.0097 | 0.0084 |
| bond angles (°) | 1.08 | 1.51 | 1.38 |
| Average temperature factors | |||
| main chain | 36.6 | 56.9 | 49.7 |
| side chains | 40.2 | 56.0 | 51.3 |
| Na+ | 46.6 | 57.9 | 61.2 |
| Mg2+ (or Co2+) | 51.5 | (57.1) | 82.9 |
| water (ligand) | 40.3 | (63.7) | (90.5) |
| Ramachandran plot | |||
| residues in most favoured regions (%) | 92.0 | 76.5 | 83.2 |
| residues in additional allowed regions (%) | 8.0 | 23.5 | 16.8 |
| Overall G factorc | 0.31 | 0.09 | 0.17 |
aR-factor = Σ||Fobs| – |Fcalc||/Σ|Fobs|.
bRfree was calculated with a fraction (5–8%) of reflections excluded from the refinement.
cG-factor is the overall measure of structure quality from PROCHECK (Laskowski et al., 1993).
The general architecture of C-TdT can be described as ring-like or doughnut-shaped (see Figure 1B), with an α-helical N-terminal domain (amino acids 163–243), an α-helical junction domain (amino acids 243–302), a central domain with a large antiparallel β-sheet (amino acids 302–450) carrying the three strictly conserved catalytic aspartate residues (Figures 1A and 2), and a C-terminal domain (amino acids 450–510) containing a small antiparallel β-sheet, which makes extensive contacts with the N-terminal domain, thus closing the ring. The four domains of C-TdT are homologous to the 8 kDa, finger, palm and thumb domains in pol β (Figure 1A). The 8 kDa domain, which can be viewed as an ‘index finger’ domain, touches the thumb to form a hole through which the incoming dNTP diffuses to reach the active site, while the primer strand lies on the palm domain, perpendicular to the axis of the hole (see Figure 6 below).

Fig. 2. Multialignment of TdT and pol µ sequences. In addition to the murine TdT (TdT_mouse) studied here, the multialignment includes the bovine (TdT_bovin), human (TdT_human), chicken (TdT_chick), Xenopus (TdT_xenla), axolotl (TdT_axolo), opossum (TdT_oposs) and trout (TdT_trout) TdT enzymes as well as the human (polm_hsap) and mouse (polm_mmus) polymerase µ. The sequences were divided into two groups: the eight TdT sequences and the two pol µ sequences. Those residues >80% conserved in each group but not in all sequences are coloured in yellow. Strictly conserved residues in each group are red; they are boxed (red) when conserved in all sequences. The sequence of human pol β (polb_hsap) is displayed but not taken into account for calculating conservation. The multiple alignment was obtained with the program Pileup (University of Wisconsin, 1997) using an opening and extension gap penalty of 8 and 2, respectively, and then displayed using the program ESPript (Gouet et al., 1999). The numbering corresponds to the murine TdT sequence. The secondary structure of C-TdT is displayed on top of the alignment as well as that of the aligned human pol β sequence (polb_hsap). Important structural features are indicated at the bottom of the alignment: the N- and C-terminal regions that are close in space are indicated by a green full circle; the three catalytic aspartate residues of motifs C and A are highlighted by full red circles, the two HhH regions by green upper triangles, and loop1 by black circles. R336, G452 and S453 are underlined, with blue upright triangles to indicate that the side chain of R336 binds to the carbonyl oxygen of G452 and helps to maintain the cis-peptide bond between G452 and S453; the two highly conserved regions to which these last three residues belong, forming together the binding site of the incoming dNTP, are singled out with blue full circles. Cysteines are indicated by a blue star. Finally, the location of the insertion present in the longer form of murine TdT is indicated by two red upright triangles.

Fig. 6. Incoming nucleotide and primer strand in the context of the molecular surface of C-TdT (drawn with Grasp; Nicholls et al., 1991). Exposed aspartate and glutamate residues are in red, while exposed arginine and lysine residue are in blue. (A) View down the oligonucleotide axis, with the incoming nucleotide in place. (B) View from the side with the final oligonucleotide model. The cavity through which the dNTP diffuses to reach the catalytic site is visible at the back.
The index finger domain corresponds to residues 160–240 (helices α1–α4) in C-TdT. Its stability seems to be reinforced by several salt bridges between Glu177 and Lys250, and Asp173 and Lys199, as well as hydrogen bonds involving Asp179 and Ser187, and Ser195 and Asp173. This domain in pol β is highly basic (net charge +10), binds to single-stranded DNA and exhibits 5′-deoxyribose phosphate (dRP) lyase activity (Beard and Wilson, 1998). This domain is acidic in murine TdT (net charge –6) as well as in all other known TdTs (net charge –4 to –6). The residues essential for pol β lyase activity have been identified (Matsumoto et al., 1998) and are not conserved in TdT (Figure 2). No dRP lyase activity has been reported for TdT.
Despite a rather low sequence identity of 22–24%, C-TdT has striking structural homology with pol β. Almost all the secondary elements of pol β are present in the three-dimensional structure of C-TdT (see Figure 2). Compared with pol β, however, the ordered part of C-TdT contains a longer N-terminal extension of 16 residues (amino acids 148–163; see Figure 1A). There are also two long insertions of 10–15 amino acids in the central domain, one between strands β3 and β4 (loop1 in Figure 1B), and the other between strands β4 and β5 (loop2 in Figure 1B). One deletion of five amino acids between strands β7 and β8 is located exactly where the second isoform of murine TdT, which results from an alternative splicing of the mRNA, has a 20 amino acid insertion (Bentolila et al., 1995; see Figure 1B). This insertion recently has been shown to modify the thermosensitivity of the enzyme but not its catalytic activity (Boulé et al., 2000).
Structure–sequence relationships
The multiple alignment of the catalytic domains of all known TdT sequences as well as the two available pol µ sequences reveals several stretches of strictly conserved residues (Figure 2). Their functional importance is highlighted by the three-dimensional structure. For instance, the conserved sequence CQR (amino acids 155–157) in the N-terminal region (green dots in Figure 2) forms a short 310 helix that interacts with both the LGL (amino acids 498–500) and RNA (amino acids 508–510) conserved sequences (green dots in Figure 2) of the C-terminus region to seal the ring-like structure of the protein.
The two longest motifs of conserved residues in the multialignment form the incoming dNTP-binding site (blue dots, Figure 2). The first of these two conserved sequences, ALLGW(T/S)GSR (amino acids 446–454), is located at the interface between the central and C-terminal domains (Figure 1B) in a hinge region involved in the open–closed conformational transition in pol β. It contains a characteristic cis-peptide bond between Gly452 and Ser453, which is partly stabilized through a direct interaction between the carbonyl group of Gly452 and the guanidinium group of Arg336. Arg336 belongs to the second dNTP-binding motif TGGFRRG (amino acids 331–337). Its mutation into alanine severely impairs TdT catalytic activity, demonstrating the importance of maintaining the cis-peptide bond at this position (Yang et al., 1994). This cis-peptide bond and its stabilization mode are also observed in pol β (Sawaya et al., 1997).
Three strictly conserved aspartate residues are known to be essential for the catalytic activity and for binding magnesium ions in the NT family. This was shown both by structural studies (Pelletier et al., 1994; Holm and Sander, 1995) and by site-directed mutagenesis experiments (Yang et al., 1994; Boulé et al., 2001). The three aspartate residues in motifs C and A (Delarue et al., 1990; Pelletier et al., 1994; see Figure 2) are located in strands β2 and β5 of the central domain, respectively (Figure 1).
Two other important structural features visible in the multialignment are the helix–hairpin–helix motifs (HhH motifs, see Figure 1A) identified in several DNA-binding proteins (Doherty et al., 1996) including pol β (green upper triangles, Figure 2). In pol β, these motifs coordinate Na+ or K+ ions which participate in the binding of the template and primer strands, and a systematic study showed a preference for cations in the order K+>Na+>Mg2+>Ca2+ (Pelletier and Sawaya, 1996). Clear peaks of electron density were indeed observed in the HhH motifs of C-TdT exactly where the K+ or Na+ ions were expected (represented in CPK mode in Figure 1B). The first HhH motif has only one strong carbonyl atom ligand coming from residue 214. This may be related to the highly non-canonical nature of this HhH motif (CφG instead of the normally strictly conserved GφG, where φ is a hydrophobic residue). The peak was assigned to a water molecule. For the second HhH motif, which is involved in the binding of the primer strand (see below), the geometry of the coordination is clearly octahedral for the four identified ligands (carbonyl atoms from residues 253, 255 and 258 and a water molecule). This peak was assigned to an Na+ ion, as there is no K+ ion in solution and its ligand geometry is not compatible with a water molecule.
TdT superimposes best with the closed form of pol β
Pol β, like several other prokaryotic and viral polymerases from the pol I and pol α families, has been shown to adopt two different conformations, open and closed (Doublié et al., 1999). By superimposing the C-TdT structure on both forms of human pol β, it is immediately apparent that TdT adopts a closed conformation (Figure 3). The r.m.s.d. is 1.7 Å for 298 superimposed Cα atoms of C-TdT with the closed conformation of pol β. The r.m.s.d. of the superposition with the open form of pol β is 2.2 Å, but with only the C-terminal 218 Cα atoms, as it is impossible to superimpose the whole structure of C-TdT with a single rotation. In comparison, all the r.m.s.ds of pairwise superpositions of the three crystal forms of TdT reported here (the native and the two binary complexes, see below) are <0.7 Å for 360 Cα atoms.

Fig. 3. C-TdT is in a closed form. Stereo view of the superimposed Cα traces of TdT (red) with the closed form of pol β (blue).
The extra 16 residues situated at the N-terminus of C-TdT (amino acids 148–163) are ‘sealing’ the closed conformation by providing additional van der Waals contacts between the N- and C-terminal domains (Figure 1). There is one hydrogen bond between the side chains of Ser151 and Gln156 within the N-terminal region, and the conserved Gln152 is interacting with the conserved Arg462 of α-helix 14. A superposition of the C-TdT atomic model onto the molecular surface of the closed form of pol β reveals an excellent surface complementarity in the region of the N-terminal extension of C-TdT (not shown).
The observation that TdT adopts a closed conformation is consistent with the following data. The last arginine residue of pol β motif A (Arg258), which is essential for the stabilization of the open conformation (Sawaya et al., 1997), is not conserved in TdT (Figure 2). The pol β transition from an open to a closed form is believed to occur upon binding of the nucleotide complementary to the nucleotide to be copied in the template strand, enhancing replication accuracy through an induced fit mechanism (Sawaya et al., 1997; Beard and Wilson, 1998). Such a transition would not be required in the case of a template-independent polymerase like TdT. Indeed, in the present crystal structure of C-TdT, the active site looks as if it were poised to react. This could explain why the two binary complexes could be assembled in the same crystal form without any major rearrangement of the protein. Finally, the transition between the open and closed form in Thermus aquaticus pol I has been correlated to the translocation step after each cycle of nucleotide incorporation (Li et al., 1998). Because TdT adopts a strictly distributive mode of synthesis (Boulé et al., 2001), such a conformational transition would not be required. It is therefore conceivable that TdT could remain permanently locked in a closed form without compromising its function.
In this respect, it is interesting to note that the structure of a translesion DNA polymerase which is the analogue of dinB of E.coli has been solved recently as a closed form in the unliganded state. This was interpreted as an indication that low fidelity and distributive DNA polymerases may not use the open–closed transition to check that the correct base is being incorporated (Zhou et al., 2001).
The nucleotide-binding site and the catalytic site
The structure of the binary complex between C-TdT and an incoming ddATP in the presence of 10 mM Co2+ has been determined at 3.0 Å resolution. Co2+ ions, known to be efficient in TdT catalysis, were used in this experiment instead of Mg2+ because of their higher X-ray scattering factors. An anomalous Fourier map readily identified two Co2+ ions (Figure 4A), which are coordinated by the two strictly conserved aspartate residues of motif C as well as by the phosphates of the incoming ddATP. The initial isomorphous difference map (not shown) revealed clear density for the three phosphates, in a conformation almost identical to that observed in pol β (Sawaya et al., 1997; see Figure 4C).

Fig. 4. The binary complex with the incoming nucleotide. (A) Electron density for the binary complex obtained by soaking TdT native crystals with CoCl2 (10 mM) and ddATP (1 mM). The anomalous difference map at 4.5 Å resolution contoured at 4.5σ is shown in cyan along with the two cobalt atoms (CPK spheres, cyan). The three residues closest to the base (Trp450, Lys403 and Asn474) are shown as ball-and-stick representations. (B) Fobs – Fcalc omit map of the final model in the incoming nucleotide-binding site region, contoured at 2.5σ. The three catalytic aspartate residues of motifs C and A as well as Arg432, which forms an ion pair with one of them, Asp434, are represented in ball-and-stick. (C) Comparison of the binding mode of the incoming nucleotide in pol β (blue) and TdT (red): the electron density of the omit map (green) displayed in (B) is superimposed on the ball-and-stick model of the incoming ddCTP (black) of the ternary complex of pol β (Sawaya et al., 1997). (D) Detailed view of the active site with the incoming ddATP in the context of the local secondary structure. The three conserved aspartates of motif C and motif A are shown, as well as the two cobalt ions (CPK spheres, cyan). The highly conserved regions among TdT sequences are coloured purple and green for the helices (α11 and α13) and cyan for the loops. Loop1 is in yellow. The C-terminus is in dark blue.
By comparison, one of the strongest peaks of the entire difference map site of the unliganded protein at 2.35 Å resolution is found between the two aspartates of motif C. Because in this case the only source of divalent ion present in the crystallization solution was 50 mM MgOAc2, a magnesium ion was assigned to this peak. These data are consistent with the observation that C-TdT adopts a closed conformation because, in pol β, a Mg2+ was also found at this position but only in the closed form (Sawaya et al., 1997).
After refinement, clear electron density is visible in the omit map for both the sugar and base moieties of the ddATP (Figure 4B). Which are the protein residues that provide contact with the ddATP molecule? The aromatic ring of Trp450 is parallel to and partially stacked with the adenine ring of the incoming nucleotide, with the CZ2 atom of Trp450 at 3.6 Å from the C8 atom of the adenine ring. The strictly conserved Asn474 (Figure 2) of the C-terminal domain is in van der Waals contact with Trp450. Interestingly, the Lys403 side chain is pointing towards the adenine ring, with the ε amino group at 4 Å above the base (Figure 4A). In addition, three positively charged residues, Lys338, Arg336 and Arg454, are pointing towards the triphosphate moiety, thereby contributing to the neutralization of the three negative charges introduced by the phosphate atoms. The sugar moiety of the incoming ddATP is bound by Trp450 on one side and close to the cis-peptide bond between Gly452 and Ser453 on the other side (see Figure 4D). Because none of the protein atoms are close to either the 2′ or the 3′ position of the ddATP sugar, no steric barrier is likely to prevent the accommodation of one or two hydroxyl groups at these positions. This structural feature could account for the unique ability of TdT to incorporate both ribo- and deoxyribonucleotides with the same efficiency (Boulé et al., 2001).
Path of the primer strand
Based on the structure of the closed ternary complex of pol β (Sawaya et al., 1997), we define three binding sites P1, P2 and P3 on the protein, for the different nucleotides of the primer strand, in the 3′–5′ direction. The isomorphous difference map of the binary complex between C-TdT and a brominated 9-mer oligonucleotide clearly shows electron density above 5σ for the phosphate groups in the P1, P2 and P3 sites (Figure 5A). In addition, the anomalous map shows density above 3σ for P1, P2 and P3 base sites at positions consistent with a regular B-DNA conformation (Figure 5A). If the primer were in an A-DNA conformation, the phosphates would still fit to the isomorphous map but the bases would be too far away from the peaks of the bromine anomalous density. The path of the primer strand is very similar in TdT and pol β. Only a minor rigid body movement of the primer strand in the pol β–DNA ternary complex is needed in order to fit the isomophous electron density of the TdT–primer binary complex (Figure 5C). The fact that only the three bases at the 3′-hydroxyl end of the primer are ordered in the binary complex is consistent with the observation that TdT has an absolute requirement for a primer containing at least three nucleotides with a 5′ phosphate (Kato et al., 1967). The remaining 5′ part of the primer is disordered in solution and is not in contact with the protein. The model with the trinucleotide placed in the P1–P3 sites was refined and a difference Fobs – Fcalc map was calculated. Contoured at 3σ, this map shows extra density reaching into the incoming nucleotide-binding site (site Incoming, see Figure 5B). This is consistent with both the isomorphous difference map, which has density at the 2.5σ level at the expected position of a 3′ phosphate group in the P1 site, and also with the anomalous difference map, which has a peak at 5σ in the incoming site where an extra base can be built (Figure 5A). The additional base clearly is not stacked with the base at the P1 site. The final model of the oligonucleotide has four nucleotides, three occupying the P3, P2 and P1 sites and one occupying the incoming site. Therefore, this TdT–primer binary complex looks as if it were an ‘enzyme–product’ complex.

Fig. 5. The binary complex with the primer strand. (A) Isomorphous density map at 3.0 Å resolution contoured at 4.0σ (magenta), showing density for the three phosphates of sites P1, P2 and P3. The final oligonucleotide model occupying these three sites is drawn as a ball-and-stick representation. The anomalous electron density coming from the anomalous signal of the bromine atoms is in green (4.0σ). (B) Fobs – Fcalc omit electron density map in the primer-binding site region (2.5σ). The last base in the Incoming binding site is not stacked with the preceding base. (C) Comparison of the binding mode of the primer strand in pol β (blue) and in TdT (red): the isomorphous Fourier difference map of the TdT binary complex (magenta) is superimposed to the primer strand of the ternary complex of pol β (black) (Sawaya et al., 1997), showing that the phosphates are located in almost the same place in the two proteins. (D) Detailed view of the stabilization of the primer phosphate at site P2, through the Na+ ion (yellow CPK sphere) located at the corner of the HhH motif, exactly as in the closed pol β structure (Pelletier et al., 1996). The electron density of the phosphate, calculated as in (a), is in magenta. Drawn with Bobscript (Esnouf, 1999).
No direct polar interactions between the protein residues and the bases are observed. The binding of the DNA primer thus relies only on interactions between the protein and the sugar–phosphate backbone, regardless of the nature of the base. All bases have much higher B-factors than the sugar–phosphate backbone (Table II). The phosphate at position P3 forms an ion pair with the strictly conserved Lys261 in the second HhH motif. It is located at the bottom of helix α6, whose macrodipole is well suited to neutralize this phosphate’s negative charge. The phosphate at position P1 is close to Arg432, a residue also involved in an ion pair with the catalytic Asp434. The phosphate at position P2 interacts with the Na+ ion of the second HhH motif (Figure 5D), as described for the pol β closed ternary complex (Sawaya et al., 1997) and for several DNA-binding proteins (Doherty et al., 1996).
Model for the ternary complex
The TdT active site contains the three conserved acidic residues that are conserved in all polymerases (Delarue et al., 1990). These residues are superimposable on the three catalytic residues of pol β. The analysis of the ddATP–C-TdT binary complex shows two divalent cations within the active site, in the position expected from the superimposed pol β ternary complex (Figure 4). These data suggest that the catalysis of nucleotide polymerization by TdT proceeds via the two metal ion mechanism (Steitz and Steitz, 1993) shown to be valid for all structurally known DNA polymerases. However, the limited resolution of the crystallographic data (3.0 Å) prevents us from discussing the nucleotidyl transfer mechanism in atomic detail.
Using the binary complexes structures described above (Figure 6), a model can be built for the ternary complex. The 3′ oxygen atom in the P1 site is at 3.6 Å from the α-phosphate of the incoming dNTP in the superimposed experimental structures of the two binary complexes of C-TdT. Therefore, some rearrangement must occur in the ternary complex, as compared with the binary complex with the incoming ddATP. A small movement is sufficient to superpose the ddATP molecule to the last base of the ‘enzyme–product’ complex. In the protein, Asn474 of the conserved DNH motif, Trp450 and Lys403 are likely to undergo side chain rearrangements in this process. Due to simple steric effects, mutating Asn474 to Gln474 should have a definite effect on the incorporation of purines but less so in the case of pyrimidines.
Based on comparative kinetics studies in the presence of various divalent ions, it has been proposed that a non-catalytic cation could be involved in the interactions between TdT and its substrates (Deibel and Coleman, 1980; Chang and Bollum, 1990). Interestingly, the recent structure reported for poly(A) polymerase, an enzyme which, like TdT, is template independent but shows extreme selectivity for ATP, revealed an unexpected third Mn2+ ion bound to the N7 of an incoming 3′ dATP. This interaction was proposed to play a role in the nucleotide substrate specificity of the enzyme (Martin et al., 2000). Although there is no Co2+ ion close to the N7 of adenine in the crystal structure of the ddATP–TdT binary complex, an additional cation could be brought in during the assembly of the ternary complex to establish a bridge between the base at the 3′-hydroxyl end of the primer strand and the incoming base. Based on the TdT structures reported here, the strictly conserved His400 could participate, through a simple Chi-1 rotation, in the coordination of such an additional cation located between the 3′ base of the primer strand and the incoming base. His475 is another possibility. The other ligands of this cation would be N7 of purines or N3 of pyrimidines, as seen in several metal–nucleotide crystal structures (Swaminathan and Sundaralingam, 1979) and RNA structures (Cate and Doudna, 1996). In accordance with in vitro experiments showing that micromolar amounts of Zn2+ added to Mg2+ increase the efficiency of both purine and pyrimidine incorporation and that Co2+ is better than Mg2+ + Zn2+ for pyrimidine incorporation (Chang and Bollum, 1990), at this bridging position Zn2+ would be best suited for purine–purine interactions and Co2+ for pyrimidine– pyrimidine interactions.
The case of loop1
Loop1 is a 16 residue loop (amino acids 384–399) which adopts a lariat-like conformation (Figure 1B). It is absent in pol β. Loop1 is stabilized by at least three different interactions, which all involve strictly conserved residues. First, the beginning and end points of the loop are tied by a hydrogen bond between the hydroxyl group of Thr384 and the carbonyl atom of Asp399. Secondly, a direct hydrogen bond connects the side chain of His475 and the carbonyl atom of Lys394 of loop1. Thirdly, a water molecule bridges the hydroxyl group of Ser392 and the side chain of Asp473 (Figure 7A). The DNH motif (amino acids 473–475; black circles in Figure 2), which participates in the stabilization of loop1, is in close contact with helix α13 at the interface between the central and the C-terminal domains, very close to Trp450 in the nucleotide-binding site (Figure 1B).
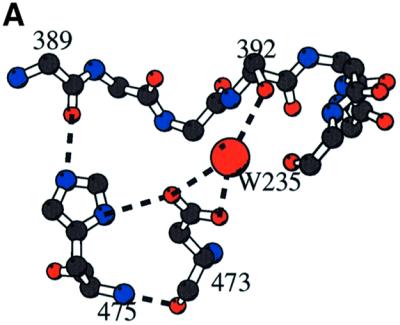
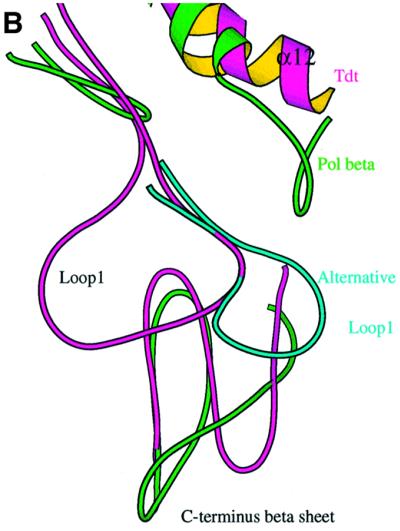

Fig. 7. Loop1. (A) Stabilization of loop1 through a central water molecule connecting Asp473 of the β–turn–β region of the C-terminal domain with Ser392 of loop1. The direct interaction of the side chain of His475 of the DNH motif with the carbonyl atom of Lys394 of loop1 is also shown. (B) Comparison of C-TdT and pol β in the vicinity of loop1. Loop1 is shown as a Cα trace (magenta) with its characteristic lariat-like structure. The underlying β-sheet of the C-terminal domain is also shown (magenta), superimposed with the same domain of the closed form of pol β (green). The five-residue insertion of pol β compared with TdT observed in this region is thus made visible. There is a deletion in TdT just before helix α12, as compared with pol β, making room for an alternative conformation of loop1 (cyan). (C) Molecular surface of TdT drawn with Grasp (Nicholls et al., 1991) together with the putative model of a primer–template duplex, derived from the human pol β ternary complex (Sawaya et al., 1997). The primer strand is coloured according to the chemical nature of its atoms, while the template strand is in dark blue. The exposed surface due to residues from loop1 is in magenta. It is clear from this view that the duplex part of the template strand cannot be accommodated by TdT.
Examination of a model template–primer duplex in the context of the C-TdT molecular surface, in a conformation inferred from the superimposed pol β ternary complex (Sawaya et al., 1997), shows that the accommodation of the template strand would be severely hindered by the presence of loop1 (Figure 7C). In order to enter the active site, the last three or four base pairs of the DNA duplex would have to melt. The energetic cost for disrupting the Watson–Crick hydrogen bonds (∼10 kcal/mol) could be compensated by the direct interaction of the primer strand with the protein. The fact that TdT does not use 3′-hydroxyl groups efficiently when they are recessed within partially double-stranded DNA or at the end of blunt DNA (Bollum, 1974) is consistent with such a scenario.
By rotating loop1 around an axis that runs parallel to strands β3 and β4, an alternative conformation of this loop can be modelled, which would make room for the template–primer (Figure 7B). Considering the path that the template–primer would thus take, the following observations can be made: there is a stretch of conserved residues D(H/C)FQKCF (amino acids 399–405) in strand β4 just after loop1 (Figure 2), which could interact with the region of the template strand annealed to the primer. Furthermore, the molecular surface of TdT below loop1 displays a good complementarity to the single-stranded part of the template–primer. This region contains three strictly conserved positively charged residues, at positions 458, 461 and 480, which could bind phosphate ions. It is curious that all these residues have been conserved throughout evolution, without any apparent evolutionary advantage. One possibility is that they might become accessible and interact with a template strand in vivo through a displacement of loop1 triggered by interactions with accessory proteins. Whether or not this would allow TdT to accommodate double-stranded DNA and possibly replicate DNA requires further experimental investigation.
Interestingly, we note that the other template-independent polymerase, poly(A) polymerase, also a member of the NT family, is devoided of loop1.
Comparison of TdT with polymerase µ
A novel DNA polymerase called pol µ has been discovered recently in humans (Dominguez et al., 2000) and mice (Aoufouchi et al., 2000). According to the authors, human pol µ exhibits terminal deoxynucleotidyltransferase activity preferentially on template–primer substrates and is able to copy a template DNA strand, albeit with very low accuracy. The pol µ and TdT sequences analysed pairwise display between 42.5 and 46.5% overall identity (Figure 2), implying that their three-dimensioanl structures are very similar (Chothia and Lesk, 1986). Given that the structural features involved in the sealing of C-TdT in a closed form are conserved in pol µ (Figure 2), it can be predicted that the pol µ core enzyme will also be locked in a closed form and that it has a distributive mode of polymerization. Pol µ has an 18 amino acid insertion that corresponds to loop1 in TdT. The molecular interactions accounting for the conformational stabilization of loop1 in the crystal structure of C-TdT involve residues not conserved in pol µ. Therefore, it is very unlikely that this loop in pol µ will adopt the same conformation as in TdT. Since the sequence of loop1 in pol µ is rich in histidine and cysteine residues, it is possible that a zinc ion might participate in the stabilization of an alternative conformation of loop1. Whether a difference in the organization of the loop in TdT and pol µ could account for their nucleic acid substrate preferences (single-stranded versus double-stranded DNA) remains to be addressed experimentally.
Conclusion
The crystal structure of the unliganded C-TdT was found to be in a closed conformation. Only minor rearrangements occur in the two binary complexes with either a single-stranded DNA or an incoming nucleotide. The presence of two Co2+ ions in the catalytic site suggests that the general two metal ion mechanism (Steitz and Steitz, 1993) is also valid for TdT. Interestingly, the three nucleotides at the 3′-hydroxyl end of the primer strand adopt a helical B-DNA conformation. The presence of a long lariat-like loop readily explains why TdT cannot accommodate a template strand. The lack of specificity towards the base being incorporated derives from an absence of specific contacts with the protein. Further structural studies of the ternary complex will be needed to confirm the presence of an additional cation near the last base of the primer and the incoming dNTP.
Materials and methods
Data collection and phasing
Crystals were grown in the conditions recently reported (Sukumar et al., 2000) for C-TdT purified as described in Boulé et al. (1998). All data were processed using DENZO (Otwinowski and Minor, 1997). A number of heavy atoms derivatives were collected, with statistics shown in Table I. The best derivative was obtained with EtHgCl for which anomalous information is also available. The positions of heavy atoms were obtained with HASSP (Terwilliger and Eisenberg, 1987). The MIRAS map was subjected to solvent flattening with an envelope representing 50% of the unit cell, leading to an increase of the figure of merit from 0.49 to 0.72 at 3.0 Å resolution. Heavy atom parameters were then refined against solvent-flattened phases with MLPHARE (CCP4, 1994). The model of pol β was placed in the resulting electron density map using a phased translation function as implemented in AMORE (Navaza and Saludjan, 1997).
Model building and refinement
The model was built gradually using O (Jones et al., 1991) or Quanta (Molecular Simulations, Inc.). The refinement was conducted with CNS (Brünger et al., 1998) using standard protocols: bulk solvent corrections and maximum likelihood target at maximum resolution of the data and no intensity cut-off. The model was inspected at each stage of the refinement using σA-weighed maps, simulated annealing omit maps as well as 2Fobs – Fcalc and Fobs – Fcalc maps; progress in the model refinement was judged by the decrease in the free R-factor. The stereochemistry of the final model was checked using Procheck (Laskowski et al., 1993). The final statistics of the refinement are included in Table II.
There are no residues in the disallowed or strictly forbidden regions of the Ramachandran plot. The numbering of the residues follows the sequence deposited in the Swissprot database (tdt_mouse). However, an insertion occurs between residues 441 and 442, as there is a mistake in the deposited sequence file (N.Doyen, personal communication). The coordinates of the native C-TdT have been deposited in the PDB (1JMS).
Binary complexes
The binary complexes were obtained by soaking crystals of the native proteins with either a solution containing 10 mM MgCl2 and 2 mM of a 9mer oligonucleotide (5-Br-dU9 purchased from Eurogentec, Belgium) whose purity was checked by mass spectrometry, or a solution containing 1 mM ddATP and 10 mM CoCl2, respectively; this soaking was repeated three times for 24 h, the crystals being transferred into a fresh solution of ligands each time.
The primer strand binary complex was refined with group B-factors and without the inclusion of water molecules since X-ray data could be used only to 3.0 Å resolution. The final R-factor was 24.1% and the Rfree 29.8% for all data was between 27.0 and 3.0 Å. Residues 394–397 from loop1 were not built.
Special care was needed for the Co2+-ddATP complex. First, the model had to be translated by ∼3 Å because of a difference in the c parameter of the unit cell (see Table I); this was determined by AMORE (Navaza and Saludjian, 1997). Rigid body refinement was carried out subsequently using CNS with four rigid bodies per molecule but without producing any noticeable change in the domain orientations and positions. A small number of residues were omitted in loop1 and/or modelled as alanines. Eighteen water molecules were added at the last step of the refinement. The final R-factor for the binary complex with the incoming nucleotide was 26.3% and the Rfree 30.9% for data between 27.0 and 3.0 Å resolution with a cut-off of 1.0σ. Coordinates of all atoms of the two binary complexes have been deposited in the PDB (1KDH and 1KEJ).
Acknowledgments
Acknowledgements
Thanks are due to the staff of the different beamlines ID14 at the ESRF in Grenoble (France) for help during data collection and generous beam-time allocation. J.L. wishes to thank Louison de Bertolis for his help in various stages of this work. We thank Ph.Dumas, P.Alzari and G.Bentley for useful suggestions on the manuscript. J.B.B. was supported by a CANAM fellowship, N.S. by a Cantarini fellowship, and N.J. by both an ARC and a Cantarini fellowship. This work was supported by Association pour la Recherche contre le Cancer (grant ARC 5470 to M.D.).
References
- Aoufouchi S. et al. (2000) Two novel human and mouse DNA polymerases of the polX family. Nucleic Acids Res., 28, 3684–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L. and Koonin,E.V. (1999) DNA polymerase β-like nucleotidyltransferase superfamily: identification of three new families and evolutionary history. Nucleic Acids Res., 27, 1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard J., Zhelovsky,A.M., Helmling,S., Earnest,T.N., Moore,C.L. and Bohm,A. (2000) Structure of yeast poly(A) polymerase alone and in complex with 3′dATP. Science, 289, 1346–1349. [DOI] [PubMed] [Google Scholar]
- Beard W.A. and Wilson,S.H. (1998) Structural insights into DNA polymerase β fidelity: hold it tight if you want it right. Chem. Biol., 5, R7–R13. [DOI] [PubMed] [Google Scholar]
- Bentolila L.A., Fanton d’Andon,M., Tri Nguyen,Q., Martinez,O., Rougeon,F. and Doyen,N. (1995) The two isoforms of mouse terminal deoxynucleotidyl transferase differ in both the ability to add N regions and subcellular localization. EMBO J., 14, 4221–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollum F.J. (1960) Oligodeoxyribonucleotides primers for calf thymus polymerase. J. Biol. Chem., 235, PC18–PC20. [PubMed] [Google Scholar]
- Bollum F.J. (1974) Terminal deoxynucleotidyl transferase. In Boyer,P. (ed.), The Enzymes. Vol. 10. Academic Press, Inc., New York, NY, pp. 145–171.
- Bork P., Hofmann,K., Bucher,P., Neuwald,A.F., Altschul,S.F. and Koonin,E.V. (1997) A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J., 11, 68–76. [PubMed] [Google Scholar]
- Boulé J.B., Johnson,E., Rougeon,F. and Papanicolaou,C. (1998) High-level expression of murine terminal deoxynucleotidyltransferase in E.coli grown at low temperature and overexpressing argU tRNA. Mol. Biotechnol., 10, 199–208. [DOI] [PubMed] [Google Scholar]
- Boulé J.B., Rougeon,F. and Papanicolaou,C. (2000) Comparison of the two murine isoforms of terminal deoxynucleotidyltransferase. A 20-amino acids insertion in the highly conserved carboxy-terminal region modifies thermostability but not catalytic activity. J. Biol. Chem., 275, 28984–28988. [DOI] [PubMed] [Google Scholar]
- Boulé J.B., Rougeon,F. and Papanicolaou C. (2001) Terminal deoxynucleotidyl transferase indiscriminately incorporates ribonucleotides and deoxyribonucleotides. J. Biol. Chem., 276, 31388–31393. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Callebaut I. and Mornon,J.P. (1997) From BCRA1 to RAP1: a widespread BCRT module closely associated with DNA repair. FEBS Lett., 400, 25–30. [DOI] [PubMed] [Google Scholar]
- Cate J.H. and Doudna,J.A. (1996) Metal-binding sites in the major groove of a large ribozyme domain. Structure, 4, 1221–1229. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Chang L.M.S. and Bollum,F. (1990) Multiple roles of divalent cations in terminal deoxynucleotidyltransferase reaction. J. Biol. Chem., 265, 17436–17440. [PubMed] [Google Scholar]
- Chothia C. and Lesk,A.M. (1986) The relation between the divergence of sequence and structure in proteins. EMBO J., 5, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.F.,2nd, Almassy,R.J., Hostomska,Z., Ferre,R.A. and Hostomsky,Z. (1994) 2.3 Å crystal structure of the catalytic domain of DNA polymerase β. Cell, 76, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Deibel M.R. and Coleman,M.S. (1980) Biochemical properties of purified human terminal deoxynucleotidyltransferase. J. Biol. Chem., 255, 4206–4212. [PubMed] [Google Scholar]
- Delarue M., Poch,O., Tordo,N., Moras,D. and Argos,P. (1990) An attempt to unify the structures of polymerases. Protein Eng., 3, 461–467. [DOI] [PubMed] [Google Scholar]
- Doherty A.J., Serpell,L.C. and Ponting,C.F. (1996) The helix–hairpin–helix DNA binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res., 24, 2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez O., Ruiz,J.F., Lain de Lera,T., Garcia-Diaz,M., Gonzalez,M.A., Kirchhoff,T., Martinez-A,C., Bernad,A. and Blanco,L. (2000) DNA polymerase µ, homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J., 19, 1731–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- Doublié S., Sawaya,M.R. and Ellenberger,T. (1999) An open and closed case for all polymerases. Structure Fold Design, 7, R31–R35. [DOI] [PubMed] [Google Scholar]
- Esnouf R. (1999) Extension of the program Molscript to display electron density maps. Acta Crystallogr. D, 55, 938–940. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz M. et al. (2000) DNA polymerase λ (Pol λ), a novel eukaryotic DNA polymerase with a potential role in meiosis. J. Mol. Biol., 301, 851–867. [DOI] [PubMed] [Google Scholar]
- Gilfillan S., Dierich,A., Lemeur,M., Benoist,C. and Mathis,D. (1993) Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science, 261, 1175–1178. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle,E., Stuart,D. and Metoz,F. (1999) ESPript: multiple sequence alignments in Postscript. Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1995) DNA polymerase β belongs to an ancient nucleotidyltransferase superfamily. Trends Biochem. Sci., 20, 345–347. [DOI] [PubMed] [Google Scholar]
- Ito J. and Braithwaite,D.K. (1991) Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res., 19, 4045–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjelgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kallenbach S., Goodhardt,M. and Rougeon,F. (1990) A rapid test for V(D)J recombinase activity. Nucleic Acids Res., 18, 6730–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K., Goncalves,J.M., Houts,G.E. and Bollum,F.J. (1967) Deoxynucleotide-polymerizing enzymes of calf thymus gland. II. Properties of the terminal deoxynucleotidyltransferase. J. Biol. Chem., 242, 2780–2787. [PubMed] [Google Scholar]
- Komori T., Okada,A., Stewart,V. and Alt,F.W. (1993) Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science, 261, 1171–1175. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Krayevsky A.A., Victorova,L.S., Arzumanov,A.A. and Jasko,M.V. (2000) Terminal deoxynucleotidyl transferase. Catalysis of DNA (oligodeoxynucleotide) phosphorylation. Pharmacol. Ther., 85, 165–173. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., McArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK, a program to assess the validity of crystallographic models. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lewis S.M. (1994) The mechanism of V(D)J joining: lessons from molecular, immunological and comparative analyses. Adv. Immunol., 56, 27–150. [DOI] [PubMed] [Google Scholar]
- Li Y., Korolev,S. and Waksman,G. (1998) Crystal structure of open and closed forms of binary and ternary complex of the large fragment of T.aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J., 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K.N., Gangi-Peterson,L., Sorscher,D.H., Wang,J., Gathy,K.N., Mahajan,N.P., Reeves,W.H. and Mitchell,B.S. (1999) Association of terminal deoxynucleotidyl transferase with Ku. Proc. Natl Acad. Sci. USA, 96, 13926–13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G. and Keller,W. (1996) Mutational analysis of mammalian poly(A) polymerase identifies a region for primer binding and catalytic domain, homologous to the family X polymerases and to other nucleotidyltransferases. EMBO J., 15, 2593–2603. [PMC free article] [PubMed] [Google Scholar]
- Martin G., Keller,W. and Doublié,S. (2000) Crystal structure of mammalian poly(A) polymerases in complex with an analog of ATP. EMBO J., 19, 4193–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y., Kim,K., Katz,D.S. and Feng,J.A. (1998) Catalytic center of DNA polymerase β for excision of deoxyribose phosphate groups. Biochemistry, 37, 6456–6464. [DOI] [PubMed] [Google Scholar]
- Navaza J. and Saludjian,P. (1997) Amore: a molecular replacement package. Methods Enzymol., 276, 581–593. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamics properties of hydrocarbons. Proteins, 11, 281–291. [DOI] [PubMed] [Google Scholar]
- Ollis D., Brick,P., Hamlin,R., Xuong,N.G. and Steitz,T.A. (1985) Structure of the large fragment of E.coli DNA polymerase I complexed with TMP. Nature, 313, 762–766. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pedersen L.C., Benning,M.M. and Holden,H.M. (1995) Structural investigation of the antibiotic and ATP binding sites in kanamycin nucleotidyltransferase. Biochemistry, 34, 13305–13311. [DOI] [PubMed] [Google Scholar]
- Pelletier H. and Sawaya,M.R. (1996) Metal ion binding to the helix–hairpin–helix motif in human DNA polymerase β by X-ray structural analysis. Biochemistry, 35, 12778–12787. [DOI] [PubMed] [Google Scholar]
- Pelletier H., Sawaya,M.R., Kumar,A., Wilson,S.H. and Kraut,J. (1994) Structures of of ternary complexes of rat DNA polymerase β, a DNA template–primer and ddCTP. Science, 264, 1891–1903. [PubMed] [Google Scholar]
- Pelletier H., Sawaya,M.R., Wolfe,W., Wilson,S.H. and Kraut,J. (1996a) Crystal structure of human DNA polymerase β complexed with DNA: implications for the catalytic mechanism. Biochemistry, 35, 12742–12761. [DOI] [PubMed] [Google Scholar]
- Pelletier H., Sawaya,M.R., Wolfe,W., Wilson,S.H. and Kraut,J. (1996b) Structural basis for metal ion mutagenicity and nucleotide selectivity in human DNA polymerase β. Biochemistry, 35, 12762–12777. [DOI] [PubMed] [Google Scholar]
- Peterson R.C., Cheung,L.C., Mattaliano,R.J., White,S.T., Chang,L.M. and Bollum,F.J. (1985) Expression of human terminal deoxynucleotidyl transferase in Escherichia coli. J. Biol. Chem., 260, 10495–10502. [PubMed] [Google Scholar]
- Roychoudhury R. (1972) Enzymatic synthesis of polynucleotides. Oligodeoxynucleotides with one 3′-terminal ribonucleotide as primers for polydeoxynucleotide synthesis. J. Biol. Chem., 247, 3910–3917. [PubMed] [Google Scholar]
- Sakon J., Liao,H.H., Kanikula,A.M., Benning,M.M., Rayment,I. and Holden,H.M. (1993) Molecular structure of kanamycin nucleotidyltransferase determined to 3.0-Å resolution. Biochemistry, 32, 11977–11984. [DOI] [PubMed] [Google Scholar]
- Sawaya M.R., Pelletier,H., Kumar,A., Wilson,S.H. and Kraut,J. (1994) Crystal structure of rat DNA polymerase β: evidence for a common polymerase mechanism. Science, 264, 1930–1935. [DOI] [PubMed] [Google Scholar]
- Sawaya M.R., Prasad,R., Wilson,S.H., Kraut,J. and Pelletier,H. (1997) Crystal structures of DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry, 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- Semizarov D.G., Arzumanov,A.A., Dyatkina,N.B., Meyer,A., Vichier-Guerre,S., Gosselin,G., Rayner,B., Imbach,J.L. and Krayevsky,A.A. (1997) Stereoisomers of deoxynucleoside 5′-triphosphates as substrates for template-dependent and -independent DNA polymerases. J. Biol. Chem., 272, 9556–9560. [DOI] [PubMed] [Google Scholar]
- Steitz T.A. and Steitz,J.A. (1993) A general two-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA, 90, 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar N., Boulé,J.B., Expert-Bezançon,N., Jourdan,N., Lescar,J., Rougeon,F., Papanicolaou,C. and Delarue,M. (2000) Crystallization of the catalytic domain of murine terminal desoxynucleotidyl transferase. Acta Crystallogr. D, 56, 1662–1664. [DOI] [PubMed] [Google Scholar]
- Swaminathan V. and Sundaralingam,M. (1979) The crystal structures of metal complexes of nucleic acids and their constituents. CRCCrit. Rev. Biochem., 6, 245–336. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. and Eisenberg,D. (1987) Heavy atom systematic search program. Acta Crystallogr. A, 43, 6–13. [Google Scholar]
- Wang J., Sattar,A.K.M.A., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- Yang B., Gathy,K.N. and Coleman,M.S. (1994) Mutational analysis of residues in the nucleotide binding domain of terminal deoxynucleotidyltransferase. J. Biol. Chem., 269, 11859–11868. [PubMed] [Google Scholar]
- Yue D., Maizels,N. and Weiner,A.M. (1996) CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. RNA, 2, 895–908. [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Morera,S., Bates,P.A., Whitehead,P.C., Hainbucher,K., Nash,R.A., Sternberg,M.J., Lindahl,T and Freemont,P.S. (1998) Structure of an XRCC1 BRCT domain: a new protein–protein interaction module. EMBO J., 17, 6404–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.L., Pata,J.D. and Steitz,T.A. (2001) Crystal structure of a DinB lesion bypass polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol. Cell, 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Zhu C., Bogue,M.A., Lim,D.S., Hasty,P. and Roth,D.B. (1996) Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell, 86, 379–389. [DOI] [PubMed] [Google Scholar]