

Abstract
The large hepatitis B virus (HBV) surface protein (LHBs) and C-terminally truncated middle size surface proteins (MHBst) form the family of the PreS2 activator proteins of HBV. Their transcriptional activator function is based on the cytoplasmic orientation of the PreS2 domain. MHBst activators are paradigmatic for this class of activators. Here we report that MHBst is protein kinase C (PKC)-dependently phosphorylated at Ser28. The integrity of the phosphorylation site is essential for the activator function. MHBst triggers PKC-dependent activation of c-Raf-1/Erk2 signaling that is a prerequisite for MHBst-dependent activation of AP-1 and NF-κB. To analyze the pathophysiological relevance of these data in vivo, transgenic mice were established that produce the PreS2 activator MHBst specifically in the liver. In these mice, a permanent PreS2-dependent specific activation of c-Raf-1/Erk2 signaling was observed, resulting in an increased hepatocyte proliferation rate. In transgenics older than 15 months, an increased incidence of liver tumors occurs. These data suggest that PreS2 activators LHBs and MHBst exert a tumor promoter-like function by activation of key enzymes of proliferation control.
Keywords: hepatitis B virus/hepatocellular carcinoma/signal transduction/surface proteins/transgenics
Introduction
In addition to causing acute and chronic hepatitis, hepatitis B virus (HBV) is considered to be a major etiological factor in the development of human hepatocellular carcinoma (HCC). Almost all HBV-associated HCCs studied so far harbor chromosomally integrated HBV DNA (Beasley et al., 1981). HBV DNA also does not integrate at specific sites of the host genome. Thus a common cis-effect onto flanking cellular genes can be excluded as a general mechanism of HBV-associated carcinogenesis.
However, integrated HBV DNA can encode two types of transcriptional activators: the already well studied HBx (for a recent review see Murakami, 1999) and the PreS2 activators LHBs (large hepatitis B virus surface protein) and MHBst (C-terminally truncated middle size surface proteins) (Kekulé et al., 1990; Hildt et al., 1996a).
The sequence encoding the PreS2 activators is localized in the HBV surface gene. The surface gene consists of a single open reading frame divided into three coding regions: preS1, preS2 and S, each starting with an in-frame ATG codon. Through alternate translational initiation at each of the three AUG codons, a large (LHBs; PreS1 + PreS2 + S), a middle size (MHBs; PreS2 + S) and a small (SHBs; S) envelope glycoprotein can be synthesized. The activator function of the surface protein requires the cytoplasmic orientation of the PreS2 domain (the minimal functional unit) that occurs in the case of MHBst (Hildt et al., 1995) and in a fraction of LHBs (Bruss et al., 1994). In contrast, full-length MHBs displays no transcriptional activator function: here the PreS2 domain directs into the lumen of the endoplasmic reticulum (ER). The generation of functional MHBst from MHBs requires deletion of the 3′ end of the preS2/S gene encoding at least the last 70 amino acids corresponding to the hydrophobic region III of the S domain during the integration process (Lauer et al., 1992). Deletions of 3′ sequences of the preS2/S gene generating functional MHBst activators were found in one-third of the integrates investigated so far (Schlüter et al., 1994; Zhong et al., 1999). This fact and the presence of complete preS/S genes in some other systematically studied integrates indicate the biological significance of the PreS2 activators.
Previous reports have described that transgenic mice that specifically overproduce LHBs in the liver develop liver tumors (Chisari et al., 1989). Up to now, intracellular retention of LHBs, resulting in a situation analogous to storage disease, and subsequent permanent inflammatory processes were considered to cause the tumors in these transgenics. The physiological situation of these mice is closely related to that of HBV carriers, who strongly overproduce HBsAg. In the livers of these patients, development of ground glass hepatocytes can be observed. In light of the recent observations that LHBs functions as a transcriptional activator (Hildt et al., 1996a), the question arose of whether this activator function could confer LHBs-dependent development of HCC.
MHBst proteins are paradigmatic for the family of PreS2 activators. However, in contrast to LHBs, the MHBst activators are produced only in very small amounts even if the expression is driven by strong promoters. This permits an analysis of signal transduction pathways in the absence of effects based on overexpression and intracellular accumulation of LHBs. In this study, we used, in particular, MHBst76 (truncated at amino acid 76) corresponding to the MHBst activator encoded by the integrate of the human hepatoma cell line huH4 (Kekulé et al., 1990). MHBst76 belongs to the ER-localized subtype of MHBst activators (Hildt et al., 1993). To exclude unequivocally any effects such as ER-overload based on the ER membrane association of MHBst76, MHBst63 that belongs to the non-membrane associated cytosolic- localized subtype was used as an appropriate control. Elsewhere, MHBst63 and MHBst76 have identical functions (Hildt et al., 1995). The molecular basis of MHBst-dependent transcriptional activation, however, remained enigmatic.
Results
The PreS2 activator MHBst is a phosphoprotein
In a recent report, mass spectrometry of MHBst76 provided preliminary evidence for the existence of a phosphorylated fraction of MHBst proteins (Hildt et al., 1996b). To analyze this in more detail, highly purified hexahistidine (His6)-tagged MHBst76 derived from Sf9 cells was subjected to two-dimensional gel electrophoresis. The two-dimensional gel shows that Sf9 cell-derived His6-MHBst76 consists of at least three differently charged populations (Figure 1A). The identity of these three spots with His6-MHBst76 was confirmed by western blot analysis (Figure 1B). To investigate whether the more negatively charged spots represent phosphorylated His6-MHBst76, acid phosphatase treatment was carried out. Subsequent western blot analysis of the two-dimensional separation showed that the two more negatively charged spots disappeared, reflecting their phosphatase sensitivity (Figure 1C). To prove unequivocally that phosphorylation of MHBst takes place in a liver cell system in vivo, labeling of His6-MHBst- or His6-MHBs-producing HepG2 cells with [32P]orthophosphate was performed. The autoradiograph of the affinity-precipitated proteins shows that both the ER-localized MHBst76 (Figure 1D, lane 2) and the cytoplasmic MHBst63 (Figure 1D, lane 4) are phosphoproteins (Figure 1D). In the case of full-length MHBs, no specifically phosphorylated forms were detectable (Figure 1D, lane 7). Further mutants analyzed in this experiment are described below. These results demonstrate that MHBst activator proteins, in contrast to MHBs, are phosphorylated.
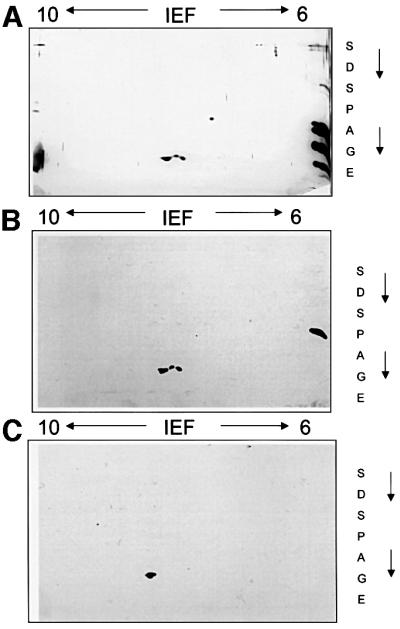
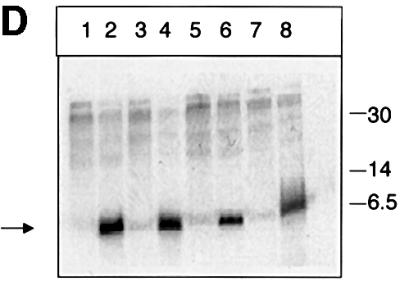
Fig. 1. MHBst is a phosphoprotein. (A) Highly purified His6-MHBst76 was derived from Sf9 cells. A 40 µg aliquot was subjected to isoelectric focusing under denaturing conditions using immobiline gel strips covering a range of pH 6–10. The separation in the second dimension was performed using a 15% Laemmli gel. The gel was silver stained. (B) Western blot analysis of highly purified His6-MHBst76 derived from Sf9 cells after two-dimensional separation as described in (A) using a PreS2-specific antiserum (F124). (C) Western blot analysis of highly purifed His6- MHBst76 derived from Sf9 cells using a PreS2-specific antiserum (F124). The sample were subjected to acid phosphatase treatment prior to two-dimensional separation. (D) Autoradiograph of Ni-NTA-precipitated and SDS–PAGE-separated His6-MHBst76 (lane 2), His6-MHBst63 (lane 4), His6-MHBs (lane 7), His6-MHBst76mutI (lane 3), His6-MHBst76mutII (lane 6), His6-MHBst63mutI (lane 5) and His6-MHBst63mutII (lane 8) derived from cellular lysates of [32P]orthophosphate-labeled, transfected HepG2 cells. Cells transfected with the cloning vector served as negative control (lane 1). The autoradiograph shows that in contrast to full-length MHBs, the functional MHBst activators are phosphoproteins.
The PreS2 activator MHBst is phosphorylated at Ser27/28
Western blot analysis using a phosphoserine- and a phosphotyrosine-specific antibody demonstrated the presence of phosphoserine in MHBst (data not shown), but provided no evidence for tyrosine phosphorylation.
To study phosphorylation of various MHBst proteins in more detail, an in vitro system was used. Purified Escherichia coli-derived proteins were incubated with isolated cytoplasm and [γ-32P]ATP. Analysis was performed by SDS–PAGE followed by autoradiography. His6-MHBst76 characterized by a mol. wt of ∼6.5 kDa (Figure 2A, lanes 1 and 3) is phosphorylated efficiently under these conditions. In the case of the negative control (Figure 2A, lane 4), His6-MHBst76 was replaced by an unrelated His6-tagged protein (His6-grb2). To exclude any autophosphorylation activity of His6-MHBst76, the cytoplasm was replaced by homogenization buffer. The autoradiograph indicates that His6-MHBst76 possesses no autophosphorylation activity (Figure 2A, lane 2) and excludes phosphorylation by contaminating bacterial kinases. A series of C- or N-terminal deletion mutants His6-MHBst63; His6-MHBst52, His6-MHBst11–76 (starts at amino acid 11 of the PreS2 region, truncated at amino acid 76) and His6-MHBst23–76, and a structural mutant MHBst52* (the amphipatic α-helix between amino acids 41 and 52 was converted to a β-sheet conformation; Hildt et al., 1995) were purified from a eubacterial expression system and used for mapping the phosphorylation site. The autoradiograph (Figure 2B) shows that neither deletion of the hydrophobic transmembrane region I (MHBst63, Figure 2B), further C-terminal deletion up to amino acid 52 (MHBst52, Figure 2B) nor mutation of the amphipatic α-helix (MHBst52, Figure 2B) abolishes the phosphorylation of MHBst. Even N-terminal deletions in the case of MHBst11–76 and MHBst23–76 did not result in a loss of MHBst phosphorylation (Figure 2B). From these data, it can be concluded that MHBst is phosphorylated between amino acids 23 and 41. The corresponding peptide harbors two S/T clusters, one between amino acids 27 and 31 (PreS2aa 23–34: PAGGSSSGTVNP) and the other between amino acids 37 and 40 (PreS2aa 34–41: PVLTTASP). To identify the phosphorylation site more precisely, two mutants were generated. In the case of mutant I, Ser27, Ser28, Ser29 and Tyr31 were replaced by alanine (MHBst76mutI; MHBst63mutI); in the case of mutant II, Tyr37, Tyr38 and Ser40 were replaced by alanine (MHBst76mutII; MHBst63mutII).
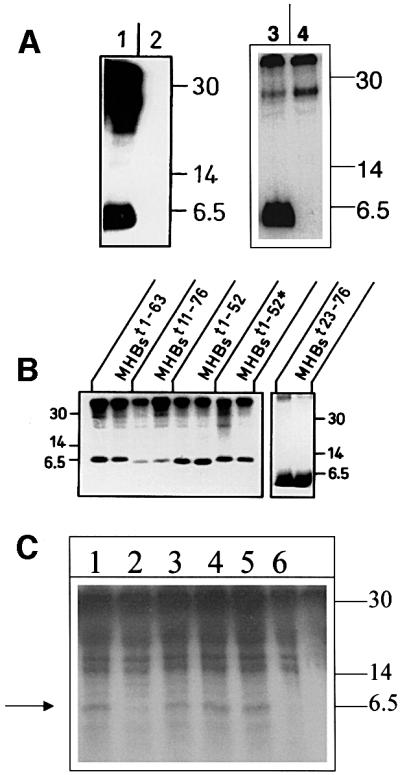

Fig. 2. MHBst is phosphorylated between amino acids 27 and 31. (A) Left: autoradiograph of eubacteria-derived MHBst76, incubated with cytoplasm derived from CCL13 cells and [γ-32P]ATP (lane 1); as negative control, MHBst76 was incubated in the absence of lysate, which was replaced by an equal volume of homogenization buffer (lane 2). The molecular weight marker is given on the right. Right: autoradiograph of eubacteria-derived MHBst76, incubated with cytoplasm derived from CCL13 cells and [γ-32P]ATP (lane 3); as negative control, MHBst76 was replaced by His6-grb2 (lane 4). (B) Autoradiograph of in vitro phosphorylated MHBst fragments (MHBst1-63, MHBst1-52, MHBst11–76, MHBst23–76 and MHBst1-52*; here the amphiphatic α-helix between amino acids 41 and 52 was replaced by a β-sheet conformation) isolated from the eubacterial expression system. The experiments were performed in duplicate. In the case of MHBst11–76, only one-fifth of the amount of MHBst employed in the other reactions was used. (C) Autoradiograph of in vivo phosphorylated and Ni-NTA-agarose-precipitated MHBst-specific proteins: lane 1, MHBst63 mutIa; lane 2, MHBst63mutIb; lane 3, MHBst63mutIc; lane 4, MHBst63mutId; lane 5, wild-type MHBst63; and lane 6, control transfected cells. (D) CAT assay after co-transfection of HepG2 cells with reporter plasmid p3xAP-1-CAT and expression plasmid pKSVMHBst76, encoding the wild-type, or plasmids pKSVMHBst76mutI, pKSVMHBst76mutII, pKSVMHBst76mutID28, or cloning vector pKSV10 as negative control. Fold inductions are mean values from three independent transfection experiments, calculated as the ratio of the induced values to the vector control. The standard deviation is shown in the diagram.
In vivo labeling experiments with [32P]orthophosphate of HepG2 cells producing MHBstmutI/II demonstrated that mutation of the S/T cluster at amino acids 27–31 abolished the phosphorylation of MHBst76mutI (Figure 1D, lane 3) or MHBst63mutI (Figure 1D, lane 5), whereas mutation of the S/T cluster at amino acids 37–40 had no influence on the phosphorylation of MHBst76mutII (Figure 1D, lane 6) or MHBst63mutII (Figure 1D, lane 8). Western blot analysis revealed that MHBst63mutI and MHBst76mutII were produced in similar amounts as the respective wild-type proteins (data not shown).
To identify the exact phosphorylation site, a series of mutants were generated in which the following residues were replace by alanine: Ser27 in MHBst63mutIa, Ser28 in MHBst63mutIb, Ser29 in MHBst63mutIc, and Tyr31 in MHBst63mutId. In vivo phosphorylation experiments using these mutants (Figure 2C) revealed that mutation of Ser28 to alanine abolishes the phosphorylation, indicating that the phosphorylation of MHBst takes place at Ser28.
Destruction of the phosphorylation site abolishes activator function of MHBst
The PreS2 activators are known to activate transcription factor AP-1 (Hildt et al., 1995). To investigate whether the integrity of the identified phosphorylation site is a prerequisite for the functionality of the MHBst activators, reporter gene assays were performed. HepG2 cells were co-transfected with the expression vectors pKSVMHBst76 (positive control), pKSVMHBst76mutI or pKSVMHBst76mutII and with the reporter construct p3xAP-1-CAT (Lauer et al., 1992). The vector pKSV10 served as negative control. Destruction of the phosphorylation site in the case of MHBst76mutI abolishes the activator function of MHBst76 (Figure 2D) as compared with cells transfected with pKSVMHBst76 (positive control) or pKSVMHBst76mutII; here, an ∼10-fold activation of the reporter gene was observed (Figure 2D). Co-transfection of pKSVMHBst76mutID28 (Ser28 was replaced by aspartate to mimic a constitutive phosphorylation) failed to induce activation of the reporter gene.
These data demonstrate that the integrity of the identified phosphorylation site is a prerequisite for the functionality of the MHBst activators.
PKC phosphorylates MHBst
The identified phosphorylation site showed a weak homology to protein kinase C (PKC)-dependent phosphorylated sequences. To investigate a possible implication of PKC isoforms in phosphorylation of MHBst proteins, in vitro phosphorylations were performed in the presence of a PKC-specific substrate peptide. The autoradiograph (Figure 3A) shows that increasing concentrations of the peptide compete phosphorylation of His6- MHBst63. This indicates that MHBst is PKC-dependently phosphorylated.
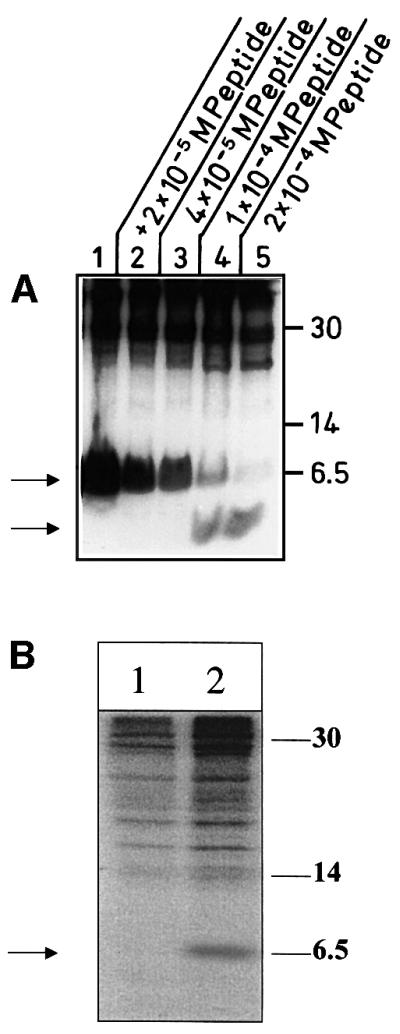
Fig. 3. Functionality of PKC is essential for phosphorylation of MHBst. (A) Competitive inhibition of in vitro phosphorylation of MHBst using increasing concentrations (2 × 10–5 M, lane 2, to 2 × 10–4 M, lane 5) of a PKC-specific peptide. In lanes 4 and 5, the lower band represents the phosphorylated peptide. Lane 1 represents the phosphorylation of MHBst63 in the absence of the peptide. (B) In vivo labeling using [32P]orthophosphate of HepG2 cells producing His6-MHBst76 (lanes 1 and 2) grown in the absence (lane 2) or presence of the PKC inhibitor Goe6976 (lane 1). His6-MHBst76 was enriched by Ni-NTA precipitation.
To analyze the relevance of classic PKC isoforms for phosphorylation of MHBst, their activity was blocked using the cPKC-specific inhibitor Goe6976. In vivo phosphorylation experiments under these conditions revealed that phosphorylation of MHBst depends on the functionality of cPKC isoforms (Figure 3B).
PKC is activated in MHBst-producing cells
Next, we analyzed whether MHBst76 affects the activity of PKC. Activation of the classic isoforms of PKC such as the liver-specific PKCα/β isoforms is associated with a translocation of PKC from the cytoplasm to the cell membrane. Immunofluorescence microscopy of a clonal mixture of HepG2 cells stably transfected with the expression plasmid pCMVMHBst76 revealed in these cells a translocation of PKCα/β from the cytoplasm to the cell membrane. In the case of control-transfected HepG2 cells, PKCα/β was found in the cytoplasm (Figure 4A and B).



Fig. 4. PKC is activated in MHBst-producing cells: inhibition abolishes MHBst-dependent activation of AP-1 and NF-κB. (A and B) Immunofluorescence of HepG2 cells stably transfected with the expression vector pCMVMHBst76 (A). Untransfected cells served as the negative control (B). For detection, a mixture of PKCα- and PKCβ-specific antisera was used. Micrographs were taken at a 400× magnification. (C) CCL13 cells were transfected with the MHBst76 expression plasmid pKSVMHBst76 or with pKSVMHBst76mutI. Transfection with pKSV10 served as the control. The activation was determined by the ratio of membrane-bound to cytosolic PKC activity. In the case of control-transfected cells, this ratio was set arbitrarily as 1. The given factors are mean values of four independent experiments. The standard deviation is shown in the diagram. (D) Transient transfection of HepG2 cells with MHBst76 expression plasmid pCMVMHBst76 and the reporter constructs p3xAP-1-CAT or p2xNF-κB-CAT. Transfection with pCDNA.3 served as the negative control. Classic PKC isoforms were inhibited by the presence of 10 nM Goe6976. Cells were harvested 40 h after transfection. CAT activity was determined as described in Materials and methods. Fold inductions are mean values from four independent transfection experiments, calculated as the ratio of the induced values to the vector control. The standard deviation is shown in the diagram.
For a quantification of PKC activation, the ratio of membrane-bound to cytosolic PKC activity was determined in MHBst76- or MHBst76mutI-producing CCL13 cells. pKSV10-transfected cells served as negative control. An ∼5-fold activation of PKC was measured in the case of MHBst76-producing cells as compared with the negative control (Figure 4C). The phosphorylation-deficient mutant MHBst76mutI did not activate PKC.
MHBst proteins induce activation of the transcription factors AP-1 and NF-κB (Lauer et al., 1992; Meyer et al., 1992). To study the relevance of PKC activation for MHBst76-dependent induction of AP-1 and NF-κB, the PKC inhibitor Goe6976 was employed. A complete loss of the MHBst76-dependent activation of the AP-1- or NF-κB-dependent reporter gene was observed (Figure 4D). These data indicate that (i) MHBst triggers activation of PKC and (ii) the functionality of PKC is essential for MHBst-dependent activation of AP-1 and NF-κB.
MHBst induces PKC-dependent activation of c-Raf-1 and Erk2
The activation of PKC can be mediated by the c-Raf-1/Erk2 signal transduction pathway (Caroll and May, 1994; Marais et al., 1998). In the case of MHBst76-producing cells, an ∼4-fold increase of c-Raf-1 activity was found (Figure 5A) as compared with the c-Raf-1 activity in control-transfected cells. As a positive control, 12-o-tetradecanoyl-phorbol-13-acetate (TPA) stimulation resulted in an ∼9-fold activation. In addition, the phosphorylation-deficient mutant MHBst76mutI, which fails to activate PKC, also caused no activation of c-Raf-1. Activation of neither B-Raf nor A-Raf could be observed in MHBst-producing cells (data not shown). To confirm that the observed MHBst-dependent activation of c-Raf-1 is triggered through PKC, Goe6976 was used to inhibit cPKC. Here, determination of c-Raf-1 activity revealed a complete loss of MHBst-dependent activation of c-Raf-1, whereas the epideraml growth factor (EGF)-dependent activation of c-Raf-1 was not significantly affected by inhibition of PKC (data not shown).

Fig. 5. MHBst induces activation of c-Raf-1and Erk2. (A and B) The activity of c-Raf-1 and Erk2 was determined by immunocomplex assays using recombinant MEK or basic myeloglycoprotein (MBP), respectively, as substrates. HepG2 cells were transfected with pCMVMHBst76 or with the phosphorylation-deficient mutant pCMVMHBst76mutI. TPA stimulation was used as a positive control. The activity of c-Raf-1 or Erk2 in control-transfected cells was set arbitrarily as 1. The given factors are mean values of three independent experiments. The standard deviation is shown in the diagram.
In the case of MHBst76-producing cells, Erk2 activity was increased ∼2.5-fold as compared with the lysate of control-transfected cells. Again, the phosphorylation-deficient mutant MHBst76mutI causes no activation of Erk2. TPA-stimulated cells served as a positive control. Here, an ∼6-fold activation was determined (Figure 5B).
In a recent report (Marais et al., 1998), it was revealed that in contrast to the receptor tyrosine kinase-mediated activation, the PKC-dependent activation of c-Raf-1 is not inhibited by a transdominant-negative mutant of Ras (pRasN17). Reporter gene assays revealed that MHBst76-dependent activation of AP-1 or NF-κB is not affected by the co-expression of RasN17 (Figure 6). Moreover, expression of transdominant-negative Ras failed to influence the MHBst76-dependent activation of c-Raf-1 or Erk2 (data not shown). The functionality of pRasN17 was demonstrated by the inhibition of HBx-dependent activation of AP-1 and NF-κB (data not shown). These results show that expression of the MHBst76 activator results in PKC-dependent activation of c-Raf-1 and Erk2.

Fig. 6. Functionality of c-Raf-1 kinase is a prerequisite for MHBst-dependent activation of AP-1 and NF-κB. HepG2 cells were transiently transfected with the MHBst76 expression plasmid pCMVMHBst76, the reporter constructs p3xAP-1-CAT or p2xNF-κB-CAT and a transdominant-negative mutant of Ras (RasN17) or c-Raf-1 (HCR13.1) or with an equal amount of the vector pMNC to equalize the total amount of DNA. Transfection with the cloning vector pCDNA.3 served as negative control. At 36 h after transfection, CAT activity was determined. Fold inductions are mean values from three independent transfection experiments, calculated as the ratio of the induced values to the vector control. The standard deviation is shown in the diagram.
MHBst fails to promote activation of AP-1 and NF-κB in cells producing a transdominant-negative mutant of c-Raf-1
In the next set of experiments, we investigated whether activation of c-Raf-1 is a prerequisite for MHBst76-dependent activation of AP-1 and NF-κB. For this purpose, reporter gene assays were performed and endogenous c-Raf-1 was inhibited by co-expression of a transdominant-negative mutant (pHCR13.1; Kölch et al., 1991) in MHBst76-producing HepG2 cells. Trans dominant-negative c-Raf-1 abolished MHBst76-dependent activation of AP-1 or NF-κB (Figure 6). These results indicate that the integrity of c-Raf-1 is essential for triggering the activation of AP-1 and NF-κB in MHBst-producing cells.
Liver-specific expression of the transgene encoding MHBst76
To study the pathophysiological relevance of the detected PKC-controlled signaling pathway, two lines of transgenic mice were established that specifically produce the PreS2 activator MHBst76 in the liver. To direct the expression of the transgene to the liver, the transgene was placed under the control of the albumin promoter. Due to the expected very low expression level of the transgene, the β-globin intron was inserted to increase the expression. To permit an efficient enrichment of MHBst from tissue lysates, MHBst was produced as a His6-tagged fusion protein. For stabilization of the mRNA, a poly(A) site was introduced at the 3′ end. A schematic representation of the transgene is given in Figure 7A.



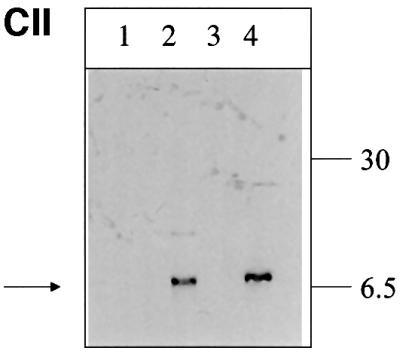
Fig. 7. Liver-specific expression of the transgene encoding MHBst76. (A) Schematic structure of the transgene encoding MHBst76. To direct the expression to the liver, the albumin promoter that was taken from the plasmid pAlb-PSX (Chisari et al., 1985) was used. To increase the expression level, the β-globin intron, derived from plasmid pSWbglob (Werner et al., 1993), was inserted. The HBV-specific fragment nt3174–3221 was fused to the sequence coding for an N-terminal His6 tag. The poly(A) site was taken from SV40, subcloned in plasmid pAK2 (Lauer et al., 1992). The HBV-specific fragment nt3174–3221 codes for MHBst76. (B) Immunofluorescence microscopy of liver sections derived from 5-month-old female MHBst76-producing mice (line 2) (tg). Sections derived from sex-matched wild-type littermates served as control (wt). The PreS2-specific mAb F124 (Neurath et al., 1987) was used for detection of MHBst76. The staining was performed using a Cy3-conjugated donkey-derived secondary antibody. The micrographs were taken at a 400× magnification. (C) I: western blot analysis of the eluates from Ni-loaded Hitrap chelating columns. The columns were loaded with lysates derived from the liver or kidney of MHBst76-producing transgenic mice or their respective littermates (5-month-old, female mice, line I). Elution was performed by a gradient of increasing imidazole concentration. The PreS2-specific mAb HBV 25-19 (Mimms et al., 1990) was used for detection of MHBst76l. II: western blot analysis, using a phosphoserine-specific antiserum (Sigma), of the eluates from Ni-loaded Hitrap chelating columns. The columns were loaded with lysates derived from the liver of wild-type mice (lanes 1 and 3) or MHBst76-producing transgenic mice (5-month-old, female mice, line I, lane 2; line II, lane 4). The western blot shows that the mouse-derived MHBst76 is a phosphoprotein.
The expression of the transgene was analyzed by immunofluorescence microscopy of liver sections derived from transgenic animals and the respective wild-type littermates stained with a PreS2-specific antiserum. An inhomogenous, specific staining of the cytoplasm can be observed, while the nucleus remains unstained, reflecting the ER localization (Hildt et al., 1993) of MHBst76 (Figure 7B).
To verify that (i) the observed staining is specific for authentic MHBst76 and (ii) the expression is liver specific, lysates derived from the liver or kidney of transgenic animals were analyzed by western blotting using the PreS2-specific antibodies HBV-25-19 or F124 (Neurath et al., 1987; Mimms et al., 1990). Under these conditions, the MHBst protein was not detectable (data not shown). Therefore, the lysates were loaded on metal-chelating columns to enrich the His6-tagged MHBst-specific proteins. The eluate was analyzed by western blotting using a PreS2-specific antiserum [monoclonal antibody (mAb) HBV25-19].
The western blot shows that MHBst76 was only detectable in the lysate derived from the liver of the transgenic animals (Figure 7C/I), confirming the liver specificity of the transgene expression. The liver-derived MHBst76 was also analyzed for phosphorylation using a phosphoserine-specific antiserum. The western blot (Figure 7C/II) confirms that MHBst76 is a phosphoprotein.
Determination of JNK2 activity and of the hsp72 level revealed that production of MHBst76, in contrast to LHBs, does not result in unspecific activation of signal transduction (see Supplementary data available at The EMBO Journal Online).
A comparable amount of MHBst76 was observed in the transgenic lines I and II. Here, the expression level seems to be sex independent (data not shown). In the case of lines I and II, no significant reduction of transgene expression was detectable over the investigated time period of 12 months.
These results demonstrate that in the two investigated transgenic mice lines, detectable, comparable, liver- specific expression of the transgene occurred.
Specific activation of the c-Raf-1/Erk2 signal and increased proliferation rate in the liver of transgenic mice producing the PreS2 activator MHBst76
The results described above demonstrate that this system is suitable to perform an in vivo analysis of signal transduction pathways specifically activated by the PreS2 activator. The activity of c-Raf-1 and Erk2 was determined by immunocomplex assays in liver-derived lysates from MHBst76- or LHBs-producing mice or the respective controls. The autoradiographs (Figure 8A and B) show a strong phosphorylation of MEK or MBP, respectively, in the case of lysates derived from LHBs- or MHBst76-producing mice as compared with the control. These results confirm that the PreS2 activator is able to trigger a significant specific activation of the c-Raf-1/Erk2 signaling pathway in vivo that is not subjected to depletion.
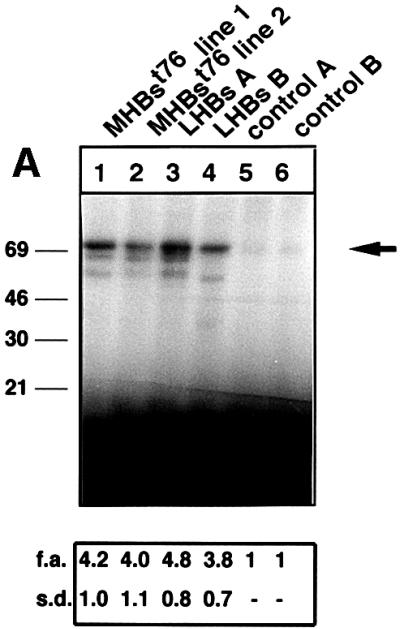
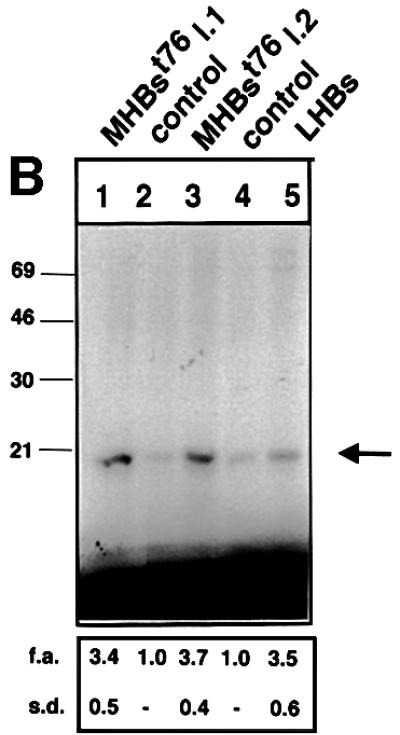
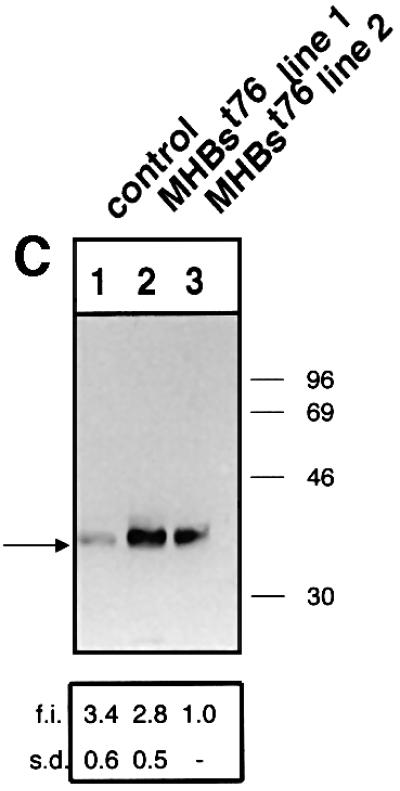

Fig. 8. MHBst76 triggers in vivo specific activation of the c-Raf-1/Erk2 signal transduction pathway resulting in an increased proliferation rate of the hepatocytes. (A) Immunocomplex assay of c-Raf-1 activity in liver-derived lysates of MHBst76-producing mice (lines 1 and 2), of LHBs-producing transgenic mice, or of the corresponding littermates as negative control. In the case of the lysates derived from LHBs- or MHBst76-producing mice, the autoradiograph shows a significant activation of c-Raf-1 as compared with the control. The activation factors (f.a.) are mean values of three independent experiments. Both f.a. and standard deviation (s.d.) are shown at the bottom of the figure. (B) Immunocomplex assay of Erk2 activity in liver-derived lysates of MHBst76-producing mice (lines 1 and 2), of LHBs-producing transgenic mice or of the corresponding littermates as negative control. In the case of the lysates derived from LHBs- or MHBst76-producing mice, the autoradiograph shows a significant activation of Erk2 as compared with the control. The f.a. are mean values of three independent experiments. Both f.a. and s.d. are shown at the bottom of the figure. (C) Western blot analysis of lysates derived from the liver of the two lines of MHBst76-producing transgenic mice (lanes 2 and 3) and of the corresponding littermates (lane 1) using a PCNA-specific antiserum. The western blot shows that in the case of the lysates derived from the MHBst76-producing transgenics, an elevated amount of PCNA as compared with the control was detectable. The induction factors (f.i.) are mean values of three independent experiments. Both f.i. and s.d. are shown at the bottom of the figure. (D) Immunofluorescence microscopy of liver cryosections derived from a male 6-month-old MHBst76-producing mouse (a and b). A sex- and age-matched wild-type littermate served as control. For detection of PCNA (a and c), a PCNA-specific monoclonal antibody (Santa Cruz) was used and was visualized using a Cy3-conjugated anti-mouse secondary antiserum (Dianova). The nuclei were visualized by DAPI staining (b and d).
Activation of c-Raf-1/Erk2 signaling can result in increased cellular proliferation. The proliferating cell nuclear antigen (PCNA) is a well established marker of cellular proliferation. Western blotting of liver-derived lysates (Figure 8C) as well as immunofluorescence microscopy (Figure 8D) of cryosections derived from 6-month-old transgenics using a PCNA-specific antiserum revealed an increased amount of PCNA in the case of the transgenics as compared with their wild-type littermates. These results indicate that the Pres2 activator MHBst76 indeed causes an increased cellular proliferation in vivo.
Increased incidence of liver tumors in transgenic mice producing the PreS2 activator MHBst76
The results described above indicate that the PreS2 activator MHBst76 is able to trigger the activation of a tumor promoter pathway in vivo. However, according to the literature, it is well established that the HBV genome does not harbor a classic oncogene. To analyze whether expression of the transgene coding for the PreS2 activator MHBst76 confers a transforming potential, we performed histological analysis of liver tissue. The analysis was performed over a time period of 18 months. Transgenics younger than 1 year old did not develop tumors. However, in the case of transgenics killed at the age of between 15 and 18 months, a significant number of mice with liver tumors was found (Figure 9). From 23 investigated male mice, 16 mice (69%) displayed liver tumors, while in the case of 17 killed female mice, a liver tumor was observed in 11 animals (65%). There were 2–8 tumors per animal, with a size between 2 and 6 mm. Moreover, the histological analysis revealed the existence of hyperplastic noduli. In the case of the corresponding wild-type controls, two of 14 male mice developed a tumor (14%), as did one of 11 female mice (9%). Immunohistochemistry of six tumors derived from male mice and five from female mice revealed that MHBst was detectable in each of these tumors (data not shown). These results are summarized in Table I. In contrast to the LHBs transgenic mice, there is no significant difference in the percentage of tumor development between male and female mice and the transgene is expressed in the tumor cells.

Fig. 9. Increased incidence of liver tumors in transgenics producing the activator MHBst76. van Gieson-stained (A and C) and HE-stained (B and D) sections of paraffin-embedded liver tissue derived from 15-month-old MHBst76-producing mice. These sections encompass healthy tissue and tumor tissue. In (C), infiltration in surrounding liver tissue can be observed in the case of a MHBst76 mouse with a primary liver tumor. These photographs were taken at 400× magnification. No significant histological change can be observed in the case of (D), which shows HE-stained liver tissue from a 4-month-old MHBst76-producing mouse at 100× magnification.
Table I. Tumor development in wild-type and transgenic mice killed after 15 months.
| Genotype | n | Sex | Tumor development |
|---|---|---|---|
| Transgenic | 23 | male | 16 |
| Transgenic | 7 | female | 11 |
| Wild type | 14 | male | 2 |
| Wild type | 11 | female | 1 |
These results indicate that the continuous activation of the signaling cascade promotes the development of liver tumors in an age-dependent process.
Discussion
MHBst- or LHBs-encoding sequences are found in many integrates subcloned from HBV-associated HCCs (Schlüter et al., 1994; Zhong et al., 1999). To evaluate the putative relevance of PreS2 activator function for the process of HBV-associated HCC development, a detailed in vitro and in vivo analysis of PreS2 activator function is of major biological significance.
Based on the in vitro and in vivo experiments, we propose that in the first step PreS2 interferes with PKC, resulting in activation of PKC and phosphorylation of PreS2.
The PreS2-dependent activation of PKC requires the structural integrity of the phosphorylation site. Conversion of the essential serine residues 27, 28, 29 to alanine abolishes the activator function, as does mutation of Ser28 to aspartate, mimicking phosphoserine. From this, it can be concluded that the conversion from the unphosphorylated to the phosphorylated form is not essential for the activator function. However, the proper interaction of the PreS2 domain with PKC is crucial to trigger activation of PKC. Co-expression of RasN17 does not affect the PreS2-dependent activation of c-Raf-1. This is in accordance with the described model of PKC-dependent activation of c-Raf-1 (Marais et al., 1998).
This signal is mediated subsequently to the nucleus by the c-Raf-1/Erk2 signal transduction pathway. Most importantly, based on results obtained from transgenic mice, it could be clearly concluded that the PreS2-dependent activation of the c-Raf-1/Erk2 cascade is not subjected to depletion resulting in its permanent activation. In accordance with this, an increased proliferation rate of hepatocytes was indeed observed in the MHBst-producing transgenics. However, this increased proliferation rate was not reflected in a significantly enlarged liver. In many features, the PreS2 activators seem to resemble HBx. Both HBx (Su and Schneider, 1997; Pollicino et al., 1998) and the PreS2 activator MHBst (E.Hildt, unpublished results) can confer a proapoptotic potential to hepatocytes. This could compensate for the increased proliferation rate, resulting in an elevated turnover of hepatocytes. On the other hand, there are reports describing an antiapoptotic potential of HBx (Su et al., 2001).
The observed activation of c-Raf-1/Erk2 signaling in vitro and in vivo indicates that the PreS2 activators are able to activate a classic tumor promoter pathway. During the process of aging, a variety of critical DNA mutations can occur (initiation). Due to the production of the PreS2 activator and the resulting permanent activation of proliferative pathways, these cells are positively selected (promotion). The cooperation of both processes can result in tumor formation. Moreover, accumulation of mutations in these mice can also be increased by MHBst-dependent inactivation of p53 by induction of mdm2 expression and sequestration of p53 to the ER (E.Hildt, unpublished results). In the case of the LHBs-producing transgenics, critical mutations are generated with higher incidence by permanent inflammation of liver tissue due to intracellular accumulation.
In conclusion, according to the classic model of carcinogenesis (initiation and promotion), the PreS2 activators MHBst and LHBs might act as a tumor promoter through activation of key enzymes of proliferation control, i.e. PKC isoforms α/β or c-Raf-1/Erk2 as described here.
Materials and methods
Construction of expression plasmids
For construction of eukaryotic expression vectors pCMV-MHBst76C-His6, the truncated preS2/S gene (ntHBV 3174–3221) was amplified from plasmid pTKTHBV2 (Will et al., 1985) by PCR employing primers with restriction sites for BamHI. For plasmid pCMV-MHBst63C-His6, the truncated preS2/S gene (ntHBV3174–3180) was amplified. In both cases, the coding sequence for the His6 tag fused to the C-terminus of the respective MHBst protein was introduced by the reverse primer. The amplified fragments were digested with BamHI and ligated into the BamHI-restricted and dephosphorylated expression vector pCDNA.3 (Invitrogen). For construction of pCMVMHBsC-His6, the complete preS2/S gene was amplified using primers with restriction sites for BglII. The His6 tag was introduced by the reverse primer. Amplified fragments were digested with BglII and ligated into BamHI-restricted and dephosphorylated expression vector pCDNA.3 (Invitrogen).
Construction of eubacterial expression vectors pQeMHBst76, pQeMHBst63, pQeMHBst52, pQeMHBst11–76, pQeMHBst23–76 and pQeMHBst1-52* was described in a recent report (Hildt et al., 1995). For construction of the coding sequence for the MHBstmutI mutants, the sequence ntHBV3174–3160 was amplified. The forward primer harbored at its 5′ end a BamHI restriction site, and the reverse primer a NarI restriction site. By a second PCR, fragment ntHBV60–221 (MHBst76) or ntHBV60–176 (MHBst63) was amplified. The forward primer introduced mutated codons, resulting in a replacement of Ser27, Ser28, Ser29 and Tyr31 by alanine; at the 5′ end, the forward primer contained a NarI site, and the reverse primer a HindIII site. The NarI site introduced an inversion from A–G in the wild-type protein to G–A in the mutant. The amplified fragments were digested with NarI, ligated and inserted into the BamHI–HindIII-restricted, dephosphorylated vector pQe8. The remaining mutants were constructed in an analogous manner. The fragments were inserted into BamHI-restricted vector pCDNA.3 or into the BglII-restricted vector pKSV10 (Pharmacia).
Cells and cell cultures
CCL13 and HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. For transfection, cells were seeded at a density of 0.8 × 106 cells per 60 mm plate. Stimulation of PKC was performed by addition of TPA (Sigma) to a final concentration of 100 ng/ml 30 min prior to lysis of the cells. Goe6976 was used at a final concentration of 10 nM to inhibit classic PKC isoforms.
Culture of Sf9 cells (ATCC, CRL-17-11) and infection with recombinant baculovirus encoding MHBst76 was performed as described by Hildt et al. (1995).
Cell fractionation
Cell fractionation for preparation of cytoplasmic and membrane fractions was performed by differential centrifugation as described earlier (Hildt et al., 1993).
Reporter gene assays
The chloramphenicol acetyltransferase (CAT) gene was used as reporter gene. The AP-1-dependent reporter plasmid p3xAP-1 CAT and the reporter plasmid p2xNF-κB-CAT (also designated p55A2) that harbors two binding sites for the transcription factor NF-κB were described in Lauer et al. (1994).
In the case of double transfections, cells were transfected with 2.5 µg of activator plasmid or 2.5 µg of pCDNA.3 as negative control, and 1 µg of p3xAP-1-CAT or 2.5 µg p2xNF-κB-CAT reporter plasmid. In the case of triple transfections, 2.5 µg of the transdominant-negative mutants of c-Raf-1 (HCR13.1; Kölch et al., 1991) or Ras (RasN17; kindly provided by W.Fantl, San Francisco, CA) were used. To exclude unspecific effects, the total amount of DNA in the controls was balanced by transfection with an equal amount of the cloning vector pMNC.
All these transfections were performed by lipofection, using DOTAP (Boehringer) according to the manufacturer’s instructions. The medium was changed 18 h after transfection. Cells were harvested 38 ± 4 h after transfection.
Transfections followed by an analysis of the PKC activity were performed by the calcium phosphate precipitation method, described elsewhere (Graham and van der Eb, 1973). Under these conditions, 4 µg of plasmid were used and, after 6 h, cells were exposed for 90 s to a glycerol shock and harvested 6–8 h later.
Transfection efficiencies were ∼60% as determined by analysis of enhanced green fluorescent protein (eGFP) fluorescence after transfection of peGFP (Clontech).
Transfection efficiencies were standardized with a luciferase reporter plasmid containing the luciferase gene under the control of the non-stimulatable minimal promoter of pTK/-37luc.
Each experiment was performed with at least three different preparations of CsCl gradient-purified plasmid DNA.
CAT assays
Relative CAT activity was determined with 50–100 µg of protein from total cellular lysates as described (Gorman et al., 1982). Acetylated and unreacted [14C]chloramphenicol was quantified with an isotope scanner (Berthold). Fold inductions are mean values from at least three independent transfection experiments.
Indirect immunofluorescence labeling
Staining was performed as described (Hildt et al., 1995). For detection, a mixture of PKCα/β-specific antibodies (Santa Cruz Biotechnology) diluted in phosphate-buffered saline–Tris (PBST) containing 10% bovine serum albumin (BSA) was used. Bound antibody was visualized by incubation with Cy3-conjugated anti-rabbit antibody (Dianova). A DMR fluorescence microscope (Leica) was used. In order to exclude fixation artifacts, the integrity of the fixed cells was confirmed by phase contrast microscopy of the same specimens.
Sections and staining of the paraffin-embedded, formaldehyde-fixed tissue and cryosections of the liver were obtained and processed as described previously (Saher and Hildt, 1999).
In vivo phosphorylation
HepG2 cells were seeded at a density of 0.8 × 106 cells per 60-mm-diameter plate. The cells were transfected with 6 µg of the His6-MHBst expression plasmids. At 20 h after transfection, the cells were washed twice with phosphate-free DMEM (Sigma) and incubated for 2 h in this medium. Medium was removed and replaced by 1.2 ml of phosphate-free medium, which contained 1 mCi of [32P]orthophosphate. Cells were incubated in this medium for 2 h then washed twice with phosphate-free medium. Lysis and immunoprecipitation were performed as described (Hildt et al., 1995).
In vitro phosphorylation assays
Highly purified, renaturated proteins (0.1 µg) derived from a bacterial expression system were added to 25 µl (20 µg of total protein) of the cytosolic fraction of CCL13 cells. The reaction mixture contained in addition 10 µl of 10× kinase buffer (20 mM Tris–HCl pH 7.5, 20 mM β-mercaptoethanol, 0.1 mM EGTA, 0.1 mM EDTA, 2% glycerol, 110 mM NaCl), 10 µl of 100 µM ATP and 5 µl of 2 mM MgCl2 containing 10 µCi of [γ-32P]ATP. By addition of water, the reaction mixture was brought to a final volume of 100 µl. This basic assay was varied by addition of several activators or inhibitors. Competitive inhibition of the PKC activity was performed by increasing concentrations of a PKC-specific substrate peptide (AAKIQASFRGHMARKK).
Immunocomplex assays
Cells were transfected by lipofection using 2.5 µg of pCMVMHBst76. After 12 h, cells were harvested and lysed in 20 mM Tris–HCl pH 7.5, 137 mM NaCl, 0. 2 mM EDTA, 1 mM EGTA, 10 mM sodium-β-glycerolphosphate, 50 mM sodium fluoride, 1% Triton X-100, 1 mM sodium orthovanadate, 0.25 M sucrose, 0.5 mM phenylmethylsulfonyl fluoride (PMSF; Sigma), 0.15 U/ml aprotinin and 2 µg/ml leupeptin. Insoluble material was removed by centrifugation at 20 000 g at 4°C for 15 min. Protein concentrations were determined by a Bradford assay. A-Raf, B-Raf, c-Raf-1 or Erk2 were immunoprecipitated from lysates by the respective polyclonal serum (Santa Cruz Biotechnology). The subsequent procedure was described recently (Hildt and Oess, 1999). Immunocomplex assays for determination of c-Raf-1 or Erk2 kinase activity in liver tissue were performed as described recently (Saher and Hildt, 1999).
Determination of PKC activity
Cells were fractionated 6 h after the glycerol shock in the cytoplasm and membrane fractions. PKC was enriched from both fractions using ion exchange chromatography on DEAE–cellulose columns as described by Maly et al. (1989). PKC activity was determined using a commercial assay system (PKC assay system; Amersham).
Protein analysis
Proteins were separated by SDS–PAGE (Laemmli, 1970; Schägger and von Jagow, 1987). Western blotting was performed as described by Hildt and Oess (1999). The different MHBst fragments were detected by the PreS2-specific mAbs HBV25-19 (Mimms et al., 1990) or F124 (Neurath et al., 1987). A mouse monoclonal antiserum (Santa Cruz) was used for detection of PCNA, and phosphoserine- or phosphotyrosine-specific mouse mAbs (Sigma) were used for analysis of protein phosphorylation.
Generation of transgenic mice
Animals were generated at the Laboratory Animal Research Unit of the University of Ulm and bred at the Technical University Munich under strict specific pathogen-free conditions. For production of preS2/St221 transgenic lineages, plasmid pTG17 was linearized with NotI and EcoRV, and the HBV fragment was microinjected into F2(CBA/Ca × C57BL/6J) embryos according to standard procedures. Transgenic lineages were established by backcrossing hemizygous transgenic individuals to the C57BL/6J (B6) inbred strain. Transgenic animals were distinguished from their non-transgenic littermates by PCR amplification of transgene sequences using PreS2-specific primers. In all experiments, the non-transgenic, sex- and age-matched littermates served as controls.
The LHBs-producing mice (the preS/S gene is expressed under the control of the albumin promoter) were generated by F.Chisari and co-workers and described in Chisari et al. (1985). The expression characteristics were determined with animals of the first backcross generation. For the tumor studies, we used animals of the second and third backcross.
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.

Fig. 10. Schematic representation of the interference of the PreS2-activators with intracellular signal transduction pathways.
Acknowledgments
Acknowledgements
We are indebted to C.Schommer and A.Kesel for their excellent technical assistance and we are very grateful to Dr Herfeld and D.Poehlmann (TU-Munich) for their help with histological analysis. This work was supported by grants of the Deutsche Stiftung für Krebsforschung to P.H.H. and of the Münchner Medizinische Wochenschrift to E.H.
References
- Beasley R.P., Lin,C.C., Hwang,L.Y. and Chien,C.S. (1981) Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet, 2, 1129–1133. [DOI] [PubMed] [Google Scholar]
- Benn J. and Schneider,R.J. (1994) Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl Acad. Sci. USA, 91, 10350–10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruss V., Lu,R., Thomssen,R. and Gerlich,W. (1994) Post translational alterations in the transmembrane topology of the hepatitis B virus large envelope protein. EMBO J., 13, 2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroll M. and May,W.S. (1994) Protein kinase C-mediated serine phosphorylation directly activates raf-1 in murine hematopoietic cells. J. Biol. Chem., 269, 1249–1256. [PubMed] [Google Scholar]
- Chisari F.V., Pinkert,C.A., Milich,D.R., Filippi,P., McLachlan,A., Palmiter,R.D. and Brinster,R.L. (1985) A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science, 230, 1157–1160. [DOI] [PubMed] [Google Scholar]
- Chisari F., Klopchin,K., Moriyama,T., Pasquinelli,C., Dunsford,H., Sell,H., Pinkert,C., Brinster,R. and Palmiter,R. (1989) Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell, 59, 1145–1157. [DOI] [PubMed] [Google Scholar]
- Gorman C.M., Moffat,L.F. and Howard,P.H. (1982) Recombinant genomes which express chloramphenicol acetyl transferase in mammalian cells. Mol. Cell. Biol., 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F.L. and van der Eb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus DNA. Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Hildt E. and Oess,S. (1999) Identification of Grb2 as a novel binding partner of tumor necrosis factor (TNF) receptor I. J. Exp. Med., 189, 1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildt E., Urban,S., Lauer,U., Hofschneider,P.H. and Kekulé,A.S. (1993) ER-localization and functional expression of the HBV transactivator MHBst. Oncogene, 8, 3359–3367. [PubMed] [Google Scholar]
- Hildt E., Urban,S. and Hofschneider,P.H. (1995) Characterization of essential domains for the functionality of the MHBst transcriptional activator and identification of a minimal MHBst activator. Oncogene, 11, 2055–2066. [PubMed] [Google Scholar]
- Hildt E., Saher,G., Bruss,V. and Hofschneider,P.H. (1996a) The hepatitis B virus large surface protein (LHBs) is a transcriptional activator. Virology, 225, 235–239. [DOI] [PubMed] [Google Scholar]
- Hildt E., Urban,S., Eckerskorn,C. and Hofschneider,P.H. (1996b) Isolation of highly purified, functionally carboxyterminally truncated hepatitis B virus middle surface activators from eucaryotic expression systems. Hepatology, 24, 502–507. [DOI] [PubMed] [Google Scholar]
- Kekulé A.S., Lauer,U., Meyer,M., Caselmann,W.H., Hofschneider,P.H. and Koshy,R. (1990) The preS2/S region of integrated hepatitis B virus DNA encodes a transcriptional transactivator. Nature, 343, 457–461. [DOI] [PubMed] [Google Scholar]
- Kölch W., Heidecker,P., Lloyd,P. and Rapp,U. (1991) Raf-1 protein kinase is required for growth of induced NIH/cells. Nature, 349, 426–428. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lauer U., Weiß,L., Hofschneider,P.H. and Kekulé,A.S. (1992) The hepatitis B virus preS2/St transactivator is generated by 3′ truncations within a defined region of the S gene. J. Virol., 66, 5284–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer U., Weiß,L., Lipp,M., Hofschneider,P.H. and Kekulé,A.S. (1994) The hepatitis B virus preS2/St transactivator utilizes AP-1 and other transcription factors for transactivation. Hepatology, 19, 23–31. [PubMed] [Google Scholar]
- Maly K., Überall,F., Loferer,H., Doppler,W., Oberhuber,H., Groner,B. and Grunicke,H.H. (1989) Ha-ras activates the Na+/H+ antiporter by a protein kinase C independent mechanism. J. Biol. Chem., 264, 11839–11842. [PubMed] [Google Scholar]
- Marais R., Light,Y., Mason,C., Paterson,H., Olson,M. and Marshall,C. (1998) Requirement of Ras-GTP–Raf complexes for activation of Raf-1 by PKC. Science, 280, 109–112. [DOI] [PubMed] [Google Scholar]
- Meyer M., Caselmann,W.H., Schlüter,V., Schreck,R., Hofschneider,P.H. and Baeuerle,P.A. (1992) Hepatitis B virus transactivator MHBst: activation of NF-κB, selective inhibition by antioxidants and integral membrane localization. EMBO J., 11, 2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimms L.T., Floreani,M., Tyner,J., Whitters,E., Rosenlof,F., Wray,L., Goetze,A., Sarin,V. and Eble,K. (1990) Discrimination of hepatits B virus (HBV) subtypes using monoclonal antibodies to the PreS1 and PreS2 domains of the viral envelope. Virology, 176, 604–619. [DOI] [PubMed] [Google Scholar]
- Murakami S. (1999) Hepatitis B virus X protein: structure, function and biology. Intervirology, 42, 81–99. [DOI] [PubMed] [Google Scholar]
- Neurath A.R. et al. (1987) Antibodies to synthetic peptides from the PreS1 and PreS2 regions of one subtype of the hepatitis B virus (HBV) envelope protein recognize all HBV subtypes. Mol. Immunol., 24, 975–980. [DOI] [PubMed] [Google Scholar]
- Pollicino T., Terradillos,O., Lecoeur,H., Gougeon,M.L. and Buendia, M.A. (1998) Pro-apoptotic effect of the HBV X gene. Biomed. Pharmacother., 52, 363–368. [DOI] [PubMed] [Google Scholar]
- Saher G. and Hildt,E. (1999) Activation of c-Raf-1 kinase signal transduction pathway in α(7) integrin-deficient mice. J. Biol. Chem., 274, 27651–27657. [DOI] [PubMed] [Google Scholar]
- Schägger H. and von Jagow,G. (1987) Tricine-sodium dodecyl sulfate polyacrylamide gel electrophoresis for the separation of proteins in the range from 1–100 kDa. Anal. Biochem., 166, 368–379. [DOI] [PubMed] [Google Scholar]
- Schlüter V., Meyer,M., Hofschneider,P.H., Koshy,R. and Caselmann,W.H. (1994) Integrated hepatitis B virus X and 3′ truncated preS/S sequences derived from human hepatomas encode functionally active transactivators. Oncogene, 9, 3335–3344. [PubMed] [Google Scholar]
- Su F. and Schneider,R.J. (1997) Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc. Natl Acad. Sci. USA, 94, 8744–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F., Theodosis,C.N. and Schneider,R.J. (2001) Role of NF-κB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J. Virol., 75, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S., Weinberg,W., Liao,X., Peters,K.G., Blessing,M., Yuspa,S.H., Weiner,R.L. and Williams,L.T. (1993) Targeted expression of a dominant-negative FGF receptor mutant in the epidermis of transgenic mice reveals a role of FGF in keratinocyte organization and differentiation. EMBO J., 12, 2635–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will H., Cattaneo,R., Dardi,G., Deinhardt,F., Schelkens,H. and Schaller,H. (1985) Infectious hepatitis B virus from cloned DNA of known nucleotide sequence. Proc. Natl Acad. Sci. USA, 82, 891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S., Chan,J., Yeo,W., Tam,J.S. and Johnson,P.J. (1999) Hepatitis B envelope protein mutants in human hepatocellular carcinoma tissues. J. Viral Hepat., 6, 195–202. [DOI] [PubMed] [Google Scholar]