

Abstract
The presence of two protein-tyrosine phosphatase (PTP) domains is a striking feature in most transmembrane receptor PTPs (RPTPs). The function of the generally inactive membrane-distal PTP domain (RPTP-D2) is unknown. Here we report that an intramolecular interaction between the spacer region (Sp) and the C-terminus in RPTPα prohibited intermolecular interactions. Interestingly, stress factors such as H2O2, UV and heat shock induced reversible, free radical-dependent, intermolecular interactions between RPTPα and RPTPα-SpD2, suggesting an inducible switch in conformation and binding. The catalytic site cysteine of RPTPα-SpD2, Cys723, was required for the H2O2 effect on RPTPα. H2O2 induced a rapid, reversible, Cys723-dependent conformational change in vivo, as detected by fluorescence resonance energy transfer, with cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) flanking RPTPα-SpD2 in a single chimeric protein. Importantly, H2O2 treatment stabilized RPTPα dimers, resulting in inactivation. We propose a model in which oxidative stress induces a conformational change in RPTPα-D2, leading to stabilization of RPTPα dimers, and thus to inhibition of RPTPα activity.
Keywords: dimerization/FRET/oxidative stress/regulation/RPTP
Introduction
Protein-tyrosine phosphorylation is of major importance for cell proliferation, differentiation, migration and transformation within higher eukaryotic organisms. The regulation of tyrosine phosphorylation levels is mediated by the antagonistic activities of protein-tyrosine kinases (PTKs) and protein-tyrosine phosphatases (PTPs) (Hunter, 1995). A lot of work has been directed at elucidating the regulation of PTKs. In contrast, little is known about the mechanism of regulation of the PTPs (Neel and Tonks, 1997; den Hertog, 1999). The receptor PTPs (RPTPs) form a large subfamily of PTPs, next to the cytosolic ones. Genetic studies in Drosophila and Caenorhabditis elegans indicate a fundamental role for RPTPs in axon path finding and fibroblast growth factor (FGF) signaling (Desai et al., 1996; Krueger et al., 1996; Kokel et al., 1998; Garrity et al., 1999). Mouse knockout studies have provided insight into the function of several RPTPs in vertebrates (Van Vactor et al., 1998; den Hertog, 1999) and demonstrate that the RPTPs have important and specific functions. However, little is known about the regulation of their PTP activity.
RPTPs contain variable extracellular domains. It is tempting to speculate that RPTPs are regulated by extracellular ligands, like the RPTKs. To date, ligands have been found for only a few RPTPs, and only RPTPβ/ζ was reported to be regulated by its ligand, pleiotrophin (Meng et al., 2000). Therefore, other means of regulation may exist for RPTPs. Interestingly, RPTPα was recently found to be regulated during mitosis by GRB2 binding and serine phosphorylation (Zheng and Shalloway, 2001).
RPTPs contain a single transmembrane domain and in most cases two conserved intracellular PTP domains (D1 and D2) separated by a so-called spacer region (Krueger et al., 1990). Interestingly, the membrane-proximal PTP domain (RPTP-D1) contains most if not all the activity (Wang and Pallen, 1991). Several reports suggest that the RPTP-D1 activity of RPTPα and CD45 is negatively regulated by dimerization (Desai et al., 1993; Jiang et al., 1999). The crystal structure of RPTPα-D1 shows a direct reciprocal interaction of a helix–loop–helix ‘wedge’ structure in the juxtamembrane region with the catalytic site of the opposing monomer (Bilwes et al., 1996). Moreover, RPTPα and CD45 are inhibited by forced dimerization, which is dependent on the wedge (Majeti et al., 1998; Jiang et al., 1999). The topology of the two RPTP-D1s in the dimer is crucial for inhibition of activity, since active dimers were identified as well (Jiang et al., 1999). Finally, RPTPα dimerizes constitutively in cells, as detected using cross-linkers and fluorescence resonance energy transfer (FRET), and multiple domains are involved in dimerization, including the transmembrane domain (Jiang et al., 2000; Tertoolen et al., 2001). RPTP dimerization may not be a general phenomenon, since dimers were not found in the crystal structure of LAR-D1D2 and RPTPµ-D1 (Hoffmann et al., 1997; Nam et al., 1999).
The membrane-distal PTP domains, RPTP-D2s, have striking features. They are largely conserved in sequence and are inactive (or much less active than RPTP-D1s). Furthermore, RPTP-D2s have a highly conserved three-dimensional structure (Nam et al., 1999; A.M.Bilwes and J.P.Noel, personal communication). Mutations in only two residues, which are otherwise highly conserved in active RPTP-D1s, are responsible for the low activity in RPTP-D2s (Lim et al., 1998; Buist et al., 1999; Nam et al., 1999). The conservation of inactive RPTP-D2s raises the question of their biological function. Several studies indicate that RPTP-D1s and RPTP-D2s interact, either intramolecularly or intermolecularly. However, the function of the RPTP-D1–D2 interaction is still unclear. RPTPδ-D2 was found to directly inhibit RPTPσ-D1 activity concomitant with binding to the juxtamembrane region of RPTPσ-D1 (Wallace et al., 1998). Inter- and intramolecular interactions between CD45-D1 and CD45-D2 may negatively regulate CD45-D1–CD45-D1 homodimerization, leading to activation of CD45-D1 (Felberg and Johnson, 1998). We recently showed binding of RPTPα to various RPTP-D2s, involving multiple binding sites, suggesting that cross-talk between RPTPs may be a broad mechanism of regulation. The involvement of the wedge in RPTP-D1–RPTP-D2 interactions suggested a possible role for D2s in the regulation of RPTP-D1–D1 dimerization (Wallace et al., 1998; Blanchetot and den Hertog, 2000). The juxtamembrane region of RPTPµ was shown to interact intramolecularly with both PTP domains of RPTPµ, thus regulating RPTPµ-D1 activity (Feiken et al., 2000). Finally, the crystal structure of the complete cytoplasmic domain of LAR provided some structural evidence for D1–D2 interactions. LAR-D1 sits on top of LAR-D2 with extensive contacts through the spacer region containing a four-residue-long ‘linker’ (Nam et al., 1999). Despite a number of reports on interactions between RPTP-D1s and RPTP-D2s, whether and how these interactions are regulated is still largely unknown.
In the present study, we show that the spacer region (the region between D1 and D2; Sp) and the C-terminal region of RPTPα interacted intramolecularly with each other to form a ‘closed’ conformation, which was unable to interact intermolecularly with RPTPα. Binding of RPTPα-SpD2 to RPTPα was inducibly restored by stress factors like H2O2, UV and heat shock. In addition, the catalytic site cysteine in RPTPα-D2 was required for H2O2-induced binding. Using FRET, we demonstrate that H2O2 evoked a conformational change in RPTPα-SpD2, which was rapid and reversible. In vitro, H2O2 induced a conformational change as well, suggesting a direct effect. The change in conformation was concomitant with stabilization of RPTPα dimers and complete inactivation of RPTPα activity. In conclusion, we provide evidence that intra- and intermolecular interactions of RPTPα are regulated by extracellular stimuli, including oxidative stress, and suggest a mechanism for regulation of RPTP complex formation and function.
Results
RPTPα interacts with RPTPα-D2
We have previously reported that RPTPα binds to multiple RPTP-D2s, including RPTPα-D2, LAR-D2, RPTPσ-D2, RPTPδ-D2 and RPTPµ-D2 (Blanchetot and den Hertog, 2000). An interesting feature of the interaction is the difference in affinity of RPTP-D2s for either full-length HA-tagged RPTPα (HA-RPTPα) or HA-RPTPα-ΔD2, a construct lacking part of the spacer region and the second domain (Figure 1A). Indeed, as depicted in Figure 1B, less myc-tagged RPTPα-D2 (Mt-RPTPα-D2) bound to full-length HA-RPTPα than to HA-RPTPα-ΔD2, while similar amounts of HA-RPTPα and HA-RPTPα-ΔD2 were expressed. This result suggests that the intrinsic RPTPα- D2 in full-length RPTPα had a negative effect on the binding of other RPTP-D2s, which may be due to occupation of the binding site by the intrinsic RPTPα- D2, or to intrinsic RPTPα-D2 holding RPTPα in a ‘closed’ conformation, thereby restricting binding to other RPTP-D2s.

Fig. 1. The spacer and the C-terminal region of RPTPα together block binding to RPTPα. (A) Schematic representation of the constructs used here. HA, HA tag; ED, extracellular domain; D1, RPTP-D1; spacer, region between RPTP-D1 and RPTP-D2; D2, RPTP-D2; Ct, C-terminus; Myc, Myc tag. (B–D) Co-immunoprecipitation experiments. 293 cells were transiently co-transfected with the constructs indicated. HA-tagged RPTPα was immunoprecipitated using anti-HA antibodies (12CA5), resolved on SDS–PAGE gels and blotted. The gels were probed with anti-Myc antibody (9E10; top panels) and anti-HA (12CA5; middle panels). Expression of Myc-tagged RPTPα-D2s (Mt-RPTPα-D2s) in the lysates was monitored in the bottom panels. (B) Co-transfection of Mt-RPTPα-D2 alone (–), with full-length HA-tagged RPTPα (FL) or mutant RPTPα lacking the second domain (ΔD2). (C) Co-transfection of HA-RPTPα-ΔD2 with various Mt-RPTPα-D2 constructs [depicted in (A)]. (D) Co-transfection of full-length HA-RPTPα with Mt-RPTPα-D2, -SpD2 or SpD2Y772F.
Intramolecular interaction of the spacer region with the C-terminal helix of RPTPα-SpD2
In order to understand better the binding observed between RPTPα-D1 and RPTPα-D2, we used a panel of deletion mutants. The longest form of RPTPα-D2 (RPTPα-SpD2) encompassed a large part of the spacer region, Sp, the region between D1 and D2 (Figure 1A). To our surprise, this construct did not co-immunoprecipitate with HA-RPTPα (Figure 1C). Deletion of the C-terminus of RPTPα-SpD2 restored binding to HA-RPTPα (Figure 1C) despite the fact that deletion of the C-terminus in RPTPα-D2, without the spacer region, led to a decrease in binding (compare the first and second lane, Figure 1C) (Blanchetot and den Hertog, 2000). These results indicate that the presence of both the spacer and the C-terminal region prohibits binding to RPTPα. A single mutation exactly at the interface between the spacer region and the C-terminal helix, Tyr772 to Phe, partially restored binding to HA-RPTPα, thus partially reproducing the effect of a deletion (Figure 1D). Presumably, Tyr772 stabilized the ‘closed’ conformation. RPTPα-SpD2 was not phosphorylated on tyrosine (data not shown), indicating that the effects of mutation of Tyr772 were not due to lack of tyrosine phosphorylation. Moreover, mutation of Tyr789, an in vivo phosphorylation site (den Hertog et al., 1994), did not affect binding between RPTPα-SpD2 and RPTPα (data not shown). In conclusion, our results suggest that RPTPα-D2 exists in a ‘closed’ conformation. Alteration of the intramolecular interaction between the spacer and the C-terminal region by deletion or subtle mutation switches RPTPα-SpD2 to an ‘open’ conformation, allowing binding to RPTPα in an intermolecular fashion.
H2O2, UV and heat shock induced binding of RPTPα-SpD2 to RPTPα
We next investigated whether RPTPα-SpD2 could be induced to bind to RPTPα in response to a panel of stimuli. Oxidative stress like H2O2, UV, as well as heat shock strongly induced binding of RPTPα-SpD2 to RPTPα (Figure 2A). Similarly, RPTPα-SpD2 bound to RPTPα-ΔD2 after H2O2, UV or heat shock (Figure 2A and data not shown). Other stimuli, like serum, tumor necrosis factor-α, lysophosphatidic acid (LPA), epidermal growth factor or 12-O-tetradecanoylphorbol 13-acetate (TPA), did not induce binding (Figure 2A and data not shown). Pre-treatment of transfected cells with N-acetyl-cysteine (NAC), a free radical scavenger, blocked UV-induced RPTPα-SpD2 binding to RPTPα (Figure 2B), indicating that the effect of UV is mediated by the production of free radicals. H2O2-induced binding was strong and rapid, reaching a maximum within 1 min of stimulation (Figure 2C), suggesting a direct effect of oxidative stress on RPTPα. As long as H2O2 was present in the medium, binding was observed (Figure 2C). Removal of H2O2 by replacing the medium led to a reduction in binding. After 120 min, no binding was detected anymore, showing that it was reversible, although dissociation was much slower than association. Note that H2O2-induced binding was not affected by the medium replacement itself (Figure 2C, last lane). In conclusion, we show that intermolecular interactions between RPTPα and RPTPα-SpD2 were rapidly induced by oxidative stress factors like H2O2 and UV, required the production of free radicals and were reversible.

Fig. 2. Binding of RPTPα-SpD2 to RPTPα is induced by oxidative stress and is reversible. (A) 293 cells were co-transfected with HA-RPTPα or HA-RPTPα-ΔD2 and Mt-RPTPα-SpD2. After stimulation with (final concentration) 10% newborn calf serum (NCS), pervanadate (VO4; 1 mM VO4 + 1 mM H2O2), 1 mM H2O2, 50 ng/ml TPA, UV (200 J/m2) or heat shock (HS; 42°C) for the time indicated, HA-RPTPα was immuno precipitated. After separation on SDS–PAGE and blotting of the gel, the blot was probed with anti-Myc (top panel) and anti-HA (middle panel) antibodies. Equal expression of the Myc-tagged RPTP-D2s was monitored in the lower panel. (B) 293 cells were transiently transfected with HA-RPTPα and Mt-RPTPα-SpD2, pre-treated (+) or not (–) with 10 mM NAC for 16 h and subjected to 100 or 200 J/m2 UV radiation, and left to recover for 10 min. Immunoprecipitation and immunoblotting were as in (A). (C) Transiently transfected 293 cells were treated with H2O2 (1 mM) for the time indicated. Alternatively, after 10 min of stimulation with H2O2, the medium was replaced with fresh pre-warmed medium for the time indicated (Recovery). Note that media change by itself did not affect H2O2-induced binding (last lane). Immunoprecipitation and immunoblotting were as in (A).
Involvement of the catalytic site cysteine of RPTPα-SpD2 in H2O2-induced binding
Since oxidative stress induced binding of RPTPα-SpD2 to RPTPα, and since it has long been proposed that free radicals act on PTPs by oxidation of their catalytic site cysteine (Knebel et al., 1996; Denu and Dixon, 1998; Denu and Tanner, 1998; Lee et al., 1998), we mutated Cys723 in RPTPα-SpD2. Interestingly, mutation of Cys723 to Ser in RPTPα-SpD2 completely abolished H2O2-induced binding to RPTPα (Figure 3A). The same result was obtained when Cys723 was mutated to Ala (data not shown). Mutation of Cys723 in full-length RPTPα also abolished H2O2-induced binding to RPTPα-SpD2 (Figure 3B), presumably because full-length RPTPα-C723S was locked in a ‘closed’ conformation. Mutation of another residue in the second catalytic site (R729K) or mutation of the first catalytic site cysteine (C433S) of RPTPα had no or hardly any effect on H2O2-induced binding to RPTPα-SpD2 (Figure 3B). These results suggest that Cys723 is specifically and directly involved in sensing free radicals, leading to the conformational change in RPTPα-SpD2 necessary to allow binding to RPTPα. Along the same lines, H2O2 increased RPTPα-D2 (without spacer) binding to RPTPα, but not to RPTPα-C433/723S (Figure 3C), suggesting that opening of full-length RPTPα is dependent on Cys723. However, Cys723 is not necessary for direct binding since a mutant RPTPα-D2-C723S (without the spacer region) was still able to bind to RPTPα (data not shown). In conclusion, we show that Cys723, the catalytic site cysteine of RPTPα- D2, plays a direct role in the H2O2-induced intermolecular interaction between RPTPα and RPTPα-SpD2.

Fig. 3. H2O2-induced binding of RPTPα-SpD2 to RPTPα is dependent on catalytic site Cys723. (A) 293 cells were co-transfected with HA-RPTPα and wild-type Mt-RPTPα-SpD2 (wt) or mutant Mt-RPTPα-SpD2 with Cys723 mutated to Ser (C723S). The cells were treated with H2O2 (1 mM, 15 min) (+) or left untreated (–). HA-RPTPα was immunoprecipitated, separated on SDS–PAGE gels and blotted. The blot was probed with anti-Myc (top panel) and anti-HA (middle panel) antibodies. Expression of the Mt-RPTP-D2s in the lysate was monitored in the lower panel. (B) Mt-RPTPα-SpD2 was co-transfected in 293 cells with full-length wild-type HA-RPTPα (wt) or mutants, as indicated. The cells were stimulated with H2O2 (1 mM, 15 min). Immunoprecipitation and immunoblotting for (B–D) were carried out as in (A). (C) Mt-RPTPα-SpD2 was co-transfected with full-length HA-RPTPα (wt), HA-RPTPα-C433S/C723S or PSG5 (–) and stimulated with H2O2 (1 mM, 15 min). (D) 293 cells were transiently co-transfected with HA-RPTPα and various Mt-RPTPα-SpD2 mutants with Cys to Ser mutations, as indicated, and treated with H2O2 (1 mM, 5 min).
Free radical-induced oxidation of proteins may lead to the formation of disulfide bonds. It is noteworthy that H2O2 did not induce non-specific covalent intermolecular bonds between RPTPα-SpD2 and RPTPα since monomeric proteins were found after SDS–PAGE separation under non-reducing conditions (data not shown). We investigated whether other cysteines in the vicinity of Cys723S were involved. Two cysteines are located close to the reactive Cys723, Cys634 and Cys734, both at ∼9 Å. Cys634 is highly conserved in all PTP domains and Cys734 is divergent between PTP domains. Mutation of the conserved Cys634 to Ser had no effect on the H2O2-induced binding between RPTPα and RPTPα-SpD2. However, mutation of Cys734 to serine induced constitutive binding independently of H2O2 (Figure 3D). These results suggest that Cys734 may be involved in the oxidative stress response, possibly by forming a disulfide bridge with Cys723 leading to a structural distortion of RPTPα-SpD2, which may form the basis for the conformational change.
Detection of conformational changes using FRET
FRET between two green fluorescent protein (GFP) derivatives has become widely used to detect protein– protein interactions (Cubitt et al., 1995; Bastiaens and Pepperkok, 2000) and conformational changes of peptides (Miyawaki et al., 1997; Nagai et al., 2000). We used FRET to test the hypothesis that H2O2 induced a conformational change in RPTPα-SpD2. The N- and C-termini of RPTPα-SpD2 are very close together, according to the crystal structure (A.M.Bilwes and J.P.Noel, personal communication), similar to LAR-SpD2 (Nam et al., 1999). CFP and YFP were fused to the N- and C-terminus of RPTPα-SpD2, respectively (Figure 4A). The emission spectra of single transfected cells were determined after excitation at 430 nm, a wavelength that only excited CFP. As expected from the structural data, FRET was detected between CFP and YFP in single-cell measurements, revealing the close proximity of the two fluorophores and consequently of the interaction between the spacer region and the C-terminus (Figure 4C). Interestingly, addition of H2O2 dramatically reduced FRET, as detected by a reduction in the YFP emission and a concomitant increase in CFP emission (reduction in energy transfer from CFP to YFP leads to enhanced direct CFP emission), indicating that a conformational change was indeed induced in RPTPα-SpD2 (Figure 4C and D). H2O2-induced changes in FRET were rapid, reaching maximal effects after 1–2 min, similar to the binding experiments (Figure 4D). Interestingly, the catalytic site cysteine mutant (C723S) was insensitive to H2O2 in that no changes in FRET and thus in conformation were observed in response to H2O2 (Figure 4E and F), consistent with the binding experiments (Figure 3). The change in FRET was specific since H2O2 had no effect on the fluorescence properties of CFP and YFP themselves (Figure 4B). Continuous dual wavelength excitation measurement of FRET indicated that the conformational changes were very rapid, reaching half-maximal levels in ∼30 s (Figure 4G). Similar to the interaction between RPTPα and RPTPα-SpD2, the conformational change was reversible and with much slower kinetics (Figure 4G and H). As a control, we fused CFP or YFP to the C- and N-termini of RPTPα-SpD2, generating CC and YY, respectively (Figure 4A). Following co-transfection of CC and YY, we did not detect any FRET (Figure 4I), demonstrating that FRET, observed in CFP–RPTPα- SpD2–YFP, was due to an intramolecular interaction.

Fig. 4. H2O2-induced conformational change in RPTPα-D2. (A) Schematic representation of the constructs used here: CFP–RPTPα-SpD2–YFP2.1 (WT), CFP–RPTPα-SpD2-C723S–YFP2.1 (C723S), CFP–RPTPα-SpD2–CFP (CC) and YFP–RPTPα-SpD2–YFP (YY). Sp, spacer region; Ct, C-terminus. (B) The spectral properties of a single 293 cell, transfected with CFP alone (excitation at 430 nm, emission maximum at 480 nm), or YFP2.1 alone (excitation at 490 nm, emission maximum at 525 nm), before (rest) and after a wash with fresh medium (wash) and during incubation in medium containing 1 mM H2O2 for the periods of time indicated. (C) Emission spectra of a single transfected 293 human embryonic kidney cell (excitation 430 nm), expressing wild-type CFP–RPTPα-SpD2–YFP2.1 (WT). H2O2 treatment (1 mM) as indicated. (D) Time course of the ratio of the emission intensities at 525 and 480 nm from (C). (E) Emission spectra as in (C) of a single cell expressing mutant CFP–RPTPα-SpD2-C723S–YFP2.1 (C723S). (F) Time course of the ratio of the emission intensities at 525 and 480 nm from (E). (G) Real-time FRET analysis was performed on a single cell expressing CFP–RPTPα-SpD2–YFP2.1 (WT) by continuous dual excitation wavelength measurements [excitation at 430 and 490 nm, emission at 535(30) nm; for details see Tertoolen et al., 2001]. Treatment with 1 mM H2O2 and subsequent recovery were followed. The half-time of decay was calculated (τ = 30 s). (H) A single cell expressing CFP–RPTPα-SpD2–YFP2.1 (WT) was treated with 1 mM H2O2 for 10 min, the medium was replaced by regular medium and emission spectra were obtained at regular intervals. The emission ratio at 525 and 480 nm is plotted against time. (I) Emission spectra of a single cell expressing both CC and YY (excitation at 430 nm, red trace; and to control for YFP expression, excitation at 490 nm, blue trace). All single-cell measurements were repeated at least three times. Representative experiments are depicted here.
The very rapid and total change in conformation induced by H2O2 suggested a direct effect of H2O2 on RPTPα-SpD2. To test this hypothesis, transiently transfected cells were lysed and the cell lysates were then tested for FRET in a spectrophotometer. Under these conditions, FRET was still observed (Figure 5A). Stimulation of cells with H2O2 prior to lysis (in vivo) led to loss of FRET in the lysate. Addition of H2O2 directly to the lysate (in vitro) also led to a large decrease in FRET (Figure 5B), suggesting that the effect of H2O2 was direct. Taken together, using FRET in vivo and in vitro, we show that oxidative stress directly provoked reversible changes in the conformation of RPTPα-SpD2 in a catalytic site cysteine-dependent fashion.
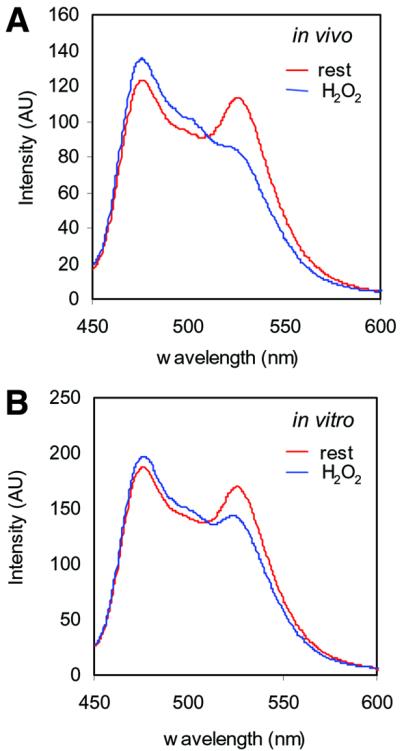
Fig. 5. The effect of H2O2 on RPTPα-D2 is direct. (A) 293 cells were transfected with wild-type CFP–RPTPα-SpD2–YFP2.1 and lysed in cell lysis buffer. Emission spectra were determined for these lysates (excitation 430 nm). The cells had been left untreated (rest) or were stimulated with 1 mM H2O2 for 5 min prior to lysis (in vivo). (B) As in (A), emission spectra were determined for cell lysates. The cell lysates were left untreated (rest) or treated with 1 mM H2O2 (in vitro). The experiments were repeated at least three times and representative measurements are depicted here.
H2O2-induced dimerization of full-length RPTPα
The formation of RPTPα dimers has been reported using chemical cross-linkers on the one hand and FRET on the other (Jiang et al., 2000; Tertoolen et al., 2001). However, co-immunoprecipitation experiments using differently tagged RPTPα were always unsuccessful. For instance, we were unable to reproducibly co-immunoprecipitate myc-tagged RPTPα (Mt-RPTPα) with HA-tagged full-length RPTPα, suggesting that RPTPα dimers were loosely associated and dissociate during the immunoprecipitation procedure. Since H2O2 affected intermolecular interactions between RPTPα, we tested co-immunoprecipitation following H2O2 treatment. Stimulation with H2O2 led to a large increase in the amount of Mt-RPTPα that co-immunoprecipitated with HA-RPTPα (Figure 6A), suggesting stabilization of dimers by intermolecular interactions. UV had the same effect as H2O2 on RPTPα complex formation, which was blocked by pre-incubation with NAC (data not shown). Mutation of the second domain catalytic site cysteine severely reduced H2O2-induced complex formation (Figure 6A). In addition, co-immunoprecipitation of full-length RPTPα was reversible after the removal of H2O2 (data not shown). Cys433 in RPTPα-D1 appeared to be involved in H2O2-induced dimerization as well, since mutation of both catalytic cysteines led to complete loss of H2O2-induced binding (Figure 6A). Mutation of Cys433 by itself had only minor effects on the H2O2-induced complex formation (data not shown). In conclusion, we show that H2O2 led to more stable complex formation of full-length RPTPα in a D2 catalytic site cysteine-dependent fashion.

Fig. 6. Oxidative stress stabilized RPTPα dimerization. (A) 293 cells were co-transfected with HA-RPTPα full length [wild-type (WT) or with the catalytic site cysteine single mutant (C723S) or double mutant (C433/C723S)] and Myc-tagged full-length RPTPα (Mt-RPTPα). Cells were treated with H2O2 (1 mM, 10 min) (+) or left untreated (–). After anti-HA immunoprecipitation and immunoblotting, the blot was probed with anti-Myc (top panel) and anti-HA (middle panel) antibodies. Equal expression of the Mt-RPTPα in the lysate was monitored in the lower panel. (B) 293 cells were transfected with HA-RPTPα, treated with H2O2 (1 mM, 10 min) (+) or left untreated. Membrane proteins were cross-linked using the non-cell-permeable cross-linker BS3. An immunoblot of whole-cell lysates is shown with the position of RPTPα monomers and dimers indicated. (C) HA-RPTPα-SpD2 was co-transfected with Mt-RPTPα-D1, Mt-RPTPα-D2 or both. H2O2 treatment (10 min, 1 mM), immunoblotting of anti-HA tag immunoprecipitates and lysates were performed as indicated.
We used the non-cell-permeable cross-linker bis[sulfosuccinimidyl]suberate (BS3) to investigate whether H2O2 induced a large increase in dimer formation. Figure 6B shows that H2O2 did not affect the extent of BS3-mediated cross-linking of RPTPα, suggesting that H2O2 does not induce de novo dimerization, but rather leads to stabilization of existing dimers. It is noteworthy that no higher order protein complexes were detected after H2O2 treatment and BS3-mediated cross-linking, indicating that H2O2 did not induce non-specific aggregation of RPTPα.
In order to obtain insight into the mechanism underlying H2O2-induced stabilization of RPTPα dimers, we investigated binding of RPTPα-SpD2 to RPTPα-D1 and RPTPα-D2. Following H2O2 treatment, RPTPα-SpD2 bound to both RPTPα-D1 and RPTPα-D2 (Figure 6C). This result was surprising, since previously we had not been able to detect direct D2–D2 interactions in the yeast two-hybrid system or using GST–RPTPα-D2 binding assays. Apparently, the individual RPTPα-D2 domains did interact, which may be due to the fact that only small epitope tags were fused to RPTPα-SpD2, instead of larger GAL4 or GST proteins. Interestingly, co-transfection of HA-RPTPα-SpD2 with Mt-RPTPα-D1 and Mt-RPTPα- D2 demonstrated that PTPα-D2 bound preferentially to Mt-RPTPα-D2 (Figure 6C, compare top panel with bottom panel). Taken together, we demonstrate here that H2O2 treatment induced stabilization of RPTPα dimers, which appears to be mediated by D2–D2 interactions.
Inactivation of RPTPα activity in vivo by H2O2 is partially dependent on Cys723
Since H2O2 induced the formation of stable RPTPα complexes, we investigated the effect of H2O2 on RPTPα activity. To this end, HA-RPTPα and mutants were immunoprecipitated and assayed in a PTP assay, using pNPP as a substrate. In order to reduce experimental variation and to get significant PTP activity above background level, several dishes of (stimulated) transfected cells were collected and pooled. Addition of H2O2 in vivo led to reproducible complete inactivation of RPTPα activity (Figure 7). Surprisingly, H2O2 only partially reduced the activity of mutant RPTPα-C723S and RPTPα-ΔD2 (40–60%) (Figure 7). Yet, addition of H2O2 (1 mM) to immunoprecipitated RPTPα-C723S completely inhibited its activity, like wild-type RPTPα (Figure 7). Wild-type RPTPα and RPTPα-C723S are equally sensitive to H2O2 treatment in vitro, in that different H2O2 concentrations inhibit wild-type and mutant RPTPα to the same extent (e.g. 0.1 mM H2O2 led to a 70% reduction in PTP activity of both wild-type and C723S; data not shown). These results correlate with the observation that wild-type RPTPα formed much more stable dimers than RPTPα-C723S, and suggest that H2O2-induced RPTPα dimers are inactive. In conclusion, we show that the effect of H2O2 on the catalytic activity of RPTPα-D1 is dependent on the catalytic site cysteine of RPTPα-D2, which is likely to be due to the H2O2-induced conformational change in RPTPα-D2.

Fig. 7. Oxidative stress-induced change in RPTPα activity. HA-RPTPα wild type (WT), HA-RPTPα-C723S (C723S) or HA-RPTPα-ΔD2 (ΔD2), lacking D2, were expressed in 293 cells. Cells were stimulated with H2O2 (1 mM, 5 min) (vivo), HA-RPTPα proteins were immunoprecipitated and PTP activity was assayed using pNPP as a substrate. Alternatively, H2O2 (1 mM) was added after immunoprecipitation of RPTPα from unstimulated cells (vitro). The activity was corrected for RPTPα expression.
Discussion
More and more evidence shows that RPTP-D2s interact with RPTPs. In some cases, RPTP-D2s have been shown to regulate RPTP activity. However, whether and how the interactions are regulated is not known. Here, we propose a model for the regulation of the interaction between RPTPα-D2 and RPTPα in which intramolecular interactions between the spacer and the C-terminal region of RPTPα-SpD2 lock RPTPα-SpD2 in a ‘closed’ conformation, prohibiting intermolecular binding to RPTPα. We show that oxidative stress induced a conformational change, which was accompanied by more stable RPTPα dimers and inactivation of RPTPα activity. Taken together, our results suggest a mechanism by which intra- and intermolecular interactions of RPTPα are regulated by oxidative stress.
The change in conformation was directly detected in vivo and in vitro using FRET between two GFP derivatives fused N- and C-terminally to RPTPα-SpD2. FRET between GFP derivatives is a valuable tool to study in vivo protein–protein interactions. Recently, FRET has been used to detect conformational changes in peptides as a result of calcium binding (Miyawaki et al., 1997) or PKA phosphorylation (Nagai et al., 2000). Here, we used FRET to detect a conformational change in a complete protein domain, and we followed the dynamics of this conformational change in living cells in real time (Figure 4). Analysis of FRET between GFP derivatives, fused to an entire protein domain, is a new approach to study the dynamics of conformational changes in proteins.
Extracellular stimuli, such as H2O2, UV and heat shock, induced a conformational change followed by intermolecular interactions between RPTPα in vivo. The catalytic site cysteine of RPTPα-SpD2 was necessary for H2O2-induced binding to RPTPα. The involvement of the catalytic site cysteine of RPTPα-D2 is very interesting. PTP catalytic site cysteines have been shown to be preferential targets of free radicals, and their oxidation a potential reversible mode to directly regulate PTP activity (Knebel et al., 1996; Denu and Tanner, 1998; Fauman et al., 1998). H2O2 induced a reversible disulfide bridge between the catalytic site cysteine and a nearby cysteine in LMW-PTP, Cdc25A and KAP (Caselli et al., 1998; Fauman et al., 1998; Song et al., 2001). Importantly, the formation of a disulfide bridge induced detectable structural changes outside the catalytic pocket of LMW-PTP (Caselli et al., 1998). A similar intramolecular disulfide bridge was suggested for several other PTPs, including SHP-1 (Pregel and Storer, 1997; Denu and Tanner, 1998). However, disulfide bridges have been excluded for PTP-1B (Lee et al., 1998) and VHR PTPs (Denu and Tanner, 1998), which preferentially form sulfenic acid after oxidative stress. Our results suggest that next to the catalytic site Cys723, Cys734 (at ∼9 Å from Cys723) may be involved in the conformational change induced by oxidative stress, possibly through the formation of a disulfide bridge. Interestingly, in the H2O2-sensing transcription factor OxyR, two cysteines, 17 Å apart, were found to form a disulfide bridge after oxidative stress, resulting in significant structural changes (Choi et al., 2001), suggesting that the distance of ∼9 Å between Cys723 and Cys734 may not per se prohibit disulfide bridge formation. Alternatively, Cys734 may be involved otherwise in the H2O2-induced conformational change. Furthermore, we cannot exclude the possibility that mere oxidation of Cys723 to sulfenic acid triggers the conformational change. More work will be required to show definitively the involvement of a disulfide bridge or of other modifications, like sulfenic acid formation in RPTPα-D2.
Strikingly, all the effects of oxidative stress on RPTPα shown here are dependent on the catalytic site cysteine of RPTPα-D2. These effects include: (i) conformational changes in RPTPα-D2; (ii) binding of RPTPα-D2 to RPTPα; (iii) formation of a stable RPTPα dimer complex; and (iv) complete inactivation of RPTPα. This suggests that all these effects may be linked, which is further supported by the kinetics of these effects (rapid induction within 1 min and slow recovery in ∼2 h).
We propose a multistep regulatory mechanism for RPTPα as a result of oxidative stress (Figure 8). Before stimulation, an equilibrium exists between monomeric and dimeric RPTPα. RPTPα dimerizes constitutively and extensively in living cells (Jiang et al., 2000; Tertoolen et al., 2001), suggesting that the dimeric state may be preferred. Whether dimeric RPTPα is active in the pre-stimulation situation depends on the rotational coupling in the dimers (Jiang et al., 1999). Oxidative stress leads to rapid oxidation of catalytic sites in PTPs, partially inactivating PTP activity (Denu and Tanner, 1998; Lee et al., 1998). RPTPα is completely inactivated following H2O2 treatment, while inactivation of RPTPα-C723S (and RPTPα-ΔD2) is partial, suggesting that additional events are responsible for complete inactivation of wild-type RPTPα. Oxidative stress rapidly induces a conformational change in D2, but not D2-C723S. This conformational change induces intermolecular interactions of RPTPα- D2, with D2–D2 interactions being preferred. We have previously demonstrated that many domains in RPTPα are involved in RPTPα dimerization, including the transmembrane domain (Jiang et al., 2000; Tertoolen et al., 2001). The oxidative stress-induced D2–D2 interactions may drive stabilization of the dimers, facilitating additional interactions in the dimer, including for instance D1–wedge interactions (Bilwes et al., 1996), which may be responsible for complete inactivation of RPTPα. Reduction of the catalytic site cysteine in D1 in the highly reducing intracellular milieu is fast (Lee et al., 1998). The kinetics of the conformational change and SpD2 binding indicate that reversion to the pre-stimulation state is slow, suggesting that RPTPα activity is only slowly returning to pre-stimulation levels too. Our model suggests that oxidative stress leads to complete inactivation of RPTPα activity, allowing efficient propagation of phosphotyrosine-dependent signaling.
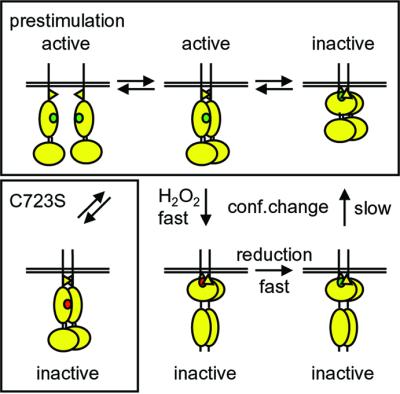
Fig. 8. A model for regulation of RPTPα by oxidative stress. Under normal conditions, RPTPα monomers and dimers are in equilibrium (pre-stimulation). Whether the dimers are active or inactive depends on their rotational coupling. Oxidative stress (e.g. H2O2) leads to rapid inactivation by oxidation of the catalytic site cysteine in D1 (green to red). Oxidative stress also induces a conformational change in D2, which may drive the formation of stabilized dimers, perhaps through D2–D2 interactions. The stabilized dimers are completely inactive. Mutant RPTPα-C723S lacks a conformational change, does not form stabilized dimers and is not inactivated completely following oxidative stress. After reduction, RPTPα slowly returns to the pre-stimulation state following a conformational change in D2. See text for details.
Activation of multiple RPTKs leads to local H2O2 production, suggesting that RPTK activation itself may lead to complete and/or sustained RPTPα inactivation. We were unable to detect any effect of growth factors or other known stimuli that produce free radicals on RPTPα-SpD2 binding capacity or conformational changes. This is most likely due to the local and low production of H2O2 in response to growth factors. RPTPα-SpD2 is cytoplasmic and may, therefore, not be localized correctly to perceive ligand-induced H2O2 production. UV is more potent and induces massive tyrosine phosphorylation, which is thought to be mediated by free radicals, leading to inactivation of PTPs (Lee et al., 1998; Gross et al., 1999). H2O2 has been known for a long time for its mitogenic effect, which may be mediated by its inhibitory effect on PTPs (Knebel et al., 1996; Bae et al., 1997). The fact that H2O2 regulates the interaction between RPTP-D2s and RPTPs provides an interesting new insight into the mode of action of H2O2, as well as into the function of the conserved RPTP-D2s.
RPTPs interact with (multiple) RPTP-D2s, and inter- and intramolecular interactions may regulate RPTP activity. The presence of an extended N-terminal region forming a helix interacting extensively with the C-terminal helix, as observed in RPTPα-D2, is a conserved structural feature found in most PTP domains crystallized to date, including RPTP-D1s and RPTP-D2s, and to a lesser extent cytosolic PTPs. Competition in binding between PTP domains and spacer regions or C-terminal regions has been described for CD45 and RPTPµ, in which the presence of an extended N-terminal region prohibited intermolecular interactions in favor of intramolecular interactions (Feiken et al., 2000; Hayami-Noumi et al., 2000). Interestingly, these interactions also affected the activity of the PTPs (Felberg and Johnson, 1998; Feiken et al., 2000; Aricescu et al., 2001). It will be very interesting to see whether these interactions in RPTPµ and CD45 are regulated by any stimulus as well.
Most RPTP-D2s contain a catalytic site cysteine, suggesting that the model for oxidative stress-induced regulation of RPTP dimerization and activity we propose here (Figure 8) may be applicable to other RPTPs. However, the crystal structure of LAR suggests that D1–D1 interactions are prohibited due to steric hindrance by LAR-D2, therefore opposing the dimerization model suggested by the crystal structure of RPTPα-D1 (Bilwes et al., 1996; Nam et al., 1999). Regulated changes in the conformation of D2 and in the interaction between D1 and D2 may unify both crystal structures in that a specific stimulus affecting the conformation of D2 may allow for D1–D1 interactions to occur in LAR. Confirmation that RPTP topology can change will require the crystallization of other full cytoplasmic regions of RPTPs, before and after stimulation. In this respect, it would be very interesting to see the structural arrangement of the two PTP domains of RPTPα, and the extent of the conformational changes induced by H2O2.
We have previously suggested cross-talk between RPTPs, involving RPTP-D1–D2 interactions (Blanchetot and den Hertog, 2000). The existence of stimulus-dependent binding between domains of RPTPs raises the possibility of regulated cross-talk, in which one or two (distinct) stimuli induce specific interactions between RPTPs. Such reshuffling of RPTP complexes may lead to specific (de)phosphorylation of (other) substrates, thus triggering a new set of downstream events. Knowledge of regulated interactions between (distinct) RPTP-D1s and RPTP-D2s, as well as between full-length RPTPs, may help to understand the mechanism of regulation and cooperation. In conclusion, we show that oxidative stress affected RPTPα’s conformation, interactions and activity, which led us to propose a model for the regulation of RPTPα. In the future, it will be interesting to extend this model to other RPTPs, which may lead to the identification of distinct stimuli that regulate RPTP complex formation and function.
Materials and methods
Constructs
PSG5-13-HA-RPTPα full length, deletions and point mutations, and PCS+MT (myc-tagged vector) RPTPα-D2 (amino acids 537–793) were described previously (Blanchetot and den Hertog, 2000). RPTPα-SpD2 (amino acids 504–793) was cloned in PCS2+MT by PCR. PCS2+MT RPTPα-SpD2ΔC-t was made in the same way as RPTPα-D2ΔC-t, by cloning a NcoI–EcoRI fragment in PCS2+MT, deleting residues 774–792 and the endogenous stop codon, which led to replacement of the RPTPα sequence by some vector sequence, leaving the overall size of the protein virtually identical. Point mutations were made by site-directed mutagenesis, and verified by sequencing.
Cell cultures and transfections
293 human embryonic kidney (HEK) cells were routinely grown in DF medium supplemented with 7.5% fetal calf serum. Cells were transfected using the standard calcium phosphate method. Briefly, 10-cm dishes were transfected with a total of 20 µg of DNA. The next day, the medium was refreshed, and left another 16 h before harvesting.
Immunoprecipitation and immunoblotting
Subconfluent (stimulated) cells were washed twice with ice-cold phosphate-buffered saline, and lysed in cell lysis buffer CLB (50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 1% TX-100 and protease inhibitors, including benzamidine, aprotinin and leupeptin) for 20 min on ice, harvested, and centrifuged at 14 000 g for 15 min. Heat shock was carried out by placing the dish in a 42°C pre-warmed waterbath, and UV treatment by placing an open dish in a Stratalinker (Stratagene). Cross-linking was performed using BS3 (Pierce) as described (Tertoolen et al., 2001). Lysates were added to 12CA5 antibodies that had been pre-coupled to protein A–Sepharose beads. After 2 h of incubation, the beads were carefully washed four times with HNTG buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 0.1% TX-100, 10% glycerol), mixed with 2× Laemmli buffer and loaded on a 7.5% SDS–PAGE gel. The proteins were transferred to PVDF membrane using a semidry transfer system. After Coomassie Blue staining, the membrane was blocked for 1 h with 5% milk in TBS–Tween (50 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween-20) at room temperature, incubated with the first antibody [anti-HA tag, 12CA5; or anti-Myc tag, 9E10 (Santa Cruz)] for 1.5 h, washed four times with TBS–Tween, incubated with horseradish peroxidase-conjugated anti-mouse antibody (Transduction Laboratories) for 1 h, washed four times with TBS–Tween, and developed using enhanced chemiluminescence.
PTP activity assays
After immunoprecipitation (see above) supplemented with two washes with PTP buffer (0.1 mM succinic acid pH 6.0, 150 mM NaCl, 1 mM EDTA), the beads were incubated with 200 µl of PTP buffer containing 10 mM pNPP and left at 30°C. After sufficient time (15–60 min), aliquots (80 µl) were taken and the reaction was stopped by addition of NaOH (100 µl, 2 M) and the OD415 nm measured. Several aliquots were taken at different times of incubation. The phosphatase activity was corrected for the amount of RPTPα present in the phosphatase assay. Aliquots of the phosphatase assay were taken and loaded on an SDS–PAGE gel. The gel was blotted and RPTPα was quantified using the Attophos detection system (Amersham) and a FluorImager (Molecular Dynamics).
FRET analysis
A Leitz orthoplan upright microscope (Leitz GMBH, Wetzlar, Germany) was used, equipped with an epi-illumination fluorescence detection system and a temperature-controlled specimen holder at 33°C. Measure ments were made in a buffer containing 145 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose and 0.5% bovine serum albumin. As an excitation source, a SPEX Fluorolog (SPEX Industries, Edison, NJ) with two excitation monochromators was used. Excitation of CFP and YFP was performed at 430(8) and 490(8) nm, respectively. For the spectral analysis of CFP and the FRET measurements, a filter cube fitted with a 455 nm dichroic mirror was used. The YFP spectra were measured with a dichroic mirror >510 nm. Spectral data were recorded with an integration time of 0.5 s/nm, slit 8 nm. Real-time dual excitation measurements were made by alternating excitation at 430 and 490 nm and analysis of YFP emission at 535(30) nm, essentially as described (Tertoolen et al., 2001). All measurements were made with a Leitz 50× NA 1.0 water immersion objective. The spectra from cell lysates were collected at room temperature using a Perkin Elmer LS50B (UK) and a quartz ultra micro cell (120 µl; Hellma, Rijswijk, The Netherlands), excitation 430(10) nm.
293 cells were cultured on glass coverslips and transfected with the appropriate construct. The constructs used for the FRET measurements were CFP–RPTPα-SpD2–YFP2.1, mutant CFP–RPTPα-SpD2-C723S– YFP2.1, CFP–RPTPα-SpD2–CFP and YFP–RPTPα-SpD2–YFP. CFP, YFP2.1 and RPTPα-SpD2 (residues Ser504–Lys793) were amplified by PCR, introducing convenient restriction sites, and cloned into pCS2+. The resulting constructs were checked by sequencing. YFP2.1 contains two extra point mutations (Val68 to Leu and Glu69 to Lys) that render YFP less sensitive to pH changes (Miyawaki et al., 1999). Importantly, the original YFP is highly sensitive to H2O2 treatment, which induced decreases in YFP emission of up to 50% (C.B., L.G.J.T. and J.d.H., unpublished), while YFP2.1 is practically insensitive to H2O2 (Figure 4B).
Acknowledgments
Acknowledgements
We thank John Overvoorde for his technical assistance, and Astrid van der Sar and Jaap van Hellemond for helpful discussions. These investigations were (in part) supported by the Research Council for Earth and Life sciences (ALW) with financial aid from the Netherlands Organization for Scientific Research (NWO).
References
- Aricescu A.R., Fulga,T.A., Cismasiu,V., Goody,R.S. and Szedlacsek,S.E. (2001) Intramolecular interactions in protein tyrosine phosphatase RPTPµ: kinetic evidence. Biochem. Biophys. Res. Commun., 280, 319–327. [DOI] [PubMed] [Google Scholar]
- Bae Y.S., Kang,S.W., Seo,M.S., Baines,I.C., Tekle,E., Chock,P.B. and Rhee,S.G. (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem., 272, 217–221. [PubMed] [Google Scholar]
- Bastiaens P.I. and Pepperkok,R. (2000) Observing proteins in their natural habitat: the living cell. Trends Biochem. Sci., 25, 631–637. [DOI] [PubMed] [Google Scholar]
- Bilwes A.M., den Hertog,J., Hunter,T. and Noel,J.P. (1996) Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization. Nature, 382, 555–559. [DOI] [PubMed] [Google Scholar]
- Blanchetot C. and den Hertog,J. (2000) Multiple interactions between receptor protein-tyrosine phosphatase (RPTP) α and membrane-distal protein-tyrosine phosphatase domains of various RPTPs. J. Biol. Chem., 275, 12446–12452. [DOI] [PubMed] [Google Scholar]
- Buist A., Zhang,Y.L., Keng,Y.F., Wu,L., Zhang,Z.Y. and den Hertog,J. (1999) Restoration of potent protein-tyrosine phosphatase activity into the membrane-distal domain of receptor protein-tyrosine phosphatase α. Biochemistry, 38, 914–922. [DOI] [PubMed] [Google Scholar]
- Caselli A., Marzocchini,R., Camici,G., Manao,G., Moneti,G., Pieraccini,G. and Ramponi,G. (1998) The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem., 273, 32554–32560. [DOI] [PubMed] [Google Scholar]
- Choi H., Kim,S., Mukhopadhyay,P., Cho,S., Woo,J., Storz,G. and Ryu,S. (2001) Structural basis of the redox switch in the OxyR transcription factor. Cell, 105, 103–113. [DOI] [PubMed] [Google Scholar]
- Cubitt A.B., Heim,R., Adams,S.R., Boyd,A.E., Gross,L.A. and Tsien,R.Y. (1995) Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci., 20, 448–455. [DOI] [PubMed] [Google Scholar]
- den Hertog J. (1999) Protein-tyrosine phosphatases in development. Mech. Dev., 85, 3–14. [DOI] [PubMed] [Google Scholar]
- den Hertog J., Tracy,S. and Hunter,T. (1994) Phosphorylation of receptor protein-tyrosine phosphatase α on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J., 13, 3020–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu J.M. and Dixon,J.E. (1998) Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol., 2, 633–641. [DOI] [PubMed] [Google Scholar]
- Denu J.M. and Tanner,K.G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry, 37, 5633–5642. [DOI] [PubMed] [Google Scholar]
- Desai D.M., Sap,J., Schlessinger,J. and Weiss,A. (1993) Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell, 73, 541–554. [DOI] [PubMed] [Google Scholar]
- Desai C.J., Gindhart,J.G.J., Goldstein,L.S. and Zinn,K. (1996) Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell, 84, 599–609. [DOI] [PubMed] [Google Scholar]
- Fauman E.B., Cogswell,J.P., Lovejoy,B., Rocque,W.J., Holmes,W., Montana,V.G., Piwnica-Worms,H., Rink,M.J. and Saper,M.A. (1998) Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell, 93, 617–625. [DOI] [PubMed] [Google Scholar]
- Feiken E., van Etten,I., Gebbink,M.F., Moolenaar,W.H. and Zondag,G.C. (2000) Intramolecular interactions between the juxtamembrane domain and phosphatase domains of receptor protein-tyrosine phosphatase RPTPµ. Regulation of catalytic activity. J. Biol. Chem., 275, 15350–15356. [DOI] [PubMed] [Google Scholar]
- Felberg J. and Johnson,P. (1998) Characterization of recombinant CD45 cytoplasmic domain proteins. Evidence for intramolecular and intermolecular interactions. J. Biol. Chem., 273, 17839–17845. [DOI] [PubMed] [Google Scholar]
- Garrity P.A., Lee,C.H., Salecker,I., Robertson,H.C., Desai,C.J., Zinn,K. and Zipursky,S.L. (1999) Retinal axon target selection in Drosophila is regulated by a receptor protein tyrosine phosphatase. Neuron, 22, 707–717. [DOI] [PubMed] [Google Scholar]
- Gross S., Knebel,A., Tenev,T., Neininger,A., Gaestel,M., Herrlich,P. and Bohmer,F.D. (1999) Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J. Biol. Chem., 274, 26378–26386. [DOI] [PubMed] [Google Scholar]
- Hayami-Noumi K., Tsuchiya,T., Moriyama,Y. and Noumi,T. (2000) Intra- and intermolecular interactions of the catalytic domains of human CD45 protein tyrosine phosphatase. FEBS Lett., 468, 68–72. [DOI] [PubMed] [Google Scholar]
- Hoffmann K.M., Tonks,N.K. and Barford,D. (1997) The crystal structure of domain 1 of receptor protein-tyrosine phosphatase µ. J. Biol. Chem., 272, 27505–27508. [DOI] [PubMed] [Google Scholar]
- Hunter T. (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell, 80, 225–236. [DOI] [PubMed] [Google Scholar]
- Jiang G., den Hertog,J., Su,J., Noel,J., Sap,J. and Hunter,T. (1999) Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-α. Nature, 401, 606–610. [DOI] [PubMed] [Google Scholar]
- Jiang G., den Hertog,J. and Hunter,T. (2000) Receptor-like protein tyrosine phosphatase α homodimerizes on the cell surface. Mol. Cell. Biol., 20, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel A., Rahmsdorf,H.J., Ullrich,A. and Herrlich,P. (1996) Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J., 15, 5314–5325. [PMC free article] [PubMed] [Google Scholar]
- Kokel M., Borland,C.Z., DeLong,L., Horvitz,H.R. and Stern,M.J. (1998) clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev., 12, 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger N.X., Streuli,M. and Saito,H. (1990) Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J., 9, 3241–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger N.X., Van Vactor,D., Wan,H.I., Gelbart,W.M., Goodman,C.S. and Saito,H. (1996) The transmembrane tyrosine phosphatase DLAR controls motor axon guidance in Drosophila. Cell, 84, 611–622. [DOI] [PubMed] [Google Scholar]
- Lee S.R., Kwon,K.S., Kim,S.R. and Rhee,S.G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem., 273, 15366–15372. [DOI] [PubMed] [Google Scholar]
- Lim K.L., Kolatkar,P.R., Ng,K.P., Ng,C.H. and Pallen,C.J. (1998) Interconversion of the kinetic identities of the tandem catalytic domains of receptor-like protein-tyrosine phosphatase PTPα by two point mutations is synergistic and substrate-dependent. J. Biol. Chem., 273, 28986–28993. [DOI] [PubMed] [Google Scholar]
- Majeti R., Bilwes,A.M., Noel,J.P., Hunter,T. and Weiss,A. (1998) Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science, 279, 88–91. [DOI] [PubMed] [Google Scholar]
- Meng K., Rodriguez-Pena,A., Dimitrov,T., Chen,W., Yamin,M., Noda,M. and Deuel,T.F. (2000) Pleiotrophin signals increased tyrosine phosphorylation of β β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc. Natl Acad. Sci. USA, 97, 2603–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A., Llopis,J., Heim,R., McCaffery,J.M., Adams,J.A., Ikura,M. and Tsien,R.Y. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature, 388, 882–887. [DOI] [PubMed] [Google Scholar]
- Miyawaki A., Griesbeck,O., Hein,R. and Tsien,R.Y. (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc. Natl Acad. Sci. USA, 96, 2135–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y., Miyazaki,M., Aoki,R., Zama,T., Inouye,S., Hirose,K., Iino,M. and Hagiwara,M. (2000) A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nature Biotechnol., 18, 313–316. [DOI] [PubMed] [Google Scholar]
- Nam H.J., Poy,F., Krueger,N.X., Saito,H. and Frederick,C.A. (1999) Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell, 97, 449–457. [DOI] [PubMed] [Google Scholar]
- Neel B.G. and Tonks,N.K. (1997) Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol., 9, 193–204. [DOI] [PubMed] [Google Scholar]
- Pregel M.J. and Storer,A.C. (1997) Active site titration of the tyrosine phosphatases SHP-1 and PTP1B using aromatic disulfides. Reaction with the essential cysteine residue in the active site. J. Biol. Chem., 272, 23552–23558. [DOI] [PubMed] [Google Scholar]
- Song H., Hanlon,N., Brown,N.R., Noble,M.E., Johnson,L.N. and Barford,D. (2001) Phosphoprotein–protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol. Cell, 7, 615–626. [DOI] [PubMed] [Google Scholar]
- Tertoolen L.G., Blanchetot,C., Jiang,G., Overvoorde,J., Gadella,T.W.J., Hunter,T. and den Hertog,J. (2001) Dimerization of receptor protein-tyrosine phosphatase α in living cells. BMC Cell Biol., 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vactor D., O’Reilly,A.M. and Neel,B.G. (1998) Genetic analysis of protein tyrosine phosphatases. Curr. Opin. Genet. Dev., 8, 112–126. [DOI] [PubMed] [Google Scholar]
- Wallace M.J., Fladd,C., Batt,J. and Rotin,D. (1998) The second catalytic domain of protein tyrosine phosphatase δ (PTPδ) binds to and inhibits the first catalytic domain of PTPσ. Mol. Cell. Biol., 18, 2608–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. and Pallen,C.J. (1991) The receptor-like protein tyrosine phosphatase HPTP α has two active catalytic domains with distinct substrate specificities. EMBO J., 10, 3231–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.M. and Shalloway,D. (2001) Two mechanisms activate PTPα during mitosis. EMBO J., 20, 6037–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]