

Abstract
The E-cadherin-based adherens junction (AJ) is essential for organogenesis of epithelial tissues including the liver, although the regulatory mechanism of AJ formation during development remains unknown. Using a primary culture system of fetal hepatocytes in which oncostatin M (OSM) induces differentiation, we show here that OSM induces AJ formation by altering the subcellular localization of AJ components including E-cadherin and catenins. By retroviral expression of dominant-negative forms of signaling molecules, Ras was shown to be required for the OSM-induced AJ formation. Fetal hepatocytes derived from K-Ras knockout (K-Ras–/–) mice failed to form AJs in response to OSM, whereas AJ formation was induced normally by OSM in mutant hepatocytes lacking both H-Ras and N-Ras. Moreover, the defective phenotype of K-Ras–/– hepatocytes was restored by expression of K-Ras, but not by H-Ras and N-Ras. Finally, pull-down assays using the Ras-binding domain of Raf1 demonstrated that OSM directly activates K-Ras in fetal hepatocytes. These results indicate that K-Ras specifically mediates cytokine signaling for formation of AJs during liver development.
Keywords: E-cadherin/homophilic adhesion/K-Ras/liver organogenesis
Introduction
Organogenesis involves massive cell proliferation and a series of differentiation and maturation processes. While these processes are cell autonomous to some extent, they are influenced profoundly by extracellular signals such as soluble factors and membrane-bound proteins. These signals activate multiple intracellular signaling pathways and induce the expression of genes that affect cell proliferation and differentiation. A number of soluble factors including hormones and cytokines are known to play a crucial role in the development of various tissues. One example is the transforming growth factor-β (TGF-β) family, whose members are involved in the determination of cell fate during a very early stage of development (Conlon et al., 1994; Winnier et al., 1995). In contrast, direct cell–cell interaction, particularly homophilic adhesion, becomes more important during later stages of development, as cells start organizing the tissue architecture.
At least three types of intercellular architecture connect adjacent cells; adherens junctions (AJs), tight junctions and desmosomes. AJs, predominant in epithelial tissues and neurons, are composed of cadherin and catenins (Nose et al., 1988; Takeichi, 1995; Drubin and Nelson, 1996; Gumbiner, 1996). Cadherin is a transmembrane protein responsible for Ca2+-dependent homophilic interaction through its extracellular domain (Nose et al., 1988; Takeichi, 1995). Since forced expression of E-cadherin, the epithelial prototype, in E-cadherin-negative cells such as fibroblasts results in cell aggregation, expression of a specific cadherin molecule is likely to be a key determinant of direct cell–cell interaction (Nagafuchi and Takeichi, 1988; Takeichi, 1988). The cytoplasmic domain of cadherin binds to β-catenin, which in turn associates with α-catenin. β-catenin acts as a bridge connecting cadherin to α-catenin, which is required for linkage between AJs and the actin cytoskeleton (Nagafuchi et al., 1994; Drubin and Nelson, 1996; Gumbiner, 1996). The association of the cadherin–catenin complex with the actin cytoskeleton is necessary for tight AJ structure (Nagafuchi et al., 1994; Takeichi, 1995).
By using epithelial cell lines, evidence is accumulating that a number of intracellular molecules participate in the regulation of cell adhesion. The Rho family proteins, Rho, Rac and Cdc42, are known to regulate cell adhesion and cytoskeletal organization (Hall, 1998). Rho is involved in the association of AJ with the actin cytoskeleton through control of actin reorganization (Braga et al., 1997; Takaishi et al., 1997). On the other hand, Rac and Cdc42 regulate formation of the cadherin–catenin complex by acting on α- and β-catenins, and also modulate association of the complex with the actin cytoskeleton (Braga et al., 1997; Kuroda et al., 1997; Takaishi et al., 1997). The complex also associates with protein kinases and phosphatases that regulate phosphorylation of β-catenin (Kypta et al., 1996; Aicher et al., 1997; Ozawa and Kemler, 1998; Rosato et al., 1998; Roura et al., 1999), implicating these molecules in the regulation of AJ formation. However, many questions regarding AJ formation remain unexplored.
A typical example is the formation of AJs during liver development (Stamatoglou et al., 1992). In adult liver, E-cadherin and β-catenin are expressed abundantly and localized at cell–cell junctions, indicating that E-cadherin-based AJs tightly connect adjacent hepatocytes. In contrast, despite the fact that fetal hepatocytes at late gestation express these proteins at a level comparable with adult hepatocytes, they are distributed diffusely throughout the cell membrane (Stamatoglou et al., 1992; Kamiya et al., 1999; our unpublished observations). These observations suggest that fetal hepatocytes are not connected tightly to each other by AJs and that there is a mechanism that regulates formation of AJs during liver development. However, it has not been studied how AJ formation is regulated developmentally and what extracellular and intracellular molecules are involved in this process. While established cell lines have been used for the study of AJ formation, they do not provide a model to study the developmental regulation of AJ formation. An in vitro system that recapitulates the developmental process is required to address such questions.
Liver development proceeds through multiple stages and is influenced by extracellular signals. In mice, liver primodium appears at embryonic day 8–9 (E8–9) (Gualdi et al., 1996). At E11, the liver accepts hematopoietic cells originating from the yolk sac and the aorta–gonad– mesonephros (AGM) region and supports embryonic hematopoiesis until birth (Medvinsky et al., 1996; Mukouyama et al., 1998). After birth, the liver acquires many metabolic functions and starts forming the architecture of liver lobules. We previously showed that liver development is stimulated by oncostatin M (OSM), an interleukin-6 (IL-6) family cytokine (Kamiya et al., 1999). In fetal liver at mid-gestation, OSM is expressed in CD45+ hematopoietic cells, whereas the OSM receptor is expressed in fetal hepatocytes, suggesting that OSM is a paracrine factor in fetal liver (Kamiya et al., 1999). In a primary culture of fetal hepatocytes from E14.5 murine embryos, OSM at a physiological concentration in the presence of dexamethasone (Dex) promotes functional maturation of fetal hepatocytes to neonatal liver cells as evidenced by expression of various liver enzymes, glycogen accumulation, ammonia clearance and lipid accumulation (Kamiya et al., 1999; Kojima et al., 2000). These results suggest that this in vitro system mimics the process of in vivo liver development.
OSM manifests its functions through the OSM receptor, which consists of the OSM-specific subunit (OSMRβ) (Lindberg et al., 1998; Tanaka et al., 1999) and gp130, the common signal transducer of IL-6 family cytokines (Hirano et al., 1997), and it activates multiple intracellular signaling molecules including STAT3, Ras, PI3K and MAPK in various cell types including fetal hepatocytes (Taga and Kishimoto, 1997; Ito et al., 2000). Our recent studies using a retrovirus-mediated gene transfer system demonstrated that STAT3 plays major roles in the expression of liver-specific genes as well as the accumulation of glycogen induced by OSM (Ito et al., 2000). In contrast, while Ras was shown to regulate expression of OSM-responsive genes negatively, its precise function in liver development remains unknown (Ito et al., 2000). Interestingly, we also noted that hepatic differentiation in response to OSM caused dramatic changes in the morphology of fetal hepatocytes (Kamiya et al., 1999). In particular, homophilic cell–cell contact became more apparent in a manner similar to that seen in differentiated hepatocytes. These observations indicate that OSM may also regulate cell adhesion(s) in fetal hepatocytes to organize cellular architecture.
In this study, by taking advantages of our in vitro system that recapitulates hepatic differentiation, we investigated the extracellular and intracellular signaling mechanism of AJ formation during liver development. Here we show that Dex up-regulates protein levels of AJ components including E-cadherin and catenins, while OSM promotes their localization at cell–cell junctions of lateral membranes. Interestingly, OSM-triggered E-cadherin localization depends specifically on K-Ras, but not other Ras or Rho family proteins. Thus, our results not only demonstrate a novel biological function of K-Ras but also shed light on the regulatory mechanism of E-cadherin-based AJs during liver development.
Results
Morphological changes of fetal hepatocytes upon induction of differentiation
As we demonstrated previously, stimulation of fetal hepatocytes with 10 ng/ml of OSM in the presence of 1 × 10–7 M Dex promotes their differentiation as evidenced by the induction of differentiation marker genes [tyrosine aminotransferase (TAT) and glucose-6-phosphatase (G6Pase)] for the neonatal liver and by the up-regulation of glycogenic activity (Kamiya et al., 1999; Ito et al., 2000). Besides these functional parameters of liver development, we also noted that OSM in combination with Dex induced marked morphological changes as compared with Dex alone (Figure 1A). In the presence of OSM and Dex (OSM/Dex), the cytosol was highly granulated and the nucleus was clear and round. Moreover, cell–cell contact became clearer in the OSM/Dex-stimulated cells, raising the interesting possibility that cell adhesion is strengthened by OSM and Dex. To test this possibility, we analyzed the ultrastructure of contact sites of cells treated with Dex or OSM/Dex (Figure 1B and C). When cells were cultured with Dex alone, no obvious adhesion structure was found between adjacent cells, even though a junctional zone was visible (Figure 1B). In contrast, an electron-dense structure, which resembles the morphology of AJs, was observed along with the apical part of lateral membranes in the cells cultured with OSM/Dex (Figure 1C), and desmosome-like structures were also observed in the junctional zone (Figure 1C, inset). In addition, a number of microvilli were found on the apical membranes (Figure 1C, lower panel) and there were glycogen and lipid granules in the cytoplasm (Figure 1C). These observations suggest that fetal hepatocytes treated with OSM/Dex develop a well-polarized epithelial morphology similar to that in mature hepatocytes and that OSM in combination with Dex modulates formation of cell adhesion(s) during the differentiation.

Fig. 1. Morphological changes induced by OSM. Fetal hepatocytes derived from murine liver at E14.5 were cultured for 6 days with Dex or OSM/Dex. (A) Phase contrast images of cultured hepatocytes. OSM/Dex-stimulated cells exhibited tight cell–cell contact, a dense cytoplasm and a clear and round nucleus. (B and C) Ultrastructure of fetal hepatocytes. Cells cultured with Dex (B) or OSM/Dex (C) were processed for electron microscopy. Upper panels: junction areas between adjacent cells. Arrows indicate junctional zones between adjacent cells. Markers shown are Lg, lipid granule; Gg, glycogen granule; If, intermediate filament. The insert in (C) shows desmosome-like structures that are indicated by arrows. Lower panels: apical membrane domain. Mv, microvilli.
OSM and Dex regulate AJ formation in a distinct manner
In order to elucidate the molecular basis for regulation of cell adhesion by these external signals, we examined the subcellular localization and expression of cell adhesion molecules by immunofluorescence and biochemical analyses. In the absence of OSM and Dex, E-cadherin was expressed weakly and distributed diffusely (Figure 2A). When cells were incubated with Dex alone, expression of E-cadherin was markedly increased, although the pattern of subcellular localization was not significantly altered. Interestingly, stimulation with both OSM and Dex dramatically changed the distribution of E-cadherin, inducing its localization at cell–cell junctions (Figure 2A). Moreover, a view of the x–z axis revealed that E-cadherin was concentrated in the apical parts of lateral membranes in the presence of OSM and Dex (Figure 2B, arrowhead). Such localization of E-cadherin at junctional zones in lateral membranes was also observed in cells treated with OSM alone, although the protein level of E-cadherin was very low (Figure 2A). These results suggest that Dex up-regulates the protein expression of E-cadherin, while OSM modulates its subcellular localization. In addition, immunostaining of β-catenin revealed that the protein was regulated similarly by Dex and OSM (Figure 2C and data not shown). As in the case of immunostaining, western blot analysis of total protein extracts demonstrated that Dex increased protein levels of E-cadherin and catenins (Figure 3A). Importantly, however, OSM did not change the protein levels regardless of whether the cells were stimulated with Dex or not (Figure 3A), indicating that OSM specifically mobilizes these proteins to cell–cell junctions without affecting their protein levels.

Fig. 2. OSM induces formation of adherens junctions, but not tight junctions. Fetal hepatocytes were cultured for 6 days without OSM/Dex (No factor) or with OSM, Dex or OSM/Dex. The localization of cell adhesion molecules was examined by immunofluorescence analysis. (A and B) Subcellular distribution of E-cadherin in cultured hepatocytes. Cells were fixed and stained with anti-E-cadherin antibody. (A) x–y views; (B) x–z views of the cells. Bars and arrows indicate the bottom and top of the cells, respectively. E-cadherin was highly concentrated at the apical parts of lateral membrane (arrowheads). (C and D) Intracellular localization of β-catenin and ZO-1. Cells were fixed and stained with antibodies against β-catenin (C) or ZO-1 (D). (E) Co-localization of E-cadherin and the actin cytoskeleton. Cells were stained with anti-E-cadherin antibody (green, left panels) and rhodamine–phalloidin (red, middle panels). Each image was overlaid (yellow, right panels). Upper panels: Dex-treated cells. Lower panels: OSM/Dex-treated cells. Scale bars: 10 µm.

Fig. 3. Expression of AJ components and subcellular localization of E-cadherin. Fetal hepatocytes were cultured for 6 days with stimulation as indicated in the figure. (A) Total protein levels of AJ components in fetal hepatocytes. Cell extracts from each culture were analyzed by western blotting using antibodies against E-cadherin, and α-, β- and γ-catenins. The location of each protein is indicated by an arrow. (B) Detergent solubility assay in cultured hepatocytes. Cell extracts were fractionated into soluble (S) and insoluble (I) fractions as described in Materials and methods. E-cadherin protein levels in each fraction were determined by western blotting using anti-E-cadherin antibody. The abundance of E-cadherin in each fraction was estimated by quantitative analysis using the NIH image program, and the ratios of [S] or [I] in [S + I] at each culture condition are shown.
Since it was known that the formation of the tight AJ complex reduces the solubility of E-cadherin in a mild cell lysis condition (Angres et al., 1996; Potempa and Ridley, 1998), we next examined the solubility of E-cadherin in different culture conditions (Figure 3B). In the culture conditions of no factor and OSM alone, nearly 100% of E-cadherin was present in the soluble fraction obtained by 0.5% NP-40 extraction and no E-cadherin was found in the insoluble fraction. Stimulation of the cells with Dex alone resulted in an increase of total E-cadherin and the accumulation of E-cadherin in the insoluble fraction (17%), and OSM/Dex further increased the percentage of insoluble E-cadherin up to 32%. Consistent with the results of solubility assays, a small portion of the actin cytoskeleton stained by phalloidin was co-localized with E-cadherin even in Dex-stimulated cells. Importantly, OSM/Dex strongly induced co-localization of actin fibers with E-cadherin at cell–cell junctions (Figure 2E), suggesting that a tight AJ complex was formed at the junctional zones on stimulation with OSM/Dex. Since all these characteristics are a hallmark of the AJ structure, our results suggest that OSM and Dex promote formation of the AJ complex, at least partly, through the regulation of the subcellular localization and protein expression of AJ components, respectively.
Finally, we analyzed the expression and localization of the ZO-1 protein, a key component of tight junctions (Stevenson et al., 1986), in different culture conditions (Figure 2D). As with the components of AJ, the protein level of ZO-1 was also up-regulated by Dex. However, ZO-1 was localized at cell junction sites even in the absence of OSM and Dex, and neither expression levels nor the distribution of ZO-1 was affected by OSM (Figure 2D). These results suggest that the formation of tight junctions is independent of OSM signaling and that in the presence of Dex alone hepatocytes are connected to each other by cell adhesion structures including tight junctions but not E-cadherin-based AJs.
Ras is essential for OSM-induced E-cadherin localization at cell–cell junctions
OSM activates STAT3 and Ras, two major signaling components of IL-6 family cytokines, in various cell types including fetal hepatocytes (Fukada et al., 1996; Taga and Kishimoto, 1997; Ito et al., 2000). To investigate the OSM signaling pathway that regulates AJ formation, we expressed mutants of STAT3 and Ras in fetal hepatocytes and examined their effects on OSM-induced E-cadherin localization. Since ΔSTAT3 is a dominant-negative mutant of STAT3 that lacks the C-terminal transactivation domain, western blotting with antibody against the N-terminal region of STAT3 verified the expression of ΔSTAT3 as a protein smaller than the endogenous STAT3 in fetal hepatocytes (Figure 4I). Expression of ΔSTAT3, however, had no effect on E-cadherin localization at cell–cell junctions in cells cultured with OSM/Dex (Figure 4C). In contrast, expression of RasN17, a dominant-negative form of Ras, completely blocked AJ formation in response to OSM, i.e. E-cadherin was distributed throughout the cell membrane even in the presence of OSM and Dex (Figure 4D and I). Likewise, the OSM-induced β-catenin localization was inhibited by RasN17, but not by ΔSTAT3 (data not shown). These results indicate that Ras is vital for OSM-triggered E-cadherin/β-catenin localization at cell–cell junctions in fetal hepatocytes.

Fig. 4. Effects of dominant-negative forms of signal molecules on OSM-triggered E-cadherin localization at cell–cell junctions. Fetal hepatocytes were infected on day 0 with the pMIG retroviral vector carrying no signaling molecule (A, B and E), ΔSTAT3 (C), RasN17 (D), RhoN19 (tagged with myc) (F), RacN17 (G) or Cdc42N17 (tagged with Flag) (H), and cultured for 6 days with Dex (A) or OSM/Dex (B–H). Viral infection was verified by green fluorescence of GFP (left panels). The localization of E-cadherin in each culture was examined by immunofluorescence analysis using anti-E-cadherin antibody (middle panels). Merged images are shown in the right hand panels. Scale bars: 10 µm. (I) Ectopic expression of dominant-negative molecules in fetal hepatocytes. Cell extracts from each culture were analyzed by western blotting using specific antibodies. ΔSTAT3 is detected as a smaller band than the endogenous STAT3. Expression of RasN17 and RacN17 was verified by augmented expression of Ras and Rac1, respectively (asterisk; non-specific band). Expression of RhoN19 (tagged with myc) and Cdc42N17 (tagged with Flag) was determined by immunoblotting using antibodies against myc and Flag, respectively. Because expression levels of endogenous STAT3 were comparable in each sample, STAT3 was used as an internal control. (J) Inhibitory effect of dominant-negative forms of the Rho family proteins on c-fos transcription in NIH-3T3 cells. To confirm whether retrovirus vectors carrying dominant-negative forms of the Rho family are functional, NIH-3T3 cells infected with these retrovirus vectors were transiently transfected with a reporter plasmid linked to the c-fos promoter and pRL plasmid and cultured with or without 2% serum for 24 h. Luciferase activity was determined using the Dual Luciferase Reporter Assay System.
The Rho family proteins, Rho, Rac and Cdc42, are known to be necessary for regulation of the E-cadherin-mediated AJs in MDCK cells and keratinocytes, which constitutively express E-cadherin (Braga et al., 1997; Kuroda et al., 1997; Takaishi et al., 1997). To examine whether the Rho family proteins are required for OSM-triggered localization of E-cadherin, we constructed pMIG retrovirus vectors carrying dominant-negative forms of Rho, Rac and Cdc42 (RhoN19, RacN17 and Cdc42N17). We first examined their function in NIH-3T3 cells, in which c-fos transcription by serum stimulation is known to be regulated by the Rho family proteins (Alberts et al., 1998; Kim and Kim, 1998; Wang et al., 1998). Expression of these mutants suppressed c-fos induction, indicating that these mutants function as a dominant-negative molecule (Figure 4J). Although expression of these mutants in fetal hepatocytes was verified by western blotting (Figure 4I), none of these mutants had any effect on the OSM-induced E-cadherin localization (Figure 4E–H). These data suggest that the Rho family proteins are not actively involved in the OSM signaling for AJ formation in fetal hepatocytes. These results indicate that Ras is a major OSM-mediated intracellular signal for AJ formation.
A novel role for K-Ras in OSM-triggered E-cadherin localization
The classical Ras family consists of H-Ras, N-Ras and K-Ras. It was shown that knockout (KO) mice lacking both H-Ras and N-Ras develop normally and are fertile (Umanoff et al., 1995), whereas K-Ras KO mice die at around E15 and exhibit abnormality in various organs including the liver (Johnson et al., 1997; Koera et al., 1997). Moreover, K-Ras-deficient embryonic stem (ES) cells failed to contribute to the liver in chimeric mice generated from blastocysts injected with the ES cells (Johnson et al., 1997). These results indicate an indispensable role for K-Ras during liver development and led us to consider the possibility that K-Ras is involved specifically in OSM-mediated signaling in fetal hepatocytes. To test this possibility, we took advantage of KO mice lacking the K-Ras gene, and first examined the expression of α-fetoprotein (AFP) mRNA, a marker of immature hepatocytes (Shiojiri et al., 1991). K-Ras–/– liver at the E14.5 stage expressed AFP abundantly and the levels were comparable with wild-type and K-Ras+/– livers in the littermates (Figure 5A). These results suggest that there are cells with the characteristics of fetal hepatocytes at E14.5 in K-Ras–/– mice and that liver development before this stage may not be compromised. In the K-Ras–/– hepatocytes isolated from an individual liver at E14.5, stimulation with OSM/Dex induced mRNA expression of differentiation markers (TAT and G6Pase), and the levels of their expression were again comparable with those in wild-type cells (Figure 5B), indicating that K-Ras–/– fetal hepatocytes have a potential in response to OSM. Interestingly, however, K-Ras–/– fetal hepatocytes failed to mobilize E-cadherin to cell–cell junctions in response to OSM, while the protein level of E-cadherin was not altered in these cells (Figure 5C). In contrast, OSM did stimulate E-cadherin localization in H-Ras–/– KO cells (data not shown). Furthermore, no obvious perturbation of E-cadherin localization was found even in H-Ras–/– N-Ras–/– double KO cells (Figure 5C). To eliminate the possibility that these observations were unexpected consequences of gene disruption, we introduced the normal Ras alleles into E14.5 K-Ras–/– cells (Figure 6). E-cadherin distribution was diffuse even in K-Ras–/– hepatocytes treated with OSM/Dex (Figures 5C and 6A). In contrast, E-cadherin was clearly localized at cell–cell junctions in the cells transduced with the K-Ras allele, as shown by the arrows in Figure 6B. These data indicate that introduction of the K-Ras allele into K-Ras–/– fetal hepatocytes restored the normal distribution of E-cadherin in response to OSM. Moreover, expression of H-Ras only partially restored the response to OSM, and N-Ras had no effect at all (Figure 6C and D). These results indicate that K-Ras acts as a specific downstream mediator of OSM signaling in the regulation of E-cadherin localization in fetal hepatocytes.

Fig. 5. A critical role for K-Ras in liver development. (A) Expression of the early hepatic marker gene, α-fetoprotein (AFP). A 10 µg aliquot of total RNA extracted from wild-type, K-Ras+/– or K-Ras–/– fetal liver at E14.5 was loaded in each lane (rRNA; loading control). mRNA expression of AFP was determined by northern blotting using a DIG-labeled probe. (B) mRNA expression of differentiation markers in K-Ras–/– fetal hepatocytes. Fetal hepatocytes at E14.5 derived from wild-type, K-Ras+/– or K-Ras–/– liver were cultured for 6 days with Dex or Dex/OSM. Total RNA (10 µg) extracted from each culture was analyzed by northern blotting to investigate expression of TAT, G6Pase or GAPDH (internal control). (C) Localization of E-cadherin in K-Ras–/– fetal hepatocytes. Fetal hepatocytes derived from wild-type, K-Ras–/– or H-Ras–/–N-Ras–/– embryonic liver were cultured for 6 days with Dex or OSM/Dex and were fixed and stained with anti-E-cadherin antibody. Scale bar: 10 µm.

Fig. 6. K-Ras is required for OSM-triggered E-cadherin localization. K-Ras–/– fetal hepatocytes were infected on day 0 with the pMIG retroviral vector carrying no signaling molecule (A), K-Ras (B), H-Ras (C) or N-Ras (D), and cultured for 6 days with OSM/Dex. To investigate their effect on OSM-triggered E-cadherin localization, subcellular localization of E-cadherin was analyzed (middle panel). Left panel: fluorescence of GFP. Right panel: merged image. E-cadherin was highly concentrated at cell–cell junctional sites (arrows) in cells infected with K-Ras virus (B). Scale bar: 10 µm. (E) Ectopic expression of the Ras family proteins in fetal hepatocytes. Wild-type hepatocytes were infected with the pMIG vector carrying a Ras cDNA and cultured for 6 days with OSM/Dex. Cell lysates from each culture were subjected to western blotting using antibodies against K-Ras, H-Ras, N-Ras or STAT3 (internal control).
Activation of K-Ras in response to OSM in fetal hepatocytes
The results above indicate that K-Ras mediates OSM signaling for AJ formation; activation of K-Ras by OSM had not been demonstrated. To confirm activation of K-Ras by OSM in fetal hepatocytes, we measured the levels of GTP-bound K-Ras by pull-down assays using the Ras-binding domain (RBD) of Raf-1. Although total protein levels of K-Ras were not altered by OSM stimulation (Figure 7, lower row), GTP-bound K-Ras, the active form, was detected in fetal hepatocytes stimulated with OSM for 15 min and the level of the active form was decreased at 30 min after stimulation (upper row). This result indicates that OSM directly activates K-Ras in fetal hepatocytes.
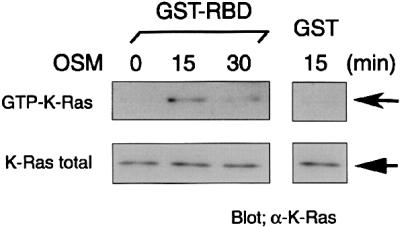
Fig. 7. Activation of K-Ras by OSM in fetal hepatocytes. Serum-starved fetal hepatocytes were stimulated with 10 ng/ml of OSM for 15 or 30 min before cell lysis. Cell lysates were subjected to affinity precipitation with GST–RBD. Protein levels of GTP-bound K-Ras and total K-Ras were determined by western blotting using a K-Ras-specific antibody. GST was used as a negative control.
Discussion
Intracellular signaling pathways activated by extracellular signals regulate many cellular events including cell proliferation, apoptosis and differentiation. In this study, we have shown a novel function of cytokine signaling, which is implicated in epithelial organization. Using the in vitro system that mimics hepatic differentiation, we have demonstrated that extracellular signals stimulate homophilic cell adhesion mediated by AJs, tight junctions and desmosomes. Interestingly, immunofluorescence analysis clearly showed that OSM in the presence of Dex induces localization of E-cadherin, β-catenin and actin at junctional zones of lateral membranes (Figure 2), suggesting that the cell polarity is established in the same way as in adult hepatocytes. Besides structures for cell–cell contact, microvilli as well as intermediate filaments appeared in the cells stimulated with OSM, as shown by Mv and If, respectively, in Figure 1C, all of which are characteristics of differentiated hepatocytes. These results indicate that OSM in the presence of Dex promotes hepatic development not only by inducing gene expression and liver functions as we showed previously, but also by forming the cellular architecture.
Dex augments the expression of AJ components, while OSM stimulates their localization at the cell–cell contact sites apparently without affecting their protein levels (Figures 2 and 3). Even in the absence of Dex, the E-cadherin protein was concentrated at the junctional zones in response to OSM (Figure 2A). Therefore, it is likely that these two extracellular signals are mediated independently; Dex up-regulates protein expression probably at the transcriptional level, and OSM modulates subcellular localization of the AJ proteins. It is noteworthy, however, that small amounts of E-cadherin and actin were localized at junctional zones in the cells stimulated with Dex alone (Figure 2E). Consistently, a significant amount of the E-cadherin protein was present in the insoluble fraction of 0.5% NP-40 extracts in this culture (Figure 3B). We speculate that high levels of protein expression induced by Dex partially lead to formation of the AJ structure at the junctional zone, though to a small extent. This idea is supported by previous observations that ectopic expression of E-cadherin was sufficient to induce formation of the AJ complex in L cells and NIH-3T3 fibroblasts that did not express endogenous E-cadherin (Nagafuchi and Takeichi, 1988; Takeichi, 1988). In contrast to such a passive mechanism, our results indicate that OSM activates a cellular system that mobilizes these proteins to the site of cell–cell contact.
AJs are a major architecture of homophilic adhesions in epithelial cells. While E-cadherin is a major component of AJs in epithelial cells, N-cadherin and LI-cadherin are also known to be expressed in hepatocytes. Our results clearly show that E-cadherin is localized at the junctional zone in response to OSM. However, the results do not exclude the possibility that another cadherin is involved in AJs in fetal hepatocytes. In fact, our results rather support this possibility. Even in the absence of OSM, fetal hepatocytes exhibited a cuboidal shape (Figure 1B), and ZO-1 as well as a part of β-catenin were found at the junctional zones (Figure 2B and C). Moreover, we found that a part of N-cadherin was localized at cell–cell contact sites, while it was distributed rather broadly (data not shown). Thus, fetal hepatocytes are linked to each other through multiple forms of adhesions, i.e. AJs by E-cadherin as well as other cadherins, tight junctions and desmosomes. Among them, OSM regulates the formation of E-cadherin-based AJs in fetal hepatocytes.
We previously described that STAT3 mediates OSM signaling for induction of hepatic differentiation markers and glycogenic activity (Ito et al., 2000). In this study, we demonstrate that OSM-induced localization of E-cadherin at cell–cell junctions requires activation of Ras but not STAT3. These results indicate that STAT3 and Ras, two major signaling pathways of OSM, play distinct roles for differentiation of fetal hepatocytes. However, excess activation of the Ras pathway by expression of RasV12, a constitutively active form of Ras, abrogated both STAT3- (Ito et al., 2000) and Ras-dependent phenotypes induced by OSM (data not shown). Thus, the balance between two intracellular signaling pathways appears to be important for induction of hepatic development.
Among the three classical Ras family proteins, K-Ras is required specifically for the OSM-induced E-cadherin-based AJ formation, because OSM-induced localization of E-cadherin was substantially decreased in K-Ras–/– hepatocytes, whereas it was unaffected in H-Ras–/– hepatocytes and even in hepatocytes lacking both H-Ras and N-Ras (Figure 5C). The inability of K-Ras–/– cells to respond to OSM is not due to a defect in the early development of the liver nor to a lack of the OSM receptor in the K-Ras–/– hepatocytes, since AFP expression was normal in E14.5 K-Ras–/– liver, and hepatocytes isolated from K-Ras–/– liver were able to express TAT and G6Pase in response to OSM (Figure 5A and B). Expression of K-Ras, but not of H-Ras or N-Ras, in K-Ras–/– cells restored the response to OSM (Figure 6). As the recovery by K-Ras was not complete, an additional component(s) might be involved in this process.
While our results indicate that among the three Ras proteins, K-Ras specifically mediates OSM signaling to induce the formation of E-cadherin-based adhesion, the molecular basis for the specificity of K-Ras currently is unknown. A structural difference in the C-terminal short stretches may provide a hint. H-Ras and N-Ras have homologous C-terminal stretches, by which both are palmitoylated (Magee and Marshall, 1999). This modification enables them to be recruited to a particular subdomain of the plasma membrane, called the caveola (Roy et al., 1999; Sternberg and Schmid, 1999). In contrast, K-Ras is not palmitoylated and is anchored to the membrane through the basic domain near the C-terminus (Hancock et al., 1990). There is no evidence so far that K-Ras is concentrated in a certain subdomain of the plasma membrane. Based on this difference, it is tempting to speculate that K-Ras stimulates a distinct array of effector molecules and thereby elicits cellular responses unique to K-Ras. It is thus possible that OSM induces the localization of E-cadherin through K-Ras by activating such unique effector proteins that are not activated by H- or N-Ras.
Affinity precipitation of the active form of K-Ras revealed that OSM rapidly activated K-Ras in fetal hepatocytes (Figure 7). However, localization of E-cadherin required much longer stimulation by OSM. Therefore, additional component(s) may be necessary for the K-Ras signaling to E-cadherin-based AJ formation. To identify the components, we investigated downstream of the Ras signaling using specific inhibitors for PI3K and MEK1, which are known to mediate Ras signaling. However, these inhibitors did not affect E-cadherin-based AJ formation in fetal hepatocytes (data not shown). To fill the gap between K-Ras activation and E-cadherin localization will be an interesting subject.
Our assay system based on a primary culture of fetal hepatocytes and retrovirus-mediated gene transfer has many advantages in the study of the molecular mechanism of liver development. Most importantly, the system allows us to analyze liver development in an individual embryo, which makes it possible to analyze the functions of the genes essential for liver development, as demonstrated for K-Ras. Furthermore, complementation by retrovirus-mediated gene transfer of the defects caused by gene targeting enables us to examine the role of the disrupted gene in liver development as well as to find a potential downstream target that is involved in the normal signaling and cellular responses. Since the mechanism of cell adhesion has been analyzed mostly in established cell lines, our system provides a novel way to address this issue in a more physiological setting.
Materials and methods
Antibodies
Mouse monoclonal antibodies against E-cadherin, α-catenin, β-catenin, γ-catenin and STAT3 were purchased from Transduction Laboratories. Mouse monoclonal antibodies against Rac1, H-Ras and myc tag, and rat monoclonal antibody against pan-Ras were from Santa Cruz. Mouse monoclonal antibodies against K-Ras and N-Ras were from Oncogene Research Products. Mouse monoclonal antibody against Flag tag was from Sigma. Mouse monoclonal antibody against ZO-1 was kindly provided by Dr S.Tsukita.
In vitro fetal hepatocyte culture
Fetal livers were isolated from E14.5 embryos of C57BL/6CrSlc mice, K-Ras KO mice or H-Ras N-Ras double KO mice. Isolated livers were dissociated with a collagenase-based dissociation buffer (liver digest medium, Gibco-BRL) followed by hemolysis with a hypotonic buffer as described previously (Kamiya et al., 1999). Dissociated cells were inoculated at a density of 2 × 104 cells/cm2 in 0.1% gelatin-coated glass bottom dishes (MatTek Corporation) or plastic dishes (Costar), and then cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) (Gibco-BRL), 2 mM l-glutamine, minimal essential medium (MEM) non-essential amino acid solution (Gibco-BRL), insulin–transferrin–selenium X (Gibco-BRL) and 1 × 10–7 M Dex (Sigma). After 4 h of inoculation, the cells were washed extensively with phosphate-buffered saline (PBS) to remove hematopoietic cells and cell debris. Culture media were changed every 2 days.
Ultrastructural analysis
The cells cultured with Dex or OSM/Dex for 6 days were fixed with a cold 2% glutaraldehyde solution (pH 7.4, in phosphate buffer) for 2 h at 4°C, and post-fixed with a 2% osmium tetroxide solution for 1 h at 4°C. Then, the cells were embedded in epoxy resin after dehydration in a graded ethanol series. Longitudinally cut ultrathin sections of the cells were stained with uranyl acetate and lead citrate, and examined under a JEM-1200EX electron microscope (JEOL, Tokyo).
Immunofluorescence analysis
Fetal hepatocytes were cultured in 0.1% gelatin-coated glass bottom dishes for 6 days without OSM/Dex (no factor) or with OSM, Dex or OSM/Dex. Cells from each culture were fixed with 4% paraformaldehyde at 4°C for 10 min, and then permeabilized with 0.2% Triton X-100 in PBS at room temperature for 15 min. Cells were incubated with mouse monoclonal antibodies against E-cadherin, β-catenin or ZO-1 at 4°C for 8 h. After incubation, cells were washed with 0.05% Triton X-100–PBS twice and incubated with phycoerythrin (PE)-, fluorescein isothiocyanate (FITC)-labeled anti-mouse IgG (Leinco Technologies, Inc.) at room temperature for 2 h. When actin filaments were stained, cells were incubated with phalloidin conjugated with rhodamine (Molecular Probes) at room temperature for 30 min. Immunofluorescence of antigens was analyzed by confocal microscopy (Leica TCS 4D).
Western blotting and detergent solubility assay
Cultured cells were lysed by cell lysis buffer (25 mM HEPES-KOH pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, leupeptin, Pefa-block, 0.2% SDS, 1% Triton X-100 and 0.5% NP-40). A 10 µg aliquot of total cell lysate was loaded for SDS–PAGE, and then transferred to an immobilon-P membrane (Millipore). The membrane was incubated with primary antibodies at room temperature for 2 h at least. It was then washed with TBS-T (10 mM Tris–HCl pH 8.0, 150 mM NaCl and 0.1% Tween-20) and incubated with anti-mouse or rat IgG conjugated with horseradish peroxidase (Amersham-Pharmacia) at room temperature for 1 h. The immune complex was visualized with the ECL system (Amersham-Pharmacia).
The detergent (NP-40) solubility assay was performed with several modifications (Potempa and Ridley, 1998). The cultured cells were lysed by NP-40 buffer (25 mM HEPES-KOH pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, leupeptin, Pefa-block and 0.5% NP-40), and centrifuged at 10 000 g for 30 min at 4°C. The supernatant was termed the soluble fraction (S). The pellet was washed with NP-40 buffer twice and resuspended in 1× Laemmli sample buffer using a vortex mixer and passing through a 27 gauge needle. The suspensions were then boiled for 10 min and centrifuged at 10 000 g for 10 min at 4°C. The supernatants were termed the insoluble fraction (I). An equal amount of each fraction was subjected to SDS–PAGE followed by western blotting with monoclonal antibody against E-cadherin. The amounts of E-cadherin in the [S], [I] and [S + I] fractions in each culture condition were estimated by quantitative analysis using the NIH Image program.
Vector construction and retroviral infection
A retrovirus vector (pMX/IRES-GFP: pMIG) carrying the internal ribosomal entry site (IRES) sequence followed by green fluorescent protein (GFP) cDNA was utilized for expression (Onishi et al., 1996). pMIG/ΔSTAT3 and RasN17 were constructed previously (Chida et al., 1999). The cDNAs encoding the dominant-negative Rho (RhoN19 tagged with myc), Rac (RacN17), Cdc42 (Cdc42N17 tagged with Flag) and wild-type H-Ras, K-Ras (a gift from Drs Y.Takai, H.Koide, T.Satoh and Y.Kaziro) or N-Ras (HSRRB) were inserted into the multicloning site of pMIG. The integrity of these vectors was determined by sequencing and by digestion with restriction enzymes.
The retroviral vectors were transfected into the retrovirus packaging cell line (BOSC23 cells) by lipofection as described previously (Kitamura et al., 1995; Ito et al., 2000). After 48 h of culture, the virus particles were enriched by centrifugation at 6000 g for 16 h at 4°C. The pellets of virus particles were resuspended in culture medium for fetal hepatocytes and the suspension filtered through a 0.45 µm filter. Fetal hepatocytes were incubated with the viral solution for 2 days and then the medium was renewed. Ectopic expression of all genes in fetal hepatocytes was determined by western blotting using specific antibodies.
Luciferase assay
NIH-3T3 cells, which were infected with retrovirus vectors carrying mutants of Rho family constructs, were transiently transfected with the luciferase reporter gene linked to the c-fos promoter using Lipofectamine plus (Gibco-BRL). As an internal control, pRL plasmid containing the Renulla luciferase gene was co-transfected. On the following day, cells were stimulated with 2% serum for 24 h. Cells were then lysed with passive lysis buffer (Promega), and luciferase activity was measured according to the technical manual for the Dual Luciferase Reporter Assay System (Promega).
Northern blotting
Total RNAs from E14.5 fetal livers and cultured cells were extracted with Trozol solution (Gibco-BRL). A 10 µg aliquot of total RNA was separated on a 1.2% agarose gel containing 2% formaldehyde and transferred onto a positive-charged nylon membrane (Boehringer-Mannheim). After UV cross-linking, the membrane was hybridized with digoxigenin (DIG)-labeled cDNA probes [AFP, TAT, G6Pase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)]. The blot was treated with anti-DIG antibody conjugated with alkaline phosphatase and then developed with CDP-star (NEB) according to the manufacturer’s instructions.
K-Ras activity assay
Fetal hepatocytes were serum-starved for 20 h and then stimulated with 10 ng/ml of OSM for 15 or 30 min. Cells were washed with cold-PBS and lysed in buffer containing 50 mM Tris–HCl pH 7.4, 200 mM NaCl, 2 mM MgCl2, 1% NP-40, 10% glycerol, 2 mM Na3VO4, leupeptin and Pefa-block. Cell lysates (1 mg) were mixed with 20 µg of GST–RBD of Raf1 pre-bound to glutathione–Sepharose 4B. Following a 2 h incubation at 4°C, the beads were collected and washed with lysis buffer. Each sample was subjected to SDS–PAGE followed by western blotting with a K-Ras-specific monoclonal antibody.
Acknowledgments
Acknowledgements
We are grateful to Drs Y.Takai, H.Koide, T.Satoh, and Y.Kaziro for providing us with the plasmids, Dr S.Tsukita for antibody, H.Saito for technical assistance, and Drs. S.Sawai, M.Tanaka and K.Sekine for helpful discussion. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a research grant from Core Research for Evolutionary Science and Technology (CREST).
References
- Aicher B., Lerch,M.M., Muller,T., Schilling,J. and Ullrich,A. (1997) Cellular redistribution of protein tyrosine phosphatases LAR and PTPσ by inducible proteolytic processing. J. Cell Biol., 138, 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts A.S., Geneste,O. and Treisman,R. (1998) Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell, 92, 475–487. [DOI] [PubMed] [Google Scholar]
- Angres B., Barth,A. and Nelson,W.J. (1996) Mechanism for transition from initial to stable cell–cell adhesion: kinetic analysis of E-cadherin-mediated adhesion using a quantitative adhesion assay. J. Cell Biol., 134, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga V.M., Machesky,L.M., Hall,A. and Hotchin,N.A. (1997) The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell–cell contacts. J. Cell Biol., 137, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chida D., Miura,O., Yoshimura,A. and Miyajima,A. (1999) Role of cytokine signaling molecules in erythroid differentiation of mouse fetal liver hematopoietic cells: functional analysis of signaling molecules by retrovirus-mediated expression. Blood, 93, 1567–1578. [PubMed] [Google Scholar]
- Conlon F.L., Lyons,K.M., Takaesu,N., Barth,K.S., Kispert,A., Herrmann,B. and Robertson,E.J. (1994) A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development, 120, 1919–1928. [DOI] [PubMed] [Google Scholar]
- Drubin D.G. and Nelson,W.J. (1996) Origins of cell polarity. Cell, 84, 335–344. [DOI] [PubMed] [Google Scholar]
- Fukada T., Hibi,M., Yamanaka,Y., Takahashi-Tezuka,M., Fujitani,Y., Yamaguchi,T., Nakajima,K. and Hirano,T. (1996) Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity, 5, 449–460. [DOI] [PubMed] [Google Scholar]
- Gualdi R., Bossard,P., Zheng,M., Hamada,Y., Coleman,J.R. and Zaret,K.S. (1996) Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev., 10, 1670–1682. [DOI] [PubMed] [Google Scholar]
- Gumbiner B.M. (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell, 84, 345–357. [DOI] [PubMed] [Google Scholar]
- Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science, 279, 509–514. [DOI] [PubMed] [Google Scholar]
- Hancock J.F., Paterson,H. and Marshall,C.J. (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell, 63, 133–139. [DOI] [PubMed] [Google Scholar]
- Hirano T., Nakajima,K. and Hibi,M. (1997) Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev., 8, 241–252. [DOI] [PubMed] [Google Scholar]
- Ito Y., Matsui,T., Kamiya,A., Kinoshita,T. and Miyajima,A. (2000) Retroviral gene transfer of signaling molecules into murine fetal hepatocytes defines distinct roles for the STAT3 and ras pathways during hepatic development. Hepatology, 32, 1370–1376. [DOI] [PubMed] [Google Scholar]
- Johnson L. et al. (1997) K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev., 11, 2468–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya A. et al. (1999) Fetal liver development requires a paracrine action of oncostatin M through the gp130 signal transducer. EMBO J., 18, 2127–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.C. and Kim,J.H. (1998) Exogenous C2-ceramide activates c-fos serum response element via Rac-dependent signalling pathway. Biochem. J., 330, 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T., Onishi,M., Kinoshita,S., Shibuya,A., Miyajima,A. and Nolan,G.P. (1995) Efficient screening of retroviral cDNA expression libraries. Proc. Natl Acad. Sci. USA, 92, 9146–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koera K., Nakamura,K., Nakao,K., Miyoshi,J., Toyoshima,K., Hatta,T., Otani,H., Aiba,A. and Katsuki,M. (1997) K-ras is essential for the development of the mouse embryo. Oncogene, 15, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Kojima N., Kinoshita,T., Kamiya,A., Nakamura,K., Nakashima,K., Taga,T. and Miyajima,A. (2000) Cell density-dependent regulation of hepatic development by a gp130-independent pathway. Biochem. Biophys. Res. Commun., 277, 152–158. [DOI] [PubMed] [Google Scholar]
- Kuroda S., Fukata,M., Fujii,K., Nakamura,T., Izawa,I. and Kaibuchi,K. (1997) Regulation of cell–cell adhesion of MDCK cells by Cdc42 and Rac1 small GTPases. Biochem. Biophys. Res. Commun., 240, 430–435. [DOI] [PubMed] [Google Scholar]
- Kypta R.M., Su,H. and Reichardt,L.F. (1996) Association between a transmembrane protein tyrosine phosphatase and the cadherin–catenin complex. J. Cell Biol., 134, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg R.A., Juan,T.S., Welcher,A.A., Sun,Y., Cupples,R., Guthrie,B. and Fletcher,F.A. (1998) Cloning and characterization of a specific receptor for mouse oncostatin M. Mol. Cell. Biol., 18, 3357–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee T. and Marshall,C. (1999) New insights into the interaction of Ras with the plasma membrane. Cell, 98, 9–12. [DOI] [PubMed] [Google Scholar]
- Medvinsky A. and Dzierzak E. (1996) Definitive hematopoiesis is autonomously initiated by the AGM region. Cell, 86, 897–906. [DOI] [PubMed] [Google Scholar]
- Mukouyama Y. et al. (1998) In vitro expansion of murine multipotential hematopoietic progenitors from the embryonic aorta–gonad– mesonephros region. Immunity, 8, 105–114. [DOI] [PubMed] [Google Scholar]
- Nagafuchi A. and Takeichi,M. (1988) Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J., 7, 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A., Ishihara,S. and Tsukita,S. (1994) The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin–α catenin fusion molecules. J. Cell Biol., 127, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose A., Nagafuchi,A. and Takeichi,M. (1988) Expressed recombinant cadherins mediate cell sorting in model systems. Cell, 54, 993–1001. [DOI] [PubMed] [Google Scholar]
- Onishi M. et al. (1996) Applications of retrovirus-mediated expression cloning. Exp. Hematol., 24, 324–329. [PubMed] [Google Scholar]
- Ozawa M. and Kemler,R. (1998) Altered cell adhesion activity by pervanadate due to the dissociation of α-catenin from the E-cadherin·catenin complex. J. Biol. Chem., 273, 6166–6170. [DOI] [PubMed] [Google Scholar]
- Potempa S. and Ridley,A.J. (1998) Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol. Biol. Cell, 9, 2185–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato R., Veltmaat,J.M., Groffen,J. and Heisterkamp,N. (1998) Involvement of the tyrosine kinase fer in cell adhesion. Mol. Cell. Biol., 18, 5762–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura S., Miravet,S., Piedra,J., Garcia de Herreros,A. and Dunach,M. (1999) Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J. Biol. Chem., 274, 36734–3640. [DOI] [PubMed] [Google Scholar]
- Roy S., Luetterforst,R., Harding,A., Apolloni,A., Etheridge,M., Stang,E., Rolls,B., Hancock,J.F. and Parton,R.G. (1999) Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nature Cell Biol., 1, 98–105. [DOI] [PubMed] [Google Scholar]
- Shiojiri N., Lemire,J.M. and Fausto,N. (1991) Cell lineages and oval cell progenitors in rat liver development. Cancer Res., 51, 2611–2620. [PubMed] [Google Scholar]
- Stamatoglou S.C., Enrich,C., Manson,M.M. and Hughes,R.C. (1992) Temporal changes in the expression and distribution of adhesion molecules during liver development and regeneration. J. Cell Biol., 116, 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg P.W. and Schmid,S.L. (1999) Caveolin, cholesterol and Ras signalling. Nature Cell Biol., 1, E35–E37. [DOI] [PubMed] [Google Scholar]
- Stevenson B.R., Siliciano,J.D., Mooseker,M.S. and Goodenough,D.A. (1986) Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol., 103, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taga T. and Kishimoto,T. (1997) Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol., 15, 797–819. [DOI] [PubMed] [Google Scholar]
- Takaishi K., Sasaki,T., Kotani,H., Nishioka,H. and Takai,Y. (1997) Regulation of cell–cell adhesion by rac and rho small G proteins in MDCK cells. J. Cell Biol., 139, 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. (1988) The cadherins: cell–cell adhesion molecules controlling animal morphogenesis. Development, 102, 639–655. [DOI] [PubMed] [Google Scholar]
- Takeichi M. (1995) Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol., 7, 619–627. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Hara,T., Copeland,N.G., Gilbert,D.J., Jenkins,N.A. and Miyajima,A. (1999) Functional reconstitution of the mouse oncostatin M receptor: molecular cloning of the mouse OSM receptor β subunit. Blood, 93, 804–815. [PubMed] [Google Scholar]
- Umanoff H., Edelmann,W., Pellicer,A. and Kucherlapati,R. (1995) The murine N-ras gene is not essential for growth and development. Proc. Natl Acad. Sci. USA, 92, 1709–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Falasca,M., Schlessinger,J., Malstrom,S., Tsichlis,P., Settleman,J., Hu,W., Lim,B. and Prywes,R. (1998) Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ., 9, 513–522. [PubMed] [Google Scholar]
- Winnier G., Blessing,M., Labosky,P.A. and Hogan,B.L. (1995) Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev., 9, 2105–2116. [DOI] [PubMed] [Google Scholar]