

Abstract
The mammalian DNA (cytosine-5) methyltransferase (Dnmt1) is involved in the maintenance of methylation patterns in the genome during DNA replication and development. The retinoblastoma gene product, Rb, is a cell cycle regulator protein that represses transcription by recruiting histone deacetylase (HDAC1). In vivo, histone deacetylase associates with Dnmt1. Here we show that Rb itself associates with human Dnmt1 (hDnmt1) independently of its own phosphorylation status. Methyltransferase activity was co-purified with Rb. The regulatory domain of hDnmt1 binds strongly to the B and C pockets of Rb (amino acids 701–872) and inhibits methyltransferase activity by disruption of the hDnmt1–DNA binary complex. Weak interaction of Rb pockets A and B with Dnmt1 was also observed. Overexpression of Rb leads to hypomethylation of the cellular DNA, suggesting that Rb may modulate Dnmt1 activity during DNA replication in the cell cycle.
Keywords: adenovirus/hDnmt1/hDnmt–DNA binary complex/methylation/Rb
Introduction
In mammalian cells the methylation status of cytosine is inherited epigenetically during DNA replication (Holliday, 1993). Such DNA methylation is involved in a variety of biological processes such as gene silencing (Bird and Wolffe, 1999), mutation (Gonzalgo and Jones, 1997), development (Li et al., 1992) and cancer (Laird and Jaenisch, 1994; Laird et al., 1995; Jones, 1996; Schmutte and Jones, 1998). Abnormal methylation patterns are often observed in cancer cells (Baylin et al., 1998; Costello et al., 2000). Such events may result from the failure of a control mechanism for the major DNA methyltransferase, Dnmt1, during the cell cycle. DNA methylation in mammalian cells is maintained during the S phase of the cell cycle by Dnmt1 (Leonhardt et al., 1992), a 184 kDa protein consisting of a regulatory (N-terminal) and a catalytic (C-terminal) domain (Figure 1A) ( Bestor et al., 1988; Yen et al., 1992). Both domains are fused by a run of 13 alternating Gly–Lys residues and can be cleaved by proteolysis of the intact enzyme with V8 protease (Bestor, 1992). A cluster of eight cysteinyl residues (CX2CX2 CX4CX2CX2), which has been shown to bind zinc ions, lies in the center of the N-terminal domain (Bestor, 1992). Another part of the N-terminal domain has been implicated in co-localizing the murine Dnmt1 with the replication machinery (Leonhardt et al., 1992). A short peptide region (TRQTTITSHFAKG) of hDnmt1 has been found to bind proliferating cell nuclear antigen (PCNA) (Chuang et al., 1997). Recently, two other regions (amino acids 653–730 and 686–812) have been shown to associate with histone deacetylase, HDAC1 (Fuks et al., 2000).

Fig. 1. Human Dnmt1 associates with Rb in vivo. (A) Diagram representing full-length hDnmt1 and Rb pockets. Amino acid numbers for the Rb A, B and C pockets are indicated. Regions associated with specific function are indicated as: PCNA, proliferating cell nuclear antigen binding; RF, replication foci directing, and Zn, zinc binding. N- and C-termini are from amino acid residues 1–1108 and 1122–1616, respectively, for hDnmt1. Conserved motifs are in roman numerals. (B) Methyltransferase activity co-immunoprecipitates with Rb in a recombinant insect cell expression system using poly(dI–dC)·poly(dI–dC) as substrate DNA. Antibodies used for immunoprecipitation are indicated at the bottom. Pre-immune serum is indicated as PI. (C) hDnmt1 co-immunoprecipitates with Rb in recombinant Sf9 cell extracts. Expression of protein is indicated by a ‘+’. Immunoprecipitation with rabbit IgG is abbreviated as IgG. Positive control retinoblastoma (C Rb) and human maintenance methyltransferase (C hDnmt1) are indicated. (D) Left panel: co-immunoprecipitation of hDnmt1 in HEK 293 cell nuclei extracts using antibody against either the N- or C-terminus of Rb. Control rabbit IgG does not bind to hDnmt1. Right panel: association of Rb and Dnmt1 in the presence (+) or absence (–) of serum using C-terminus antibody for Rb. For both panels, HEK 293 cell nuclear extract was used as a control to show expression of Rb as well as hDnmt1 in cells. Cells used for the study (NIH 3T3 and HEK 293) are indicated at the top.
The retinoblastoma gene product, Rb, is a major cell cycle regulator nuclear phosphoprotein of ∼110 kDa. It contains three pockets, A, B and C, which bind to a variety of cellular and regulatory proteins (Taya, 1997). Rb has the ability to suppress cell proliferation via cell cycle-dependent phosphorylation (Weinberg, 1995). It has been established that the growth-suppressing activity of Rb is exerted by binding and inhibiting the transcription factor E2F (Nevins, 1992; La Thangue, 1994). Rb also recruits HDAC1 to E2F and cooperates with it to repress the E2F-regulated promoter of the gene encoding the cell cycle protein cyclin E (Brehm et al., 1998; Luo et al., 1998; Magnaghi-Jaulin et al., 1998). In several cancer cells, including retinoblastoma, osteosarcoma, small-cell lung cancer and bladder cancer, inactivation of the gene for tumor suppressor proteins, such as Rb, via mutation, deletion or methylation, has been observed (Weinberg, 1991; Ohtani-Fujita et al., 1997).
In this study we have investigated the possible communication between hDnmt1, the human maintenance methyltransferase and the tumor suppressor gene product Rb. Our results show a physical interaction between hDnmt1 and Rb. Rb is also shown to compete with the assembly of the intermediate DNA–hDnmt1 binary complex that precedes DNA methylation, suggesting a role in hypomethylation of the cellular DNA in vivo. A novel mechanism in which Rb modulates hDnmt1 activity is discussed.
Results
Rb interacts with and binds to hDnmt1
We first determined whether the Rb and hDnmt1 proteins interact physically in vivo using a recombinant baculovirus expression system. Both hDnmt1 (Figure 1A) and a fragment of human Rb containing ABC pockets (amino acids 379–928; Figure 1A) were cloned in opposite directions with respect to each other in a baculovirus dual promoter expression vector, p2Bac. hDnmt1 and the Rb ABC pockets were under the transcriptional control of the p10 and polyhedrin promoters, respectively. Sf9 cells were infected with the recombinant baculovirus to express the two proteins. The cell extract was used in a pull-down assay for methyltransferase activity using an antibody that recognizes amino acid residues either at the N- or C-terminus of Rb. Both antibodies were able to pull down DNA methyltransferase activity, which could be detected by using either poly(dI–dC)·poly(dI–dC) or hemimethylated DNA as substrate compared with the pre-immune serum (Figure 1B). The antibody directed towards the C-terminus was less effective than the one specific for the N-terminus. The physical interaction between hDnmt1 and Rb was also confirmed by several other methods, such as co-immunoprecipitation and a yeast two-hybrid assay. For the co-immunoprecipitation assay, extracts from SF9 cells expressing Rb ABC and hDnmt1 were used along with the control extracts that were expressing either Rb ABC or hDnmt1 construct. Extracts were clarified with the pre-immune serum, centrifuged, and the supernatants were then incubated either with anti-hDnmt1 or anti-Rb antibodies. The antibody–protein complexes were analyzed by western blotting using either anti-Rb or anti-hDnmt1 antibodies (Figure 1C). The results indicate that the hDnmt1–Rb interaction was specific since only when both proteins were expressed in Sf9 cells could either be detected (Figure 1C). Control Sf9 cells in which only hDnmt1 or Rb was expressed showed no reactivity, confirming the interaction. A yeast two-hybrid assay system was used to probe further the nature of the interaction between Rb and hDnmt1. Yeast cells expressing both GAL4AD–hDnmt1 (amino acids 1–489) and GAL4BD–Rb (amino acids 778–808 or 778–847) were able to grow on His– plates and express β-galactosidase, confirming the interaction of both proteins in vivo (data not shown). Control GAL4BD–Rb fusion (amino acids 778–808 or 778–847) or GAL4AD–hDnmt1 fusion alone was not able to complement His–, thus were not able to grow. To prove unequivocally that hDnmt1 and Rb do interact within human cells, endogenous co-immunoprecipitations were carried out on HEK 293 nuclear extracts using antibody specific for either the N- or C-terminus of Rb (anti-Rb N or anti-Rb C). Rabbit IgG antibody was used as a control. The western blots were probed with anti-hDnmt1 N-terminus antibody (Figure 1D). Indeed, hDnmt1–Rb complexes were pulled down by anti-Rb antibodies, thus confirming that Rb interacts with hDnmt1 in human cells.
The cell cycle dependence of the interaction was tested by growing both NIH 3T3 and HEK 293 cells either in the presence or absence of serum. Nuclear extracts were used for endogenous co-immunoprecipitation. As expected, the actively growing, serum-supplemented HEK 293 cells had higher levels of hDnmt1–Rb complexes as compared with serum-starved cells, which do not divide. Thus the quantity of Rb bound to hDnmt1 was much less. The antibody used was specific for human Dnmt1 and so was unable to detect mouse Dnmt1 in NIH 3T3 cells. These results suggest a strong interaction between hDnmt1 and Rb during DNA replication and cell division.
N-terminus of hDnmt1 interacts with pockets of Rb
Glutathione S-transferase (GST) fusion proteins can be used in vitro to identify proteins or protein fragments that interact with the fusion. The fusion protein is incubated with the cell lysate and protein complexes are isolated by capture on glutathione–Sepharose beads. After separation by SDS–PAGE, the proteins associated with the fusion can be detected by western blot analysis.
To search for the interacting protein motifs between hDnmt1 and Rb, a series of deletions of hDnmt1 and Rb were generated and the interactions of these various truncated proteins were analyzed using a GST pull-down assay, followed by western blot analysis (Figure 2A, upper panel). The results with a GST–Rb fusion indicate that full-length hDnmt1 binding to Rb depended on the presence of amino acids 701/778 to 888 (Figure 2A, lower panel, lanes 4 and 5). Following longer exposure, a faint Dnmt1 band was observed in the GST–Rb 380–778 lane (data not shown). In similar experiments using the catalytically active Dnmt1 N-terminal deletion mutants hΔ336Dnmt1 and hΔ541Dnmt1, the first 336 amino acids were identified as the interacting region (Figure 2B) since only the full-length hDnmt1 and not the deletion mutants was able to form a complex with Rb. This region includes the PCNA-binding motif (amino acids 122–322) of hDnmt1 (Chuang et al., 1997).

Fig. 2. Interacting regions between Rb and hDnmt1. (A) Upper panel: the GST–Rb fusion constructs along with the amino acid numbers. Lower panel: the GST pull-down results using purified full-length hDnmt1 protein (+) and anti-N-terminal hDnmt1 antibody. (B) Upper panel: the catalytically active deletion mutants of hDnmt1. Deleted amino acids are indicated on the right. Lower panel: pull-down assay with either GST or GST–Rb ABC is indicated (+), along with the hDnmt1 and mutants at the top.
Rb inhibits the DNA methyltransferase reaction of hDnmt1
Since the primary function of mammalian Dnmt1 is to methylate newly replicated DNA, we investigated whether the binding of Rb to hDnmt1 interfered with the methyltransferase activity of the human enzyme. For these experiments full-length hDnmt1 and the methylation-proficient purified N-terminal deletion mutant enzymes lacking 336 and 541 amino acids (hΔ336Dnmt1, hΔ541Dnmt1) were used. Rb fragments fused to maltose binding protein (MBP) (Figure 3A) were incubated in an assay mixture containing hemimethylated oligonucleotides, S-adenosyl-l-[methyl-3H]methionine and a fixed concentration of either full-length recombinant hDnmt1 or mutants hΔ336Dnmt1 or hΔ541Dnmt1. The radioactive incorporation was measured and data were analyzed as described previously (Bacolla et al., 1999; Pradhan et al., 1999). Addition of MBP had no effect on DNA methylation. However, we observed a dramatic inhibition of the enzyme activity when Rb C (amino acids 701–928, B and C pockets), Rb 2 (amino acids 701–872, B and C pockets) or Rb 5 (amino acids 778–888, C pocket) was incubated with full-length hDnmt1 (Figure 3B). This inhibitory effect was greatly reduced with N-terminal mutants hΔ336Dnmt1 and hΔ541Dnmt1, although the methyltransferase inhibition patterns for both N-terminal deletion mutants were similar in the presence of Rb fragments (data not shown). A comparative methylation profile between full-length hDnmt1 and hΔ336Dnmt1 in the presence of various fragments of Rb is shown (Figure 3B). These results confirm the involvement of the first 336 amino acids of hDnmt1 in Rb binding and suggest their interaction represses methyltransferase activity. From the GST pull-down assays (Figure 2B) it appears that the inhibition is primarily mediated through protein– protein interactions of Rb 2 and Rb 5 with full-length hDnmt1. Since excess DNA substrate was used for the methyltransferase reaction, the possibility of Rb pockets binding to DNA and preventing enzyme–DNA complex formation may be ruled out. This observation was further confirmed using a mouse–prokaryotic hybrid DNA (cytosine-5) methyltransferase, Dnmt1–HhaI (Pradhan and Roberts, 2000). The N-terminal regions of mouse and hDnmt1 have >90% identity and are expected to experience similar inhibitory effects in the presence of Rb. In contrast, the prokaryotic M.HhaI enzyme does not have a regulatory domain, and was not sensitive to the presence of Rb. In the presence of GST–Rb ABC, Dnmt1–HhaI lost 50% of its methyltransferase activity (Figure 3C). These results demonstrate that it is the N-terminus of the hybrid enzyme that causes repression of methyltransferase reaction in the presence of Rb.


Fig. 3. Rb inhibits hDnmt1 via disruption of a DNA–hDnmt1 binary complex. (A) MBP–Rb fusion proteins used for methyltransferase assay. (B) Inhibition of methyltransferase activity in the presence of various fragments of Rb. The enzyme used for the assay was either full-length hDnmt1, indicated by gray bars, or a 336 amino acid deletion mutant of hDnmt1, indicated by black bars. (C) Rb ABC also inhibits a hybrid Dnmt1–HhaI methyltransferase, but not M.HhaI alone. (D) Gel-shift assay showing binary complex formation between hDnmt1 (40 nM), radiolabeled DNA (1.4 nM) and various competitor proteins (200 nM) as indicated. Arrows indicate the relative positions of various complexes. (E) Gel-shift assay with a fixed concentration of hDnmt1 (40 nM) and an increasing concentration of Rb C (0, 1, 2, 3, 4, 5, 6× molar concentration, lanes 2–9). Arrow indicates the relative position of DNA–hDnmt1 complexes. Lane 1 shows input radiolabeled oligonucleotide duplexes. ‘+’ indicates presence of protein/DNA in the reaction mix.
Rb dissociates the DNA–hDnmt1 binary complex
For the catalytic steps of the methyltransferase reaction, enzyme–DNA binary complex formation is a crucial step. To test if Rb can dissociate DNA–hDnmt1 binary complexes, gel-shift assays were carried out. Initially, a constant amount of radiolabeled hemimethylated DNA was titrated with an increased amount of enzyme (data not shown). For the entire radiolabeled DNA to form a complex with enzyme, a 30-fold molar excess of recombinant hDnmt1 (40 nM) was mixed with 1.4 nM radiolabeled DNA and incubated at 37°C for 30 min. This resulted in a DNA–hDnmt1 binary complex, as observed on a native gel (Figure 3D, lane 2). The identity of this complex was confirmed by a super-shift observed upon incubation with anti-hDnmt1 antibody (Figure 3D, lane 3). The dissociation of the complex was examined in the presence of 5-fold excess competitor proteins (200 nM). Neither the control MBP protein nor the MBP–Rb N had any effect on the binary complexes. However, the binary complexes between hDnmt1 and DNA were greatly reduced in the presence of MBP–Rb C (Figure 3D, lane 5), indicating that the Rb B C pocket is sufficient to disrupt a DNA–hDnmt1 binary complex. In a competition gel-shift assay a 1:3 molar ratio between recombinant hDnmt1 and MBP–Rb C was sufficient to reduce DNA– hDnmt1 binary complex formation by 50% (Figure 3E, lane 5). The same effect required about five to ten times more Rb pocket A and B (data not shown). The Rb 2 fragment had a very similar effect on complex dissociation as that of Rb C (data not shown). This suggests that Rb C has a much higher affinity for hDnmt1 than do the Rb AB pockets. In vivo, it is possible that all the Rb pockets either interact independently or could cooperate with each other in binding to hDnmt1.
Overexpression of Rb leads to hypomethylation of the genome and transgene activation
These observations suggest that Rb may interfere with DNA methylation during cell division and development. This hypothesis was tested by transfecting Saos-2 cells, an osteosarcoma cell line that does not express Rb (Craig et al., 1998), with either a mock adenovirus vector or one encoding Rb. This vector expresses genes in both replicative and non-replicative cells; however, the vector does not replicate, thus enabling an examination of the effect of Rb on genomic DNA in human cells. Cell extracts were examined for Rb expression using anti-Rb antibody in a western blot assay. Saos-2 transfected with adenovirus-Rb resulted in a large accumulation of Rb protein (Figure 4A, lanes 3 and 4). Expression of Rb had no effect on hDnmt1 expression in adenovirus-Rb transfected cells (Figure 4A, lanes 3 and 4) and the amount of PCNA remained the same (data not shown). Cellular DNAs were isolated 30 h post-transfection from adenovirus- and adenovirus-Rb-transfected Saos-2 cells and the isolated DNAs were analyzed for changes in methylation. We hypothesized that if Rb was capable of binding and inhibiting hDnmt1 during cell division, then cellular DNA would lose methylation following DNA replication in vivo, resulting in a greater number of unmethylated CG sites. To test this hypothesis, cellular DNAs from either adenovirus (adeno)- or adenovirus-Rb (adeno-Rb)-transfected cells were digested with McrBC endonuclease, an enzyme that cleaves DNA containing methylcytosines on one or both strands. As expected, adeno-Rb-transfected Saos-2 DNA showed greater resistance to McrBC cleavage than did adenovirus-transfected DNA, thus proving the association of hDnmt1 with Rb leads to hypomethylation of the genome in vivo (Figure 4B). These newly available unmethylated CG sites were also measured by a global methylation assay (Lin et al., 2001) using M.SssI enzyme, which recognizes the same sites for methylation as that of the mammalian DNA (cytosine-5) methyltransferase Dnmt1. M.SssI recognizes and methylates both unmethylated and hemimethylated CG sites with equal efficiency. An increase of ∼2-fold in methyl acceptance of the substrate DNA (Figure 4C) suggests the presence of a significant proportion of unmethylated and hemimethylated CG target sites in the cellular DNA of adeno-Rb- but not adeno-transfected cells, demonstrating involvement of Rb in hypomethylation of the genomic DNA. Since Rb binds to hDnmt1 and inhibits DNA methylation in vitro, it is possible that excess Rb in Saos-2 adeno-Rb-transfected cells sequesters the available hDnmt1, resulting in improper maintenance or abolition of the methylation pattern in vivo. Here we cannot rule out the possible involvement of other protein(s) that may have been activated by Rb, such as 5-methylcytosine DNA glycosidase (Zhu et al., 2001) or demethylase (Bhattacharya et al., 1999). However, the above assay does not specifically link the methyl acceptor sites on the DNA with a loss of hDnmt1 activity due to Rb–hDnmt1 complex formation. In order to examine the loss of maintenance methylation (hemimethylated CG sites) in cellular DNA, we repeated the methyltransferase assay using purified hDnmt1 in place of M.SssI. Human Dnmt1 methylates hemimethylated CGs 10- to 40-times more efficiently than unmethylated CG (Pradhan et al., 1999), thus providing a means for distinguishing unmethylated and hemimethylated sites. Methylation of control adenovirus-transfected DNA was a better substrate than adeno-Rb-transfected DNA, since methyl group incorporation on the former was 30% higher (Figure 4D). This suggests fewer hemimethylated CG target sites in the latter DNA. These results indicate that overexpression of Rb has a bearing on the methylation of the genome. Thus, Rb modulates DNA methylation via binding to target hDnmt1 and rendering it inactive in vivo.

Fig. 4. Transfections of adenovirus mock and adenovirus-Rb DNA into Saos-2 cells. (A) Western blots of the transfected Saos-2 cell extracts using anti-hDnmt1 and anti-Rb antibodies. Endogenous hDnmt1 and Rb along with adenovirus-Rb (Adeno-Rb)-transfected cell extracts are indicated. Lanes 1 and 5 represent 10 μg of HEK 293 cell extracts. HEK 293 cells do express both Dnmt1 and Rb. Note that Saos-2 cell extracts transfected with adenovirus mock (Adeno) do not express endogenous Rb, but HEK 293 and adeno-Rb do (lanes 1, 3, 4 and 5). Lanes 3 and 4 represent extracts from two independent transfections. Equal amounts of cell extracts were loaded in each lane. (B) McrBC digestion of the genomic DNA from adenovirus mock and adenovirus-Rb-transfected Saos-2 cells. The total DNAs were digested by McrBC with or without GTP as indicated above. Molecular weight markers are in kbp. The source of DNA is also indicated at the the top of the gel. (C) Global methylation assay of adeno- and adeno-Rb-transfected cellular DNA. Available CpG target sites were methylated with M.SssI in the presence of tritiated AdoMet. The amount of 3H incorporated into adeno-transfected DNA (c.p.m.) was normalized as 100% methylation. Each DNA was assayed three times in duplicate. (D) Global hemimethylation assay of adeno- and adeno-Rb-transfected cellular DNA. Available hemimethylated CpG target sites in the genome were methylated with hDnmt1 in the presence of tritiated AdoMet. The amount of 3H incorporated into adeno-transfected DNA (c.p.m.) was normalized as 100% methylation. Each DNA was assayed three times in duplicate. Note the value represented here is an overestimate, since hDnmt1 methylation values also represent the addition of a few de novo methylation events by the enzyme.
The above observations suggested that Rb could interfere with DNA methylation during DNA replication. This hypothesis was tested using an in vitro methylated pGL3-promoter vector. This vector contains an SV40 promoter upstream of the luciferase gene. Methylated reporter genes are transcriptionally inactive, thus luciferase expression is not expected. If Rb is capable of binding and inhibiting hDnmt1 during plasmid replication, the reporter plasmid will lose maintenance methylation following DNA replication in vivo and be transcriptionally active. The methylated reporter was introduced into T antigen-containing COS-7 cells that support replication of plasmid pGL3 promoter. Adeno or adeno-Rb constructs were co-transfected along with the β-galactosidase gene construct to normalize transfection efficiency. We observed suppression of fully methylated reporter gene in the absence of Rb (Figure 5A, compare Um with Me) and luciferase gene expression was ∼30–40% more in adeno-Rb-transfected cell extracts compared with adeno- transfected (Figure 5A, compare Me+Ad with Me+Ad-Rb). To elucidate the role of DNA methylation in luciferase gene expression, transfected cell DNAs were extracted and digested with the methylation-sensitive restriction enzymes MspI (CCGG) or HpaII (CCGG). MspI and HpaII are isoschizomers that differ in their methylation sensitivity (van der Ploeg et al., 1980). HpaII digestion is blocked if the internal CG is methylated, whereas MspI can cut such DNA. If Rb is capable of binding and inhibiting hDnmt1 during plasmid replication, the reporter plasmid will have poor maintenance methylation following DNA replication in vivo and will have a different methylation pattern in the presence of Rb. In Figure 5B, we observed identical methylation patterns for both MspI and HpaII digestion of unmethylated pGL3 promoter. However, methylated pGL3 promoter plasmid DNA was resistant to HpaII but not MspI, an indication of the presence of CpG methylation. When control adeno and fully methylated pGL3 promoter were co-transfected, a set of HpaII-digested bands was observed (Figure 5B, lane 6). A small portion of methylated DNA appears to have lost its methylation. This loss of methylation could be as a result of the lack of proper maintenance methylation associated with rapid replication of pGL3 promoter in T antigen-based COS-7 cells. This coincides with low levels of luciferase gene expression in adeno-transfected cells (Figure 5A, Me+Ad). In previous studies, an alteration of methylation was observed in hamster cells transgenic for adeno 12 DNA (Muller et al., 2001), suggesting methylation pattern alteration might be another factor in luciferase gene expression. Evidence of hypomethylation was observed in a co-transfection of pGL3 promoter and adeno-Rb by increased intensity of HpaII fragments (Figure 5B, lane 8). Densitometric scans of lanes 6 and 8 suggested a 30% increase in lower molecular weight bands in lane 8, demonstrating additional unmethylated CGs in HpaII sites (data not shown). Loss of methylation in pGL3 promoter plasmid (Figure 5B) was similar to that of whole genome hypomethylation (Figure 4C), suggesting hypomethylation events follow a similar mechanism. These results imply a possible role for Rb in inhibition of the maintenance methylation of genes, perhaps by sequestering the available hDnmt1.
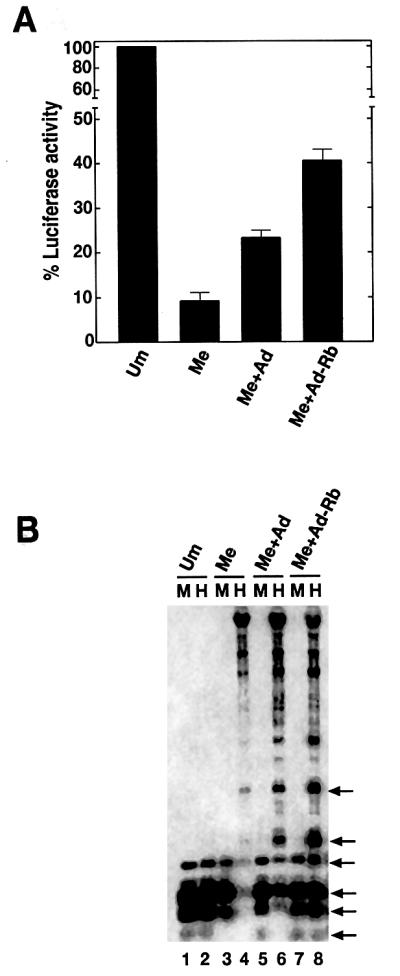
Fig. 5. Luciferase gene expression and methylation analysis of methylated pGL3 promoter (reporter gene construct) in COS-7 cells. (A) Luciferase gene expression in the transfected COS-7 cell extracts. Transfected DNAs are indicated: Um, unmethylated pGL3 promoter; Me, methylated pGL3 promoter; Me+Ad, methylated pGL3 promoter and adeno construct; and Me+Ad-Rb, methylated pGL3 promoter and adeno construct containing Rb. Luciferase expression was normalized with β-galactosidase internal control and was taken as 100% for unmethylated pGL3 promoter. (B) Changes in methylation pattern of the luciferase gene in the presence of exogenous Rb: Southern blot of total DNA probed with random-primed luciferase gene. Each lane represents either MspI (M) or HpaII (H) restriction digest as indicated. The combination of transfected DNA is as in (A). HpaII specific bands with significant change in intensity (lane 8) are indicated on the right by arrows.
Discussion
Defects in mammalian methylation patterns have severe consequences for cell survival, gene expression and development, as evident in the embryonic lethality of homozygous deletions of the mouse Dnmt1 gene (Li et al., 1992). Altered patterns of DNA methylation are often observed in cancer cells. In these cells, despite a decrease in overall DNA methylation (Gama-Sosa et al., 1983), the normally unmethylated CpG islands in the promoter region of crucial genes are densely methylated, resulting in transcriptional silencing. It is estimated that 600 out of 45 000 CpG islands are highly methylated in human tumors (Costello et al., 2000). This epigenetic coding region modification may accompany, and potentially cause, a loss of tumor suppressor gene function (Laird and Jaenisch, 1994; Jones, 1996), including p16, p15, VHL and E-cad. Each gene can be partially reactivated by demethylation using drugs, such as 5-aza-2′-deoxycytidine. It is hypothesized that in neoplastic cells the protective mechanism from abnormal methylation is lost, possibly by excess exposure to Dnmt1. An increase in methyltransferase activity was found to be target cell specific and proposed to be an early event in lung cancer (Belinsky et al., 1996). Furthermore, induced overexpression of Dnmt1 in human fibroblast tissue culture cells has been shown to induce hypermethylation of CpG islands and cellular transformation (Vertino et al., 1996).
Mammalian DNA methylation is a complex phenomenon. For correct methylation of the vertebrate genome, several protein factors and methyltransferases must work together in harmony. The major maintenance methyltransferase, Dnmt1, is evolutionarily conserved in mouse, human, Xenopus and sea urchin, suggesting a similar functional role. During DNA replication, Dnmt1 is located at the replication foci and methylates the newly synthesized daughter strands. PCNA, an auxiliary factor, facilitates loading of δ and ε DNA polymerases onto the replication foci in cycling cells. Chuang et al. (1997) have suggested that binding of hDnmt1 to PCNA, coordinates methyltransferase activity with DNA replication, and further that this binding is negatively regulated by p21. This regulation can be attributed, at least in part, to the mutually exclusive binding of p21 and hDnmt1 to PCNA. Results presented here suggest that binding of Rb to hDnmt1 will block binding of PCNA. Thus, an altered protein–protein interaction between hDnmt1 and various regulatory proteins might disrupt enzymatic function.
In our interaction studies we have identified the first 336 amino acids of hDnmt1 as the region that interacts with Rb using three biologically active (methylation-proficient) proteins: full-length hDnmt1 and deletion derivatives hΔ336Dnmt1 and hΔ541Dnmt1. Further N-terminal deletion of hDnmt1 beyond amino acid 580 abolishes methylation activity even when the catalytic domain remains, a result attributed to misfolding (Zimmermann et al., 1997). Due to the potential for global misfolding, deletions beyond this point were not used in this study. Previous studies (Robertson et al., 2000) have suggested that an alternative region of hDnmt1 (amino acids 416–913) interacts with Rb, based on experiments that utilized in vitro translated C-terminal deletions of hDnmt1 and GST–Rb fusion proteins. This region includes HDAC1 binding (amino acids 653–812) (Fuks et al., 2000). Thus, it is possible that another protein factor might have mediated Rb binding on hDnmt1. In contrast, we have used proteins expressed in vivo for interaction studies. We have also performed GST pull-down assays with GST–fusion fragments of hDnmt1. Only the fusion protein containing hDnmt1 peptide 1–334 amino acids was able to bind Rb. Along with a yeast two-hybrid assay and two different GST pull-down assays, our data indicate that the first 334 amino acids are indeed involved in the interaction with Rb.
An association between increased DNA methyltransferase activity and early events leading towards the establishment of a transformed state is documented in human cells (Kuerbitz and Baylin, 1996) and in a strain of mice genetically susceptible to lung carcinogenesis (Belinsky et al., 1996). In human colon cancer, the hDnmt1 transcript is present in 1.5- to 4-fold higher amounts than that in normal colon tissue (Lee et al., 1996; Schmutte et al., 1996). Hypomethylation was also associated with a reduced number of intestinal adenomas in mice (Laird et al., 1995). Thus, the regulation of hDnmt1 appears to be an important step in cancer progression. A simple model for the effect noted here envisages Rb acting in normal cells to regulate Dnmt1 being loaded at the replication foci. Conversely, the aberrant methylation patterns noted in tumors may result from alterations in Rb or the Rb pathway (Figure 6), alterations also noted in almost all tumor cell lines (Hanahan and Weinberg, 2000; Guan et al., 2001). Our results indicate that Rb binds to hDnmt1 and that this association can inhibit methyltransfer activity. It is not presently known if Rb alone is sufficient to modulate Dnmt1 activity or whether other transacting factors are involved in vivo. For example, some evidence suggests that p21 can negatively regulate the PCNA–hDnmt1 complex in the normal cell, potentially playing an indirect role in modulating hDnmt1 activity (Chuang et al., 1997).

Fig. 6. A proposed regulation of DNA methylation. In normal cells, hDnmt1 is regulated by Rb, thus maintenance methylation is carried out only at the hemimethylated CGs (filled lollipop, methyl cytosine) during replication, and excess Dnmt1 is sequestered by Rb. DNA in cancer cells lacking functional Rb may facilitate aberrant methylation patterns.
Human Dnmt1 also participates in methylation-independent gene silencing via transcriptional repression. Recently, Robertson et al. (2000) have shown that complex formation between Dnmt1, Rb, E2F and HDAC1 represses transcription from E2F-responsive promoters in vivo. One aspect of this repression is thought to involve Dnmt1 and HDAC1 association (Fuks et al., 2000), leading to the generation of deacetylated histones as well as an altered chromatin state, which in turn has the potential to recruit methyl DNA binding proteins at the newly replicated sites, forming a transcriptionally silenced chromatin. Indeed, co-transfection of a human ARF promoter (Robertson and Jones, 1998)-driven reporter plasmid containing two GAL-4 DNA binding sites along with GAL-4Rb and hDnmt1, or GAL-4 Dnmt1 and Rb, into human cells resulted in repression of reporter gene activity. In this assay, GAL-4 Dnmt1 protein is targeted artificially into the GAL-4 DNA binding sites of ARF promoter. However, re-isolation and methylation analysis of the transfected ARF construct did not detect methylation (Robertson et al., 2000). This shows that the hDnmt1–Rb complex is not effective methylation machinery in vivo, supporting our observation. Results reported here show that Rb can sequester the available hDnmt1 and prevent the enzyme from binding to the substrate in vitro. We suggest that such an interaction in vivo may lead to loss of maintenance methylation on hemimethylated DNA. In fact, a point mutation on Rb (C706F) inhibits hDnmt1–Rb complex formation (Robertson et al., 2000). Rb binding and regulating hDnmt1 is perhaps one of many mechanisms in which some sequences in the genome, like CpG islands, remain methylation free during normal cellular growth and development. However, during cancer progression, in the absence of Rb or hDnmt1 regulatory proteins, such sequences are methylated aberrantly.
Using steady-state kinetic approaches and in vitro inhibition studies with hDnmt1 and methylated DNA (Bacolla et al., 2001), methylated DNA binding and allosteric activation domains have been shown to reside with the N-terminal 501 amino acids. Here we show that the same region of hDnmt1 also binds to Rb. In conjunction with those studies, it was suggested that hDnmt1 enzymatic activity could be inhibited via interaction of the N-terminal domain with binding proteins (Bacolla et al., 2001), either directly or indirectly by interfering with allosteric activation. Our present studies suggest that Rb can play this role. In our previous studies with a series of mammalian–prokaryotic hybrid methyltransferases (Pradhan and Roberts, 2000) we have shown that properties of the native Dnmt1 depend on elements located in the C-terminal region, with interplay between regions both at the N- and C-terminal amino acids being crucial. The binding of a protein factor to hDnmt1 has the potential to disrupt communication between the domains, thus producing the aberrant methylation pattern observed in cancer cells. Thus, Rb may play a dual regulatory role including both transcription of E2F-responsive promoters and maintenance methylation at DNA replication via Rb–Dnmt1 interaction.
Materials and methods
Plasmids
A cDNA containing full-length human Dnmt1 (Yen et al., 1992; accession No. X63692), pBS-5.0HMT(a) (a gift from Professor S.B.Baylin), was used for hΔ336Dnmt1 and hΔ541Dnmt1 construction by inserting it into pVIC1 (Pradhan et al., 1999), a baculovirus transfer vector. Full-length hDnmt1 expression has been described previously (Pradhan et al., 1999). Human Rb ABC was obtained by PCR from a T-cell library and cloned in-frame with GST to give rise to pGXRb ABC. Rb was inserted at the BamHI–EcoRI site downstream of the polyhedrin promoter and hDnmt1 was cloned at the SmaI site downstream of the P10 promotor of p2Bac (Invitrogen). The final construct was p2Rb-HMT. All MBP fusion constructs were in-frame with pMAL-c2X (New England Biolabs). Similarly, GST fusion constructs were constructed in pGEX-5X-1 (Amersham Pharmacia). For yeast two-hybrid assays, the vectors pAS2-1 and pACT2 (Clontech) were used. Details of other constructs are available on request.
MBP/GST fusion protein expression, pull-down, methyltransferase and yeast two-hybrid assay systems
Fusion proteins were expressed in Escherichia coli BL21 or ER2502 cells. Purification of recombinant fusion proteins from the bacterial lysate followed the manufacturer’s instructions. For GST pull-down assay, GST or the GST fusion proteins was bound to glutathione–Sepharose beads. The assay was performed by pre-incubating the GST or GST fusion protein beads with 100 µg/ml bovine serum albumin (BSA) and the protein to be studied in a binding buffer (50 mM Tris pH 7.5, 28 µM ZnCl2, 0.1% Triton X-100, 220 mM NaCl, 10% glycerol) at 4°C for 4 h. Beads were washed 3–4 times with binding buffer containing 300 mM NaCl. The beads were mixed with 1× SDS–PAGE sample loading buffer (New England Biolabs) and incubated at 98°C for 5 min. The protein mixtures were separated on a 4–20% polyacrylamide gel (ISS miniplus SepraGel). The protein bands were blotted on to a PVDF membrane and probed using indicated antibodies. The methyltransferase assay was as described previously (Bacolla et al., 1999; Pradhan et al., 1999) and the final wash contained binding buffer with 0.1 M NaCl. For the yeast two-hybrid assay system, Clontech’s protocols were followed.
Cell culture, immunoprecipitation and western blot analysis
Sf9 insect cells were infected with recombinant baculovirus containing both Rb and hDnmt1 from p2Rb-HMT. Forty-eight hours post-infection the cells were lysed in binding buffer. The lysate was clarified using pre-immune serum and appropriate anti-Rb (Cell Signaling Technology) or anti-hDnmt1 Abs (New England Biolabs) were used to capture the Rb–hDnmt1 complex. The Rb–hDnmt1–antibody complex was then trapped using protein A–Sepharose beads. After three washes (binding buffer containing 300 mM NaCl), the bound proteins were denatured and separated on a denaturing gel, blotted and probed with specific antibodies. For co-immunoprecipitation, HEK 293, NIH 3T3 and Saos-2 cells were grown on MEM supplemented with 10% horse serum or Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum (FCS) respectively. For adenovirus transfection viral DNA and Saos-2 cells were co-incubated in DMEM without serum for an hour and incubated further with DMEM plus 10% FCS for protein expression. The cells were harvested after 30 h. The nuclei were purified and co-immunoprecipitation was carried out in TNN buffer (40 mM Tris–HCl pH 8.0, 120 mM NaCl, 0.5% NP-40). After clarification of the extract with appropriate control IgGs, the protein complex was captured with an appropriate agarose-conjugated antibody and analyzed by western blotting.
Gel-shift assay
32P-end-labeled oligonucleotide (1.4 nM) (36-mer hemimethylated FMR-1; Pradhan et al., 1999) and 40 nM recombinant hDnmt1 (New England Biolabs) were incubated for 30 min at 37°C in 1× binding buffer [10 mM HEPES pH 7.5, 10% glycerol, 50 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 2.5 mM MgCl2, 0.2% Triton X-100] in a 25 µl reaction volume. Protein–DNA complexes were resolved on a 4.8% native polyacrylamide gel at 4°C in 0.5× TBE (45 mM Tris–HCl, 45 mM H3BO3, 10 mM EDTA pH 8.3) at 140 V. After running the gel, it was fixed, dried and autoradiographed. For super-shift assays, anti-hDnmt1 was added to the above reaction and incubated on ice for 10 min before loading. When necessary, competitor proteins were added after the DNA–hDnmt1 reaction and incubated for another 30 min at 37°C.
Adenovirus vector for Rb gene delivery into Saos-2 cells and global methylation assay of the cellular DNA
Rb gene was cloned into adenovirus vectors with the E1 deletion and were replication defective. Inserted gene was under the control of CMV promoter/enhancer and therefore was expressed at high levels. DNAs from Saos-2 cells transfected with adenovirus or adenovirus-Rb were purified using the Qiagen kit. Ethanol-precipitated DNA was dissolved in TE buffer and quantified at A260. The methylation status of the genome was assayed using McrBC enzyme. Approximately 250 ng of cellular DNA were incubated with 20 U of enzyme with or without cofactor, 1 mM GTP. After incubation at 37°C for 3 h, the reaction mixture was separated on a 1% TBE–agarose gel, stained with ethidium bromide and visualized under UV for distribution of digested DNA fragments.
In the second stage, changes in the 5-methylcytosine content in the cellular DNA expressing Rb was measured using M.SssI and S-adenosyl-l-[methyl-3H] methionine (AdoMet) under steady-state kinetic conditions (Lin et al., 2001). DNA (100 ng) in duplicate was used as substrate for global methylation assay using M.SssI. Methylation assays were carried out at 37°C for 10 min in duplicate, with a total volume of 25 µl of reaction mixture. A typical reaction contained 5 µM AdoMet (sp. act. 15 Ci/mmole, Amersham Pharmacia Biotech), 100 ng substrate DNA and 1 nM enzyme in assay buffer (10 mM Tris–HCl pH 7.9, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT). The methylation reactions were stopped by transferring the reaction tubes to an ethanol–dry ice bath and were processed by spotting the reaction mix on DE81 paper circles (Whatman). These circles were washed sequentially with 4× 1 ml of cold 0.2 M ammonium bicarbonate, 4× 1 ml of Milli Q water and 4× 1 ml of ethanol using a manifold (Millipore) connected to a vacuum pump. The filters were dried, 2.5 ml of opti-fluor (Packard) were added to each and 3H incorporation was measured. Each experiment was repeated twice in duplicate.
For hemimethylation assay, 40 nM human Dnmt1 was used with 200 ng of cellular DNA as described above along with buffer M+. The incubation time was 30 min and the samples were processed as described (Pradhan et al., 1999).
Plasmid methylation, cell transfection and luciferase assays
The pGL3-promoter vector (Promega) was methylated using M.SssI and cold S-adenosyl-l-methionine (New England Biolabs) following the manufacturer’s instruction. The methylation status was checked using HhaI and HpaII restriction enzymes. Methylated plasmids were purified (Qiaquick nucleotide removal kit, Qiagen) before transfection. As an internal control a constant amount of pCMVβ (Clontech), a vector containing the β-galactosidase gene, was also co-transfected. Adeno or adeno-Rb was transfected along with pCMVβ. COS-7 cells grown in DMEM supplemented with 10% FCS were transfected with a mixture of DNA and FuGENE6 (Roche Molecular Biochemicals) at a ratio of 1:3 µg/µl. Thirty or forty-eight hours post-transfection, the cells were harvested, and luciferase and β-galactosidase assays carried out. β-galactosidase assay was used for normalization of transfection. Genomic DNA was isolated 30–48 h post-transfection from COS-7 cells using the Easy-DNA Kit (Qiagen). The purified DNA was digested by restriction enzymes MspI or HpaII. Digestion of a cocktail of genomic DNA plus plasmid or phage DNA was performed in parallel as a control (to monitor complete digestion). The digested DNA fragments were separated on a 1% agarose TBE gel. The DNA in the gel was blotted on to Hybond N+ and probed with a random-primed luciferase gene. The blot was washed using a standard protocol and autoradiographed.
Acknowledgments
Acknowledgements
We thank Drs R.J.Roberts, W.Jack, T.Evans and A.Bacolla for discussions and critical reviews and Professor S.B.Baylin, Johns Hopkins University, for the gift of the cDNA clones for hDnmt1 (pBKSHMT5.0). Support and encouragement from Dr D.G.Comb, New England Biolabs is much appreciated. The gifts of adenovirus and adenovirus-Rb DNA from Dr Kim, Nebraska University, is highly appreciated. This research was partly supported by a National Institute of Health Grant GM46127 to Dr R.J.Roberts.
References
- Bacolla A., Pradhan,S., Roberts,R.J. and Wells,R.D. (1999) Recombinant human DNA (cytosine-5) methyltransferase. II. Steady-state kinetics reveal allosteric activation by methylated DNA. J. Biol. Chem., 274, 33011–33019. [DOI] [PubMed] [Google Scholar]
- Bacolla A., Pradhan,S., Larson,J.E., Roberts,R.J. and Wells,R.D. (2001) Recombinant human DNA (cytosine-5) methyltransferase. III. Allosteric control, reaction order and influence of plasmid topology and triplet repeat length on methylation of the fragile X CGG.CCG sequence. J. Biol. Chem., 276, 18605–18613. [DOI] [PubMed] [Google Scholar]
- Baylin S.B., Herman,J.G., Graff,J.R., Vertino,P.M. and Issa,J.P. (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res., 72, 141–196. [PubMed] [Google Scholar]
- Bestor T.H. (1992) Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J., 11, 2611–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor T., Laudano,A., Mattaliano,R. and Ingram,V. (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol., 203, 971–983. [DOI] [PubMed] [Google Scholar]
- Belinsky S.A., Nikula,K.J., Baylin,S.B. and Issa,J.P. (1996) Increased cytosine DNA–methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl Acad. Sci. USA, 93, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.K., Ramchandani,S., Cervoni,N. and Szyf,M. (1999) A mammalian protein with specific demethylase activity for mCpG DNA. Nature, 397, 579–583. [DOI] [PubMed] [Google Scholar]
- Bird A.P and Wolffe,A.P. (1999) Methylation-induced repression—belts, braces and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- Brehm A., Miska,E.A., McCance,D.J., Reid,J.L., Bannister,A.J. and Kouzarides,T. (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature, 391, 597–601. [DOI] [PubMed] [Google Scholar]
- Chuang L.S., Ian,H.I., Koh,T.W., Ng,H.H., Xu,G. and Li,B.F. (1997) Human DNA-(cytosine-5) methyltransferase–PCNA complex as a target for p21WAF1. Science, 277, 1996–2000. [DOI] [PubMed] [Google Scholar]
- Costello J.F. et al. (2000) Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nature Genet., 25, 132–138. [DOI] [PubMed] [Google Scholar]
- Craig C. et al. (1998) Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene, 16, 265–272. [DOI] [PubMed] [Google Scholar]
- Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase DNMT1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- Gama-Sosa M.A., Slagel,V.A., Trewyn,R.W., Oxenhandler,R., Kuo,K.C., Gehrke,C.W and Ehrlich,M. (1983) The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res., 11, 6883–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalgo M.L. and Jones,P.A. (1997) Mutagenic and epigenetic effects of DNA methylation. Mutat. Res., 386, 107–118. [DOI] [PubMed] [Google Scholar]
- Guan L.S., Li,G.C., Chen,C.C., Liu,L.Q. and Wang,Z.Y. (2001) Rb-associated protein 46 (RbAp46) suppresses the tumorigenicity of adenovirus-transformed human embryonic kidney 293 cells. Int. J. Cancer, 93, 333–338. [DOI] [PubMed] [Google Scholar]
- Hanahan D. and Weinberg,R.A. (2000) The hallmarks of cancer. Cell, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Holliday R. (1993) Epigenetic inheritance based on DNA methylation. In Jost,J.P. and Saluz,H.P. (eds), DNA Methylation: Molecular Biology and Biological Significance. Birkhauser Verlag, Basel, Switzerland, pp. 452–468.
- Jones P.A. (1996) DNA methylation errors and cancer. Cancer Res., 56, 2463–2467. [PubMed] [Google Scholar]
- Kuerbitz S.J. and Baylin,S.B. (1996) Retention of unmethylated CpG island alleles in human diploid fibroblast × fibrosarcoma hybrids expressing high levels of DNA methyltransferase. Cell Growth Differ., 7, 847–853. [PubMed] [Google Scholar]
- La Thangue N.B. (1994) DRTF1/E2F: an expanding family of heterodimeric transcription factors implicated in cell-cycle control. Trends Biochem. Sci., 19, 108–114. [DOI] [PubMed] [Google Scholar]
- Laird P.W. and Jaenisch,R. (1994) DNA methylation and cancer. Hum. Mol. Genet., 3 Spec. No. 3, 1487–1495. [DOI] [PubMed] [Google Scholar]
- Laird P.W., Jackson-Grusby,L., Fazeli,A., Dickinson,S.L., Jung,W.E., Li,E., Weinberg,R.A. and Jaenisch,R. (1995) Suppression of intestinal neoplasia by DNA hypomethylation. Cell, 81, 197–205. [DOI] [PubMed] [Google Scholar]
- Leonhardt H., Page,A.W., Weier,H.U. and Bestor,T.H. (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell, 71, 865–873. [DOI] [PubMed] [Google Scholar]
- Lee P.J., Washer,L.L., Law,D.J., Boland,C.R., Horon,I.L. and Feinberg,A.P. (1996) Limited up-regulation of DNA methyltransferase in human colon cancer reflecting increased cell proliferation. Proc. Natl Acad. Sci. USA, 93, 10366–10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Bestor,T.H. and Jaenisch,R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Lin C.-H., Hsieh,S.-Y., Sheen,I.-S., Lee,W.-C., Chen,T.-C., Shyu,W.-C. and Liaw,Y.-F. (2001) Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res., 61, 4238–4243. [PubMed] [Google Scholar]
- Luo R.X., Postigo,A.A. and Dean,D.C. (1998) Rb interacts with histone deacetylase to repress transcription. Cell, 92, 463–473. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L., Groisman,R., Naguibneva,I., Robin,P., Lorain,S., Le Villain,J.P., Troalen,F., Trouche,D. and Harel-Bellan,A. (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature, 391, 601–605. [DOI] [PubMed] [Google Scholar]
- Muller K., Heller,H. and Doerfler,W. (2001) Foreign DNA integration. Genome-wide perturbations of methylation and transcription in the recipient genomes. J. Biol. Chem., 276, 14271–14278. [DOI] [PubMed] [Google Scholar]
- Nevins J.R. (1992) E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science, 258, 424–429. [DOI] [PubMed] [Google Scholar]
- Ohtani-Fujita N. et al. (1997) Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet. Cytogenet., 98, 43–49. [DOI] [PubMed] [Google Scholar]
- Pradhan S. and Roberts R.J. (2000) Hybrid mouse–prokaryotic DNA (cytosine-5) methyltransferases retain the specificity of the parental C-terminal domain. EMBO J., 19, 2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan S., Bacolla,A., Wells,R.D. and Roberts,R.J. (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification and comparison of de novo and maintenance methylation. J. Biol. Chem., 274, 33002–33010. [DOI] [PubMed] [Google Scholar]
- Robertson K.D. and Jones,P.A. (1998) The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell. Biol., 18, 6457–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K.D., Ait-Si-Ali,S., Yokochi,T., Wade,P.A., Jones,P.L. and Wolffe,A.P. (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genet., 25, 338–342. [DOI] [PubMed] [Google Scholar]
- Schmutte C. and Jones,P.A. (1998) Involvement of DNA methylation in human carcinogenesis. Biol. Chem., 379, 377–388. [DOI] [PubMed] [Google Scholar]
- Schmutte C., Yang,A.S., Nguyen,T.T., Beart,R.W. and Jones,P.A. (1996) Mechanisms for the involvement of DNA methylation in colon carcinogenesis. Cancer Res., 56, 2375–2381. [PubMed] [Google Scholar]
- Taya Y. (1997) RB kinases and RB-binding proteins: new points of view. Trends Biochem. Sci., 22, 14–17. [DOI] [PubMed] [Google Scholar]
- van der Ploeg L.H., Groffen,J. and Flavell,R.A. (1980) A novel type of secondary modification of two CCGG residues in the human γ δ β-globin gene locus. Nucleic Acids Res., 8, 4563–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertino P.M, Yen,R.W., Gao,J. and Baylin,S.B. (1996) De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell. Biol., 16, 4555–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R.A. (1991) Tumor suppressor genes. Science, 254, 1138–1146. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Yen R.W. et al. (1992) Isolation and characterization of the cDNA encoding human DNA methyltransferase. Nucleic Acids Res., 20, 2287–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B., Benjamin,D., Zheng,Y., Angliker,H., Thiry,S., Siegmann,M. and Jost,J.P. (2001) Overexpression of 5-methylcytosine DNA glycosylase in human embryonic kidney cells EcR293 demethylates the promoter of a hormone-regulated reporter gene. Proc. Natl Acad. Sci. USA, 98, 5031–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann C., Guhl,E. and Graessmann,A. (1997) Mouse DNA methyltransferase (MTase) deletion mutants that retain the catalytic domain display neither de novo nor maintenance methylation activity in vivo. Biol. Chem., 378, 393–405. [DOI] [PubMed] [Google Scholar]