

Abstract
The AT-rich interaction domain (ARID) is a DNA-binding module found in many eukaryotic transcription factors. Using NMR spectroscopy, we have determined the first ever three-dimensional structure of an ARID–DNA complex (mol. wt 25.7 kDa) formed by Dead ringer from Drosophila melanogaster. ARIDs recognize DNA through a novel mechanism involving major groove immobilization of a large loop that connects the helices of a non-canonical helix–turn–helix motif, and through a concomitant structural rearrangement that produces stabilizing contacts from a β-hairpin. Dead ringer’s preference for AT-rich DNA originates from three positions within the ARID fold that form energetically significant contacts to an adenine–thymine base step. Amino acids that dictate binding specificity are not highly conserved, suggesting that ARIDs will bind to a range of nucleotide sequences. Extended ARIDs, found in several sequence-specific transcription factors, are distinguished by the presence of a C-terminal helix that may increase their intrinsic affinity for DNA. The prevalence of serine amino acids at all specificity determining positions suggests that ARIDs within SWI/SNF-related complexes will interact with DNA non-sequence specifically.
Keywords: ARID/AT-rich interaction domains/Dead ringer/DNA recognition/structure/NMR
Introduction
The AT-rich interaction domain (ARID) is a novel DNA-binding module found in a large number of eukaryotic transcription factors that regulate cell proliferation, differentiation and development (Kortschak et al., 2000). It adopts a novel protein fold (Yuan et al., 1998; Iwahara and Clubb, 1999b) that is found in a variety of important DNA-binding proteins, including: Bright, a regulator of IgH transcription in mature B cells (Herrscher et al., 1995); Dead ringer, involved in embryogenesis (Gregory et al., 1996); Mrf-1 and Mrf-2, transcriptional modulators of the cytomegalovirus major intermediate-early promotor (Huang et al., 1996); and several other human proteins implicated in the regulation of transcription, such as DRIL1 (Kortschak et al., 1998), Jumonji (Bergelefranc et al., 1996), RPB1, RBP2 (Fattaey et al., 1993), SMCx (Agulnik et al., 1994b) and SMCy (Agulnik et al., 1994a). ARIDs are also components of SWI/SNF-related chromatin remodeling complexes involved in the global control of transcription, where they may function in gene-specific targeting (Collins et al., 1999; Vazquez et al., 1999; Nie et al., 2000). Based on primary sequence homology, they can be partitioned into at least two subfamilies: (i) minimal ARIDs, which contain an ∼80 amino acid conserved core region; and (ii) extended ARIDs, which consist of the core region and additional highly conserved amino acids at their polypeptide termini. Minimal ARIDs are distributed in all eukaryotes, while extended ARIDs are restricted to metazoans; they are most prevalent in humans, which have 11 ARID-containing proteins (Venter et al., 2001).
Dead ringer is an essential ARID-containing transcription factor involved in anterior–posterior patterning and muscle development in the Drosophila embryo. Genetic analysis indicates that it plays a critical role in segmentation, head and cephalic furrow formation by altering the expression levels of several key developmental genes, including engrailed, wingless, even-skipped, argos and buttonhead (Shandala et al., 1999). Its biochemical function in dorsal–ventral axis formation has been studied extensively, where it acts in concert with the morphogen dorsal to repress transcription of the dorsal ectoderm determining gene zerknüllt (zen). Expression of zen is controlled by an upstream silencer called the ventral repression region (VRR) (Doyle et al., 1989; Ip et al., 1991), which harbors two Dead ringer binding sites containing the nucleotide sequence TATTGAT (sites AT2 and AT3). Dead ringer binds to these sites through its extended ARID, which, in conjunction with Dorsal binding to an adjacent site, facilitates the co-operative recruitment of the global co-repressor groucho (Valentine et al., 1998). Transcription is then presumably repressed through the Sin3–Rpd3 deacetylase complex, which is recruited to the VRR by groucho interactions with Rpd3 (Chen et al., 1999). A second protein, Cut, also represses transcription from the VRR by binding to the AT2 and AT3 sites (Ip et al., 1991). However, Cut appears to repress transcription through a groucho-independent mech anism (Valentine et al., 1998). Although Cut and Dead ringer recognize the same binding site within the VRR, Cut does not contain an ARID and binds DNA through its ‘Cut repeat’ and homeodomain-binding motifs.
ARID-containing proteins exhibit a varied degree of specificity for DNA and may interact with the minor groove. The binding specificity of the minimal ARID-containing protein Mrf-2 (Whitson et al., 1999), and the extended ARID-containing proteins Bright (Herrscher et al., 1995; Kaplan et al., 2001) and Dead ringer (Gregory et al., 1996; Valentine et al., 1998; Iwahara and Clubb, 1999b) have been determined. These proteins preferentially interact with the nucleotide sequence ATT embedded within a larger AT-rich site. However, several minimal ARIDs have been shown to bind DNA in a non-sequence-specific manner (Collins et al., 1999; Dallas et al., 2000) or to sites that are not rich in AT nucleotides (Nie et al., 2000), presumably through both major and minor groove interactions (Herrscher et al., 1995; Whitson et al., 1999). NMR spectroscopy has been employed to determine the three-dimensional (3D) structures of the minimal and extended ARIDs within the Mrf-2 [Yuan et al., 1998; revised structure, Protein Data Bank (PDB) accession code 1IG6] and Dead ringer proteins (Iwahara and Clubb, 1999b), respectively. This work revealed a conserved and structurally novel DNA-binding fold comprised of six α-helices and a short β-hairpin, with the extended ARIDs decorated with additional α-helices at their N- and C-termini. However, the molecular basis of DNA recognition by this highly abundant family of eukaryotic transcription factors remains unknown, since these structures were solved in the absence of DNA. Here we report the 3D solution structure of the complex between the extended ARID from the Dead ringer protein and its 15 bp DNA-binding site. ARIDs recognize DNA through a novel mechanism involving major groove immobilization of a large loop that connects the helices of a non-canonical helix–turn–helix (HTH) motif, and through a concomitant structural rearrangement that produces stabilizing contacts from a β-hairpin. The biological implications of the structure with regard to the functions of other ARID-containing proteins are discussed.
Results and discussion
NMR structure determination of the Dead ringer–DNA complex
In order to understand how ARIDs recognize DNA, we determined the solution structure of the complex between the extended ARID of the Dead ringer protein (DRI- DBDF355L, amino acids Gly262–Gly398 of Dead ringer with leucine substituted for phenylalanine at position 355) and its cognate DNA-binding site (DRI-DBDF355L–DNA, mol. wt 26 kDa). The nucleotide sequence of the DNA in the complex contains the naturally occurring Dead ringer binding site within the VRR (the boxed sequence in Figure 1A). The DRI-DBDF355L mutant was used in the structure determination because it exhibits substantially improved NMR spectra compared with the wild-type complex. As described previously, several proton resonances proximal to the side chain of Phe355 in the wild-type complex are broadened because of chemical exchange. The replacement of this aromatic side chain with leucine narrows these line widths, presumably by reducing the degree of conformational exchange and/or by reducing the chemical shift differences between the exchanging sub-states (Iwahara et al., 2001b). The improved spectral properties of the DRI-DBDF355L–DNA complex were required for solving its high-resolution structure, since it enabled several resonances within the interface to be unambiguously assigned. The quality of the DRI-DBDF355L–DNA NMR data is demonstrated in Figure 1B, which displays selected portions of the 3D [F1] 13C,15N-filtered, [F2] 13C-edited 3D NOESY spectrum of the complex formed by 13C,15N isotopically enriched protein and unlabeled DNA (Zwahlen et al., 1997). These data indicate that the side chains of Ile350, Thr351, Ser352 and Leu355 are proximal to the major groove of the duplex.

Fig. 1. (A) Nucleotide sequence of the 15 bp DNA fragment used in the NMR structure determination of the Dead ringer ARID–DNA complex. The boxed region corresponds to the Dead ringer binding site within the VRR (Valentine et al., 1998). The nucleotides are labeled as in the 3D structure of the complex. (B) Panels from the 3D [F1] 13C,15N-filtered, [F2] 13C-edited 3D NOESY spectrum of the 13C,15N DRI-DBDF355L–DNA complex. Cross-peaks correspond to intermolecular protein–DNA NOEs. The identity of the amino acid is indicated at the top and the identity of the DNA proton is indicated next to each cross-peak. This figure displays only a limited number of the 80 intermolecular NOEs used to calculate the 3D structure of the complex. The peak at 4.63 p.p.m. corresponds to the NMR signal of H2O. (C) EMSA of the wild-type and mutant DRI-DBDF355L proteins. Lane 1, 32P-labeled cognate DNA; lanes 2 and 3, 32P-labeled cognate DNA and 1 µM of wild-type DRI-DBD in the presence of 0.5 and 2.5 µM of unlabeled non-cognate DNA, respectively. Lanes 4–6 are identical to lanes 1–3 except that the DRI-DBDF355L protein is substituted for the wild-type protein. The concentration of the 32P-labeled DNA duplex was 50 pM and contained the nucleotide sequence 5′-TGCGGATCCTGTATTGATGTGGCTGCAGTT-3′, where the underlined sequence corresponds to the duplex used in the structure determination.
We have previously shown that the NMR chemical shifts of the DRI-DBDF355L–DNA and wild-type DRI-DBD–DNA complexes are essentially identical, and that each exhibits a similar pattern of intra- and intermolecular nuclear Overhauser effects (NOEs; Iwahara et al., 2001b), indicating a similar mode of interaction with the duplex. In order to verify that the mutation does not disrupt the ability of the ARID to sequence-specifically recognize its cognate site, we performed an electrophoresis mobility-shift assay (EMSA) of the wild-type and mutant DRI-DBDF355L proteins (Figure 1C). In the presence of a 15 bp DNA fragment containing the Dead ringer binding site, both proteins form similarly shifted complexes that persist when challenged with unlabeled non-cognate DNA (Figure 1C, compare lanes 2 and 3, and 5 and 6). Therefore, the improved spectral properties of the DNA complex of the DRI-DBDF355L protein and its similar mode of sequence-specific binding make it an effective surrogate in which to study DNA recognition by ARIDs.
Structure of the ARID–DNA complex
The solution structure of the DRI-DBDF355L–DNA complex was calculated using simulated annealing methods (Figure 2A and B). A total of 3402 experimentally derived restraints were employed [2455 NOEs, 395 dihedral angles, 192 hydrogen bonds, 117 13Cα/13Cβ chemical shifts, 41 3JHNα and 23 3JPH3′ scalar couplings, 94 residual dipolar couplings measured from a protein–DNA complex partially aligned in Pf1 bacteriophage (Hansen et al., 1998) and 85 backbone 15N T1/T2 ratios (Tjandra et al., 1997)]. The co-ordinate precision of the ensemble of 20 conformers to the mean structure is 0.70 ± 0.09 Å for all heavy atoms (residues Phe265–Arg399 and bp 2–14 of the duplex) and 0.46 ± 0.13 Å for the DRI-DBDF355L backbone and all DNA heavy atoms. All of the structures exhibit good covalent geometry and have no NOE, dihedral angle, scalar or residual dipolar coupling violations >0.5Å, 5°, 2 Hz or 2 Hz, respectively. A summary of the structural and restraint statistics is presented in Table I.

Fig. 2. (A) A schematic representation of the 3D solution structure of the DRI-DBDF355L–DNA complex. Amino acids Phe265–Glu391 of the DRI-DBDF355L are shown. The protein is colored blue with the exception of the DNA contacting β-hairpin (colored green, amino acids Ile307–Val312). The DNA of the complex is formed by nucleotides Thy3–Gua14 and Cyt17–Ade28 (colored red). (B) Ensemble of 20 NMR structures of the Dead ringer DRI-DBDF355L–DNA complex shown in the same orientation as in (A). The ensemble was obtained by superimposing the backbone heavy atoms of residues Trp263–Glu391 and the heavy atoms of Thy3–Thy13 and Ade18–Ade28. The protein and DNA molecules are colored blue and red, respectively. (C) Solvent accessible surface area of the DRI-DBDF355L protein in the complex color coded by surface charge (blue and red correspond to basic and acid regions, respectively). The heavy atoms of the bound DNA molecule are colored yellow, and two orientations of the complex are displayed. (A) and (C) show the lowest energy conformer.
Table I. Structural statistics of 20 final structures and the energy-minimized average structurea.
| <SA> | (SA)best | |
|---|---|---|
| R.m.s.ds from NOE restraints (Å)b | ||
| Protein | ||
| intra-residue (561) | 0.033 ± 0.003 | 0.031 |
| sequential (|i – j| = 1) (495) | 0.048 ± 0.003 | 0.043 |
| medium range (|i – j| < 4) (450) | 0.056 ± 0.003 | 0.057 |
| long range (|i – j| ≥ 5) (468) | 0.063 ± 0.003 | 0.064 |
| DNA | ||
| intra-residue (173) | 0.069 ± 0.002 | 0.069 |
| sequential (199) | 0.026 ± 0.002 | 0.025 |
| inter-strand (29) | 0.011 ± 0.002 | 0.011 |
| protein–DNA (80) | 0.052 ± 0.007 | 0.046 |
| R.m.s.ds from hydrogen bonding restraints (Å)c | ||
| protein (102) | 0.018 ± 0.002 | 0.019 |
| DNA (90) | 0.010 ± 0.001 | 0.010 |
| R.m.s.ds from dihedral angles restraints (°) | ||
| protein (329) | 0.348 ± 0.037 | 0.365 |
| DNA (208)d | 0.043 ± 0.031 | 0.060 |
| R.m.s.ds from residual dipolar coupling restraints (Hz) | ||
| 1DNH (protein backbone) (90) | 0.77 ± 0.03 | 0.70 |
| 1DNεHε (Arg) (4) | 0.34 ± 0.32 | 0.14 |
| R.m.s.ds from 15N T1/T2 restraints | ||
| protein backbone amide (85) | 0.81 ± 0.01 | 0.80 |
| R.m.s.ds from 13C chemical shift restraints (p.p.m.) | ||
| 13Cα (117) | 1.14 ± 0.02 | 1.11 |
| 13Cβ (117) | 0.94 ± 0.02 | 0.92 |
| R.m.s.ds form 3J coupling restraints (Hz) | ||
| 3JHNHα (41) | 0.52 ± 0.02 | 0.47 |
| 3JPH3′ (23) | 1.00 ± 0.03 | 1.05 |
| Deviation from idealized covalent geometry | ||
| bonds (Å) | 0.003 ± 0.000 | 0.003 |
| angles (°) | 0.686 ± 0.005 | 0.681 |
| impropers (°) | 0.470 ± 0.015 | 0.465 |
| ELennard–Johns (kcal/mol)e | –1023 ± 13 | –1019 |
| PROCHECK results (%)f | ||
| most favorable region | 87.7 ± 1.4 | 86.4 |
| additionally allowed region | 12.2 ± 1.3 | 13.6 |
| generously allowed region | 0.0 ± 0.0 | 0.0 |
| disallowed region | 0.1 ± 0.4 | 0.0 |
| Co-ordinate precision (Å)g | ||
| protein backbone + DNA heavy atoms | 0.46 ± 0.13 | |
| protein heavy atoms + DNA heavy atoms | 0.70 ± 0.09 |
aThe notation of the NMR structures is as follows: <SA> are the final 20 simulated annealing structures; (SA)best is the lowest energy structure within the ensemble. The number of terms for each restraint is given in parentheses.
bNone of the structures exhibited distance violations >0.5 Å, dihedral angle violations >5°, coupling constant violations >2 Hz or residual dipolar coupling violations >2 Hz.
cTwo distance restraints were employed for each hydrogen bond. Hydrogen bond restraints for bp 2–17 in the DNA were used to maintain Watson–Crick base pairing.
dOne hundred and forty-two loose DNA dihedral angle restraints (α, β, γ and ε) consistent with A and B form DNA. Sixty-six dihedral angle restraints maintain S-type sugar puckering (ν1, ν2 and δ).
eValues of Lennard–Johns energy were calculated with 10 Å cut-off and constant dielectric. Interactions between bonded atoms and atoms that are bonded to a common third atom are excluded.
fLaskowski et al. (1993).
gThe co-ordinate precision is defined as the average atomic r.m.s.d. of the 20 individual SA structures and their mean co-ordinates. These values are for Dead ringer residues Phe265–Arg390 and the central 13 bp region of the DNA.
The ARID binds DNA as a monomer, recognizing the duplex through insertion of a loop and an α-helix into the major groove, and by extensive non-specific anchoring contacts to the adjacent sugar-phosphate backbone (Figure 2A). A large positively charged interaction surface envelops the duplex, burying ∼2100 Å2 of solvent accessible surface area (Figure 2C). The opposite face of the protein is comprised of acidic side chains, giving rise to a surface charge imbalance that may facilitate recognition of the negatively charged duplex. The structure of the DNA fragment in the complex is similar to B-form; the heavy atom root mean square difference (r.m.s.d.) between bp 3–13 and B-form DNA is ∼1.6 Å, and the duplex is only modestly bent, by ∼21°. The extended ARID is formed by eight α-helices (helix H1, Phe265–Glu277; H2, Pro282–Arg298; H3, Leu315–Ala324; H4, Leu328– Lys334; H5, Trp337–Gly343; H6, Ala353–Tyr364; H7, Tyr366–Lys372; H8, Pro378–Asn388) and a short β-hairpin (β-strand B1, Ile307–Met308; B2, Ser311–Val312). The protein can be envisioned as consisting of two subdomains. A C-terminal helical subdomain is formed by helices H3–H8, and interacts with the phosphodiester backbone and the major and minor grooves. An N-terminal subdomain (helices H1, H2 and the short β-hairpin) rotates upon DNA binding to form stabilizing interactions with the phosphodiester backbone of the duplex. The structure is well characterized by the NMR data, enabling numerous protein–DNA interactions to be unambiguously identified (summarized in Figure 3). A total of 16 amino acid side chains contact DNA in the complex: Arg304, Ile307, Ala309 and Lys310 from the β-hairpin region; Lys335, Gln338, Ser349, Ile350, Thr351, Ser352, Leu355, Thr356 and Arg358 within helices H5 and H6, and the intervening turn; and Asn388, Arg389 and Arg390 within the C-terminal tail.

Fig. 3. Summary of intermolecular contacts observed in the structure of the DRI-DBDF355L–DNA complex. Arrows to the bases and riboses indicate intermolecular hydrogen bonds, with the position of arrowhead denoting the acceptor. Arrows to phosphates indicate salt-bridge interactions and solid lines indicate van der Waals interactions. An interaction was scored as a salt bridge when the heavy distance of oppositely charged atoms was <4.5 Å. An interaction was classified as a hydrogen bond when the H - - - Xacceptor distance was <3.0 Å, and the angle between the Xdonor - H - - - Xacceptor was <60°. All interactions reported in the figure occur in at least 35% of the conformers. The dotted line denotes a potential water mediated indirect hydrogen bond between Thr356 and Gua9. The gap between Thr357 and Gua9 (Oγ1–O6 and Oγ1–N7 distances are 6.5 ± 0.1 Å and 6.4 ± 0.1 Å, respectively) is suitable in size to accommodate a bound water molecule (Schneider and Berman, 1995) and presumably serves as a bridge between the protein and DNA.
The AT-rich site is recognized by a large loop connecting helices H5 and H6 (the H5/H6 loop, residues Leu344–Ser352), which comprises the ‘turn’ of a non-canonical HTH motif that forms a network of base-specific hydrogen bonds to the floor of the major groove (Figure 4A). These interactions are stabilized by base-specific hydrophobic interactions between the side chains of Thr351 (H5/H6 loop), Leu355 (helix H6) and Thr356 (helix H6), which pack against the methyl groups of Thy21, Thy11 and Thy8, respectively. Additional van der Waals interactions between Gua9 and the side chain of Ile350 (H5/H6 loop) further stabilize the interface, and position the side chains of Thr351 and Ser352 above the Ade10–Thy11 base step for intermolecular hydrogen bonding. Contacts to Ade20 play a central role in recognition, since in 100% and 85% of the conformers, the hydroxyl groups of Thr351 and Ser352, respectively, accept a hydrogen bond from its N6 amine. Supplementing this predominant interaction are less frequently observed interfacial direct hydrogen bonds between the hydroxyl of Thr351 and the N6 amine (in 40% of the conformers) and N7 atom (in 45% of the conformers) of Ade10. The adjacently positioned side chain of Ser352 also donates a hydrogen bond to the O4 group of Thy21 (in 35% of the conformers) or accepts a hydrogen bond from the exocyclic N6 amine of Ade20 (in 40% of the conformers). It is important to emphasize that the heavy atom co-ordinates of the H5/H6 loop–DNA interface are precisely defined by the NMR data (Figure 2). Thus, the array of intermolecular hydrogen bonds from Thr351 and Ser352 observed in the ensemble results from the inability to define the co-ordinates of their hydroxyl protons, which are in rapid exchange with the bulk solvent. Since the H5/H6 loop–DNA interaction surface is significantly hydrated, it appears likely that all of these direct hydrogen bonds will be sampled in solution, and that these contacts will be supplemented by indirect water-mediated interfacial hydrogen bonds. The plasticity of this molecular interface is further emphasized by the retention of an elevated degree of mobility within the protein–DNA interface (Figure 5A) and by the positioning of Thr356 within helix H6, which is poised to form an indirect water-mediated hydrogen bond to the base of Gua9 (Figure 4A).

Fig. 4. (A) An expanded view of the major groove interface. The side chains of heavy atoms of Ade20–Cyt22 and Gua9–Thy11 are shown (colored black). The ensemble of backbone atoms of residues Ser349–Leu357 (colored light blue) and the side chains of DNA contacting amino acids (green) are shown. Hydrogen bonding interactions that are prevalent in the ensemble of conformers are represented as dotted lines. (B) Expanded view of the DNA interface formed by the H5/H6 loop and β-hairpin structures. The solvent accessible surface of the DNA is colored dark blue, except for the oxygen atoms of the phosphate groups, which are colored light blue. (C) The DNA interface formed by amino acids within helices H4, H6 and H8. The coloring scheme is identical to (B). All figures were generated using the lowest energy structure within the ensemble of conformers.

Fig. 5. DNA-dependent changes in the structure and backbone dynamics. (A) Plot of the (1H), 15N NOE values of the backbone amide nitrogen atoms of the DRI-DBDF355L protein in the presence (blue) and absence (red) of DNA. Regions of regular secondary structure are indicated above the plot. (B) Overlay of the backbone structures of the extended ARID in the DRI-DBDF355L–DNA complex (blue) and in the absence of DNA (Iwahara and Clubb, 1999b). The overlay was constructed by superimposing helices H3–H8 of each structure (r.m.s.d. for the backbone atoms of residues 315–388 is 1.5 Å). For clarity, helices H1 and H2 are represented as cylinders and the β-hairpin is represented with arrows. DNA binding causes a structural rearrangement in the DRI-DBDF355L protein that causes the hairpin to be displaced by ∼10 Å towards the C-terminal subdomain (for example, the Cα atoms of the DNA contacting residues Ala309 and Lys310 in the energy-minimized structures of the free and bound forms of the protein are shifted 10.2 and 12.5 Å, respectively). DNA binding also causes a large rotation of the two subdomains with respect to one another; in the energy-minimized structures, helices H1 and H2 of the N-terminal subdomain are shifted by 30.8 and 17.4° relative to the C-terminal subdomain upon binding DNA, respectively.
A pocket formed by helices H4–H6 and H8 stabilizes the major groove interface and facilitates minor groove interactions from the C-terminal tail of the protein (Figure 4B). The phosphodiester backbone on the 3′ side of the major groove (from Ade18 to Thy21) is inserted into this pocket and stabilized by electrostatic and hydrogen bonding interactions. In particular, the side chains of Gln338 and Lys342 protrude from helix H5 to interact with the phosphate group of Ade18, while the side chain of Lys335 forms a salt bridge to the phosphate of Cyt19 in the majority of conformers. Interestingly, the side chains of Arg358 and Asn388 interact with each other, linking the HTH unit to helix H8, and forming favorable contacts to the phosphate group of Ade20. The positioning of the C-terminal tail is further restricted by a salt-bridge between the side chain of Arg389 and the phosphate of Thy21, which may help position the side chain of Arg390 within the minor groove. The guanidine group of Arg390 is poised for a variety of favorable contacts with acceptor groups within the minor groove (the O2 atoms of Thy13 and Cyt19) and the adjacent phosphodiester backbone (the O4′ and phosphate group of Cyt19 and Ade20, respectively). However, these contacts are poorly defined in the structure, consistent with the high degree of mobility exhibited by this portion of the interface (Figure 5A), and are unlikely to contribute to binding specificity, since the contacts are to bases that are positioned outside of the consensus-binding site. The molecular interface is completed by contacts from the β-hairpin to the 5′ portion of the major groove interface (Figure 4C). In 90% of the conformers, the backbone amide atoms of Ala309 and Lys310 within the hairpin donate hydrogen bonds to the phosphate oxygens of Thy408. These interactions, along with salt-bridges to the adjacent phosphate groups from the side chains of Arg304 and Lys310, stabilize the docking of the ARID. The β-hairpin also directly contributes hydrophobic amino acids that pack against the H5/H6 loop, thereby stabilizing its conformation within the major groove.
DNA binding is coupled to the conformational ordering of the β-hairpin and H5/H6 loop, which are stabilized by subdomain repositioning and contacts to the duplex. Figure 5A shows a plot of the backbone amide (1H),15N heteronuclear NOE relaxation parameters of the DRI-DBDF355L protein in the presence (blue) and absence (red) of DNA. The largest changes in the conformational dynamics are observed within the H5/H6 loop and β-hairpin, which in the absence of DNA exhibit a significant amount of mobility, on the picosecond time scale, which is quenched upon binding. This is evidenced by the small magnitude of the backbone heteronuclear NOEs of amino acids within these structural elements in the DNA-free protein, and the increase in these values in the presence of DNA (Figure 5A). The H5/H6 loop and β-hairpin reside at the molecular interface, and new DNA contacts probably contribute to their immobilization (Figures 3 and 4A). However, a large structural rearrangement in the ARID also accompanies binding, stabilizing the H5/H6 loop. As shown in Figure 5B, DNA binding drives the rotation of the N-terminal subdomain (comprised of helices H1 and H2 and the β-hairpin) relative to the remainder to the polypeptide. This results in an ∼10 Å shift of the hairpin towards the H5/H6 loop and the formation of new protein–protein van der Waals contacts. In particular, DNA binding causes the side chains of Met308 and Ala309 within the hairpin to be inserted into the hydrophobic pocket formed by the side chains of Thr356 and Ile357 within helix H6 and Pro347 and Ile350 within the H5/H6 loop. These new interactions position the hairpin on the duplex, and stabilize the H5/H6 loop for sequence-specific interactions within the major groove.
Dead ringer recognizes an adenine–thymine base step
In the structure of the complex, numerous direct base-specific contacts are formed between the ARID and the Ade10–Thy11 base step (Figures 3 and 4A), indicating that these contacts play a significant role in the recognition of AT-rich DNA. In order to test this hypothesis, we measured the affinity of the wild-type DRI-DBD for its cognate site and related oligonucleotides harboring single nucleotide substitutions (Figure 6). Substitution of either nucleotide within the Ade10–Thy11 base step severely reduces the affinity of the complex (Figure 6, compare lanes 4 and 5 with lane 2); base substitutions at positions 10 and 11 result in a 16.9 ± 5.6- and 9.0 ± 5.1-fold decrease in affinity, respectively. This is consistent with the presence of specific interactions by the side chains of Thr351, Ser352 and Phe355 (Leu355 in the structure of the DRI-DBDF355L–DNA complex). In contrast, a nucleotide substitution removed from the Ade10–Thy11 interface does not affect binding (Figure 6, lane 3). In the structure of the DRI-DBDF355L–DNA complex, a hydrophobic interaction between the side chain of Leu355 and the Thy21 methyl group occurs within the Ade10–Thy11 base step and, presumably, in the wild-type protein through the phenylalanine side chain at this position. The significance of the C7 methyl group was tested by substituting the Thy21 base with deoxyuridine, which effectively removes the methyl while retaining the full complement of intermolecular hydrogen bonds to the ARID. This packing interaction is energetically significant, since this mutant exhibits a 5.3 ± 0.8-fold reduction in affinity (Figure 6, lane 6). This finding is further substantiated by the fact that the DRI-DBDF355L protein exhibits reduced affinity as compared with the wild-type protein, revealing that contacts from Phe355 are important contributors to affinity that are not completely reproduced in the DRI-DBDF355L mutant. However, Phe355 is not critical for binding specificity, since both the DRI-DBDF355L and DRI-DBD proteins bind DNA in a sequence-specific manner (Figure 1C). The highly homologous extended ARID from the Bright transcription factor exhibits reduced affinity in the presence of distamycin, which is in agreement with our finding that the side chain of Arg389 within the C-terminal tail contacts the minor groove. However, this interaction comprises only a small portion of the molecular interface, explaining why relatively high levels of distamycin are required to abolish binding (∼5 mM; Herrscher et al., 1995) compared with ‘true’ minor groove binding proteins; <10 µM distamycin is required to disrupt the binding of the TATA-box binding and the high mobility group (HMG) proteins (Chiang et al., 1994; Copenhaver et al., 1994).

Fig. 6. EMSA of binding of wild-type GST–DRI-DBD fusion protein to oligonucleotides containing mutations within the Dead ringer binding site. Lane 1, 32P-labeled cognate DNA only; lanes 2–6, GST–DRI-DBD and 32P-labeled oligonucleotides corresponding to the wild-type, mutant-1, mutant-2, mutant-3 and mutant-4 DNA sequences, respectively. The nucleotide sequence of each mutant site is listed to the right of the gel. The GST–DRI-DBD concentration was 100 nM in lanes 2–6. Dissociation constants reported in the text were measured from binding isotherms and are the average of three independent measurements (data not shown). The GST–DRI-DBD binds to its cognate site with a Kd = 28 ± 10 nM.
Insights into DNA binding by other ARIDs
ARIDs have been identified in >50 eukaryotic transcription factors that regulate cell proliferation, differentiation and development (Kortschak et al., 2000). The work presented here and the structures of the DRI-DBD and Mrf-2 proteins in the absence of DNA (Yuan et al., 1998; Iwahara and Clubb, 1999b; revised structure, PDB accession code 1IG6) suggest that there are at least three structural variants of the domain: (i) minimal ARIDs like Mrf-2 consist of a structurally conserved core domain formed by six α-helices (H2–H7 in the DRI-DBD); (ii) extended ARIDs like the DRI-DBD contain the core domain and additional α-helices at their N- and C-termini (Iwahara and Clubb, 1999b); and (iii) N-terminal extended ARIDs, which have yet to be visualized experimentally, but based on primary sequence homology, supplement the core domain only with an N-terminal α-helix (Iwahara and Clubb, 1999b). Since the central core domain forms the majority of contacts to the duplex in the DRI-DBDF355L–DNA complex, it is expected that all ARIDs will bind DNA in a similar manner, recognizing the major groove of DNA through a conserved HTH and adjacent hairpin. This conclusion is further substantiated by a primary sequence alignment, which reveals identical or conservatively substituted amino acids at positions shown to contact the duplex in the DRI-DBDF355L–DNA complex (residues colored green in Figure 7).

Fig. 7. Primary sequence alignment of several ARIDs with the Dead ringer. Amino acids that are identical or similar to residues within the DRI-DBD are indicated by bold and outlined letters, respectively. Conserved positions that contact the phosphodiester backbone or minor groove in the DRI-DBDF355L–DNA complex are denoted by green vertical bars, while conserved positions that interact with the major groove are indicated by yellow vertical bars. The position of the Phe355Leu mutation used in the structural work is indicated by an asterisk. The secondary structure of the DRI-DBD is shown above the alignment. Sixteen residues of the Dead ringer ARID are involved in DNA binding. The mouse Bright, human DRIL1, Bdp and worm T23D8.8 are expected to have the same DNA-binding specificity as that of Dead ringer, because all base-contacting residues and 11 of 12 phosphate-contacting residues are conserved. These proteins are from evolutionally distant species, suggesting that this conserved ARID–DNA interaction must be important in metazoans. The alignment was generated using the programs BLAST (Altschul et al., 1990) and Pfam (Bateman et al., 2000), which can be found online at www.ncbi.nlm.nih.gov/BLAST and www.sanger.ac.uk/Software/Pfam, respectively. Dm, Drosophila melanogaster; Mm, Mus musculs; Hs, Homo sapiens; Ce, Caenorhabditis elegans; At, Arabidopsis thaliana; Sc, Saccharomyces cerevisiae.
The structure of the complex explains why several ARID-containing proteins preferentially interact with AT-rich DNA, but suggests that many other family members will not possess this specificity. ARIDs were originally named based on the propensity of the Bright, Dead ringer and Mrf-2 proteins to bind to AT-rich DNA (Herrscher et al., 1995; Gregory et al., 1996; Valentine et al., 1998; Whitson et al., 1999). Our results indicate that this specificity is due to a series of interactions to an adenine–thymine base step, which, in the structure of the Dead ringer complex, is recognized by energetically significant contacts from the side chains of Thr351, Ser352 and Phe355. These amino acids are almost completely conserved in the Bright and Mrf-2 proteins, suggesting that similar contacts will be involved in their binding. Many other ARID-containing proteins are likely to exhibit distinct binding specificities because the length and composition of their H5/H6 loops are varied (residues colored yellow in Figure 7). Particularly significant is the finding that the OSA1, BAF120, BAF250 and p270 proteins possess serine amino acids at positions implicated in the recognition of the adenine–thymine base step. These proteins are components of SWI/SNF-related chromatin remodeling complexes in various organisms, and the absence of non-polar residues at DNA-contacting positions indicates that they will not preferentially interact with AT-rich DNA. Furthermore, it appears likely that they will bind DNA in a non-sequence-specific manner, since serine side chains can function as both acceptors and donors of hydrogen bonds, and because they are expected to reside at a portion of the molecular interface that is exposed to the solvent.
In the structure of the Dead ringer complex, the hydroxyl groups of serine and threonine are placed within a flexible loop that adapts its structure to form a network of base-specific hydrogen bonds within the major groove. This appears to be a general property of all ARIDs, since the analogous loop in the minimal ARID of the Mrf-2 protein is flexible in the absence of DNA, and presumably resides at the molecular interface (Zhu et al., 2001). In contrast, the extensive DNA interface formed by helix H8 in the extended ARIDs should be absent from other types of ARIDs based on primary sequence homology. This is supported by the observation that the analogous region within the minimal ARID of the Mrf-2 protein is structurally disordered in the absence of DNA, but is predicted to interact with DNA in the protein–DNA complex based on NMR studies (Zhu et al., 2001). In the minimal and N-terminally extended ARIDs, the primary sequence of this polypeptide segment is poorly conserved and it would thus seem unlikely that the extensive set of stabilizing contacts in the Dead ringer complex will be consistently reproduced in other ARIDs, implying that they will generally bind DNA less tightly. This conclusion is supported by the presence of additional DNA-binding domains in many minimal ARID-containing proteins (e.g. HMG domain; Riechmann et al., 2000) and by the observation that several non-extended ARIDs bind DNA in a non-sequence-specific manner.
Relationship to other DNA-binding domains
ARIDs are structurally unrelated to any other protein family, but close inspection reveals the presence of a non-canonical HTH motif (helices H5 and H6) that interacts with DNA in an unusual manner. The HTH is a ubiquitous DNA-binding motif that typically inserts the second helix, the so called ‘recognition helix’, into the major groove for base-specific hydrogen bonding (Pabo and Nekludova, 2000; Figure 8). In contrast, in the DRI-DBDF355L–DNA complex, no direct hydrogen bonds are made from its recognition helix (helix H6), rather the nine-residue preceding ‘turn’ adapts its structure upon immobilization to form several base-specific hydrogen bonds to the Ade10–Thy11 base step. Other protein–DNA complexes contain lengthy turns separating the helices of their HTH motif, but their turns either protrude into the solvent or contact the sugar-phosphate backbone exclusively. As shown in Figure 8, the prominent role of the loop in the Dead ringer complex is a direct result of the anomalous positioning of the recognition helix (helix H6), which is placed at a steep angle extending away from the duplex compared with other HTH-containing protein–DNA complexes. This unusual arrangement largely precludes extensive interactions with the duplex, allowing only van der Waals contacts from the side chains of Leu355 and Thr356, and a potential water-mediated hydrogen bond from the hydroxyl of Thr356. Interestingly, Dead ringer and the Engrailed homeodomain have the same sequence specificity (Kalionis and O’Farrell, 1993; Gregory et al., 1996), but the DNA contacts from their respective HTH units are not conserved (Kissinger et al., 1990; Billeter et al., 1993).
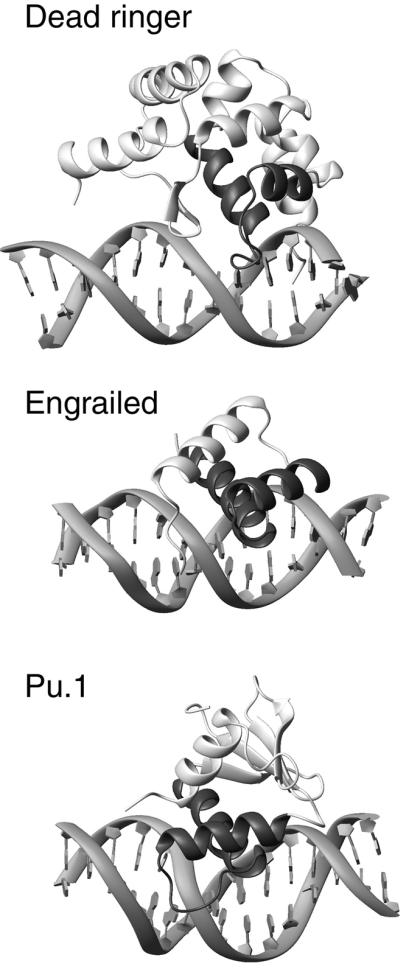
Fig. 8. Comparison of the extended ARID with other HTH-containing eukaryotic protein–DNA complexes. The structures of the Engrailed homeodomain–DNA complex (PDB accession code 3HDD) (Fraenkel et al., 1998), Pu.1 Ets domain–DNA complex (PDB accession code 1PUE) (Kodandapani et al., 1996) and Dead ringer DRI-DBD–DNA complex are shown. The HTH portion of each structure is shaded, and emphasizes the distinct positioning of this motif in the DRI-DBD–DNA complex. Although the recognition helix in the DRI-DBD–DNA complex appears to play a largely passive role in binding, similar to other HTH proteins, the side chain’s N-terminal end of the first helix of the HTH unit (helix H5) helps to stabilize the complex by interacting with DNA (Lys335 and Gln338). The turn of the HTH motif in the DRI-DBD–DNA complex forms base-specific contacts to the floor of the major groove, a function not previously ascribed to this portion of the motif.
During dorsal–ventral axis formation, Dorsal and Dead ringer bind to adjacent sites within the VRR to repress the transcription of the zen gene (Valentine et al., 1998). In order to understand this process we constructed a model of the ternary Dorsal–DRI-DBD–DNA complex based on the complex structures of Dead ringer and the close dorsal homolog Gambif1 (Cramer et al., 1999). Interestingly, the model predicts that the Dorsal and Dead ringer DNA-binding domains will interact with one another at the AT2-dl2 site, and that this interaction will be mediated by helix H8 of Dead ringer. This protein–protein interaction may partially explain Dorsal and Dead ringer’s co-operative recruitment of Groucho to the VRR (Valentine et al., 1998), as well as the observation that the spacing between the dl2 and AT2 sites is important for repression (Cai et al., 1996). It also suggests that in addition to DNA binding, the C-terminal helix within extended ARIDs may mediate protein–protein interactions.
Materials and methods
Resonance assignments of the complex
The Dead ringer DRI-DBDF355L–DNA complex was prepared as described previously (Iwahara et al., 2001b). The complex consists of 15N-labeled or 13C,15N-labeled protein and unlabeled DNA (dCCTGTATTGATGTGG, the Dead ringer binding site is underlined). Two types of NMR sample were prepared: (i) a 2.0 mM complex in 93% 1H2O, 7% 2H2O (referred to as the H2O sample); and (ii) a 1.4 mM complex in 100% 2H2O (referred to as the 2H2O sample). Both of these samples contained 20 mM deuterated Tris–HCl pH 6.7, 0.01% NaN3, 0.5 mM EDTA and 5 mM deuterated DTT. All NMR spectra were recorded at 37°C using Bruker DRX-500 and -600 MHz NMR spectrometers, except for the 3D 15N-edited NOESY spectrum (Marion et al., 1989), which was measured using a Varian 750-Unityplus spectrometer. The 1H, 15N and 13C resonances of the protein backbone were assigned using 3D HNCA, HN(CO)CA and HNCO (Grzesiek and Bax, 1992b); CBCA(CO)NH (Grzesiek and Bax, 1992a); HNCACB (Wittekind and Mueller, 1993); and HCACO spectra (Grzesiek and Bax, 1993; Zhang and Gmeiner, 1996). The side-chain resonance assignments were obtained by analyzing 3D HC(C)H-TOCSY and (H)CCH-TOCSY (Olejniczak et al., 1992; Kay et al., 1993); 3D HC(C)H-COSY (Ikura et al., 1991); and 3D C(CO)NH (Grzesiek et al., 1993a) spectra. The 12C-attached 1H resonances of the DNA were assigned using 2D [F1,F2] 13C-filtered NOESY (Iwahara et al., 2001b) and 2D [F1] 13C-filtered TOCSY (Ogura et al., 1996) spectra recorded using the 2H2O sample. The DNA 1H resonances of 14N-attached and adenine H2 protons were assigned by analyzing the 2D [F1,F2] 13C,15N-filtered NOESY and NOESY spectra of the H2O sample. The programs NMRPipe (Delaglio, 1995) and NMRView (Johnson and Blevins, 1994) were utilized for processing and analysis, respectively.
Experimental restraints for the structure calculations
NOE cross-peaks for the protein were collected from 3D 13C- and 15N-edited NOESY spectra recorded with mixing times of 80 ms (Muhandiram et al., 1993). Strong, medium and weak intensity NOE cross-peaks were converted to distance restraints of 1.8–3.0, 1.8–4.0 and 1.8–5.0 Å, respectively. The side-chain dihedral angles χ1 and χ2 were determined using 3D HNHB (Archer et al., 1991), 15N-edited ROESY, 13C-edited ROESY (Clore et al., 1991), 3D HN(CO)C (Hu and Bax, 1997), 13Cγaromatic 15N or 13C′ spin-echo difference HSQC (Hu et al., 1997), 13C′ or 15N 13Cγ spin-echo difference constant-time (CT)-HSQC (Grzesiek et al., 1993b; Bax et al., 1994) and long range 13C–13C correlation (Bax et al., 1992) spectra. 3JHNHα coupling constants were measured from a 3D HNHA spectrum (Kuboniwa et al., 1994). The program TALOS (Cornilescu et al., 1999) was utilized to obtain restraints for the backbone φ and ψ dihedral angles, with the ranges of these restraints set to either twice the standard deviation of the predicted value or to ±30°, whichever number was larger.
The DNA structure was defined using NOEs obtained from the 2D [F1,F2] 13C-filtered NOESY spectrum (Iwahara et al., 2001b) of the 2H2O sample. For 268 NOE cross-peaks that were well resolved, initial NOE build-up rates were measured from spectra recorded with mixing times of 25, 50, 75 and 100 ms. Cross-peaks were converted to inter-proton distances (rcalc) using the initial build-up rates and by reference to the intrabase cytosine H5 to H6 cross-peak (the distance between these atoms was set to 2.44 Å). The lower and upper distance bounds of restraints involving the H1′, H3′, H4′ and base protons were set to 0.90 and 1.25 rcalc, respectively. The lower and upper bounds involving the H2′ and H2′′ protons were set to 0.80 and 1.15 rcalc, respectively. Ribose sugar puckering was determined using a 2D [F1] 13C-filtered JUNSY experiment (Iwahara et al., 2001a) and confirmed by an analysis of the NOE data. Several nucleotides are predominantly in the S-type configuration, and their ν1, ν2 and δ dihedral angles were restrained to 35 ± 20°, –35 ± 20° and 150 ± 30°, respectively. A 2D (31P),1H spin-echo difference CT-NOESY experiment (Wu et al., 2001) with [F1] 13C-filtering was used to measure the 3JPH3′ coupling constants.
Intermolecular NOEs (67 out of a total of 80) were identified through an analysis of the 3D [F1] 13C,15N-filtered [F2] 13C-edited NOESY-HSQC and [F1] 13C,15N-filtered [F2] 15N-edited NOESY-HSQC spectra of the H2O complex (Zwahlen et al., 1997). Additional intermolecular NOEs were identified by analyzing 2D [F1] 13C-filtered NOESY (Iwahara et al., 2001b), 3D 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC spectra of the complex. The 15N T1, T2 and heteronuclear (1H),15N NOE experiments were recorded at 500 MHz. The refinement against the T1/T2 ratios was performed assuming an overall correlation time of 11.7 ns and D||/D⊥ of 1.5, which were obtained by inspecting the distribution of T1/T2 ratio values (Tjandra et al., 1997). Residues that exhibited heteronuclear (1H),15N NOE values <0.65 or exchange broadening were excluded from the list of restraints. Residual dipolar couplings were measured on a sample of the complex that contained 10 mg/ml Pf1 phage as a co-solute (Hansen et al., 1998). Protein backbone 1DNH and arginine 1DNεHε couplings were measured with derivatives of the TROSY experiment (Pervushin et al., 1997; Andersson et al., 1998). The 1DNH couplings ranged between –11 and 10 Hz, and the rhombicity was estimated to be 0.55 (Clore et al., 1998). The 1DNεHε couplings were normalized with respect to the 1DNH couplings and loosely restrained to compensate for their potential higher mobility. Lower and upper bound restraints for positive 1DNεHε couplings were set to the measured value and 10 Hz, respectively; negative couplings had lower and upper bounds of –11 Hz and the measured value, respectively.
Structure calculations
All of the experimental restraints (except the intermolecular NOEs) were used to calculate the structures of the protein and DNA individually using simulated annealing methods (Nilges et al., 1988) and the NIH version of X-PLOR (Brünger, 1993; Kuszewski et al., 2001). The protein and DNA structures with the lowest energies were then randomly displaced from one another and separated by 75 Å to generate 50 starting structures, which were then docked using rigid-body minimization (Clore, 2000). In this protocol, only protein residues that exhibited intermolecular NOEs were allowed to change their conformation and the components of the complex were moved with respect to one another to satisfy the intermolecular NOE restraints. The structures of each of the resultant 50 complexes were then refined using simulated annealing (starting and final temperatures were 2000 and 100 K, respectively). The values for the final force constants were as follows: NOE restraints, 30 kcal/mol Å2; distance restraints based on the initial NOE build-up rates, 60 kcal/mol Å2; dihedral angle restraints, 200 kcal/mol rad2; 3JHNHα restraints, 2.0 kcal/mol Hz2; 3JPH3′ restraints, 1.0 kcal/mol Hz2; 15N T1/T2 restraints, 1.0 kcal/mol; and backbone 1DNH restraints, 1.0 kcal/mol Hz2. Weak planarity restraints were applied during the simulated annealing with a force constant of 10 kcal/mol Å2 for 8 bp that were not in contact with the protein. Loose dihedral angle restraints were applied for the phosphate backbone to prevent problems associated with local mirror images (α = –70 ± 50°, β = 180 ± 50°, γ = 60 ± 35°, ε = 180 ± 50°, ζ = –85 ± 50°) (Omichinski et al., 1997). A conformational database potential for protein and DNA dihedral angles (Kuszewski et al., 1997) was applied using a final force constant of 1.0 kcal/mol. The measured 3JPH3′ coupling constants were directly refined against to define the ε dihedral angle (Lankhorst et al., 1984). Two distance restraints were used to define hydrogen bonds within regions of regular protein secondary structure and in Watson–Crick base pairs, and were introduced at the final stages of refinement. The program MolMol was used to generate the structure figures (Koradi et al., 1996). The final structures have been deposited in the PDB (accession code 1KQQ).
Gel mobility-shift assay
The glutathione S-transferase (GST) fusion proteins of wild-type and mutant Dead ringer ARID domains were prepared as described previously (Iwahara and Clubb, 1999a; Iwahara et al., 2001b). The 29 bp DNA fragment containing the Dead ringer binding site (5′-TGCGGATCCTGTATTGATGTGGCTGCAGTT-3′) was 32P-labeled at its 5′-termini with T4 polynucleotide kinase and [γ-32P]ATP (New England Biolabs). The protein and labeled DNA were incubated at room temperature for 30 min in 10 µl of binding buffer (20 mM Tris–HCl pH 7.5, 20 mM NaCl, 20 mM KCl, 10% glycerol, 10 µg/ml BSA, 1 mM DTT) and then separated by electrophoresis on a 8% polyacrylamide/TBE gel at 4°C. To assess binding specificity, reactions containing 50 pM 32P-labeled cognate DNA and 1 µM protein were challenged with 0.5 and 2.5 µM unlabeled competitor that did not contain the protein binding site (5′-CGAAGACGTGTTGGG-3′). The dissociation constant (Kd) of the wild-type GST–DRI-DBD protein, and single nucleotide mutants, for its cognate site was determined using the conditions described above. Each mutant oligonucleotide consisted of the 29 bp fragment and the indicated nucleotide substitution (Figure 6). Binding isotherms were generated by varying the protein concentration (0, 1, 10, 20, 40, 100, 200, 400, 1000, 2000 and 4000 nM protein), the gels were quantified using a PhosphorImager (Molecular Dynamics Inc.) and the Kd was determined by least-squares fitting using the program Sigmaplot (SPSS Science).
Acknowledgments
Acknowledgements
We thank Dr Robert Peterson for technical support and members of the Clubb laboratory for useful discussions. This work was supported by a grant from the US Department of Energy (DE-FC-03-87ER60615).
References
- Agulnik A.I., Mitchell,M.J., Lerner,J.L., Woods,D.R. and Bishop,C.E. (1994a) A mouse Y chromosome gene encoded by a region essential for spermatogenesis and expression of male-specific minor histocompatibility antigens. Hum. Mol. Genet., 3, 873–878. [DOI] [PubMed] [Google Scholar]
- Agulnik A.I., Mitchell,M.J., Mattei,M.G., Borsani,G., Avner,P.A., Lerner,J.L. and Bishop,C.E. (1994b) A novel X gene with a widely transcribed Y-linked homologue escapes X-inactivation in mouse and human. Hum. Mol. Genet., 3, 879–884. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andersson P., Annila,A. and Otting,G. (1998) An α/β-HSQC-α/β experiment for spin-state selective editing of IS cross peaks. J. Magn. Reson., 133, 364–367. [DOI] [PubMed] [Google Scholar]
- Archer S.J., Ikura,M., Torchia,D.A. and Bax,A. (1991) An alternative 3d-NMR technique for correlating backbone 15N with side chain H-β-resonances in larger proteins. J. Magn. Reson., 95, 636–641. [Google Scholar]
- Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A., Max,D. and Zax,D. (1992) Measurement of long-range 13C–13C J couplings in 20-kDa protein–peptide complex. J. Am. Chem. Soc., 114, 6923–6925. [Google Scholar]
- Bax A., Vuister,G.W., Grzesiek,S., Delaglio,F., Wang,A.C., Tschudin,R. and Zhu,G. (1994) Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol., 239, 79–105. [DOI] [PubMed] [Google Scholar]
- Bergelefranc J.L., Jay,P., Massacrier,A., Cau,P., Mattei,M.G., Bauer,S., Marsollier,C., Berta,P. and Fontes,M. (1996) Characterization of the human Jumonji gene. Hum. Mol. Genet., 5, 1637–1641. [DOI] [PubMed] [Google Scholar]
- Billeter M., Qian,Y.Q., Otting,G., Muller,M., Gehring,W. and Wutrich, K. (1993) Determination of the nuclear magnetic resonance solution structure of an Antennapedia homeodomain–DNA complex. J. Mol. Biol., 234, 1084–1093. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. (1993) X-PLOR Manual, Version 3.1. Yale University, New Haven, CT.
- Cai H.N., Arnosti,D.N. and Levine,M. (1996) Long-range repression in the Drosophila embryo. Proc. Natl Acad. Sci. USA, 93, 9309–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.Q., Fernandez,J., Mische,S. and Courey,A.J. (1999) A functional interaction between the histone deacetylase Rpd3 and the corepressor Groucho in Drosophila development. Genes Dev., 13, 2218–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S.Y., Welch,J., Rauscher,F.J.,III and Beerman,T.A. (1994) Effects of minor groove binding drugs on the interaction of TATA box binding protein and TFIIA with DNA. Biochemistry, 33, 7033–7040. [DOI] [PubMed] [Google Scholar]
- Clore G.M. (2000) Accurate and rapid docking of protein–protein complexes on the basis of intermolecular nuclear overhauser enhancement data and dipolar couplings by rigid body minimization. Proc. Natl Acad. Sci. USA, 97, 9021–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore G.M., Bax,A. and Gronenborn,G.M. (1991) Stereospecific assignment of β-methylene protons in larger proteins using 3D 15N-separated Hartman–Hahn and 13-separated rotating frame Overhauser spectroscopy. J. Biomol. NMR, 1, 13–22. [DOI] [PubMed] [Google Scholar]
- Clore G.M., Gronenborn,A.M. and Bax,A. (1998) A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson., 133, 216–221. [DOI] [PubMed] [Google Scholar]
- Collins R.T., Furukawa,T., Tanese,N. and Treisman,J.E. (1999) Osa associates with the Brahma chromatin remodeling complex and promotes the activation of some target genes. EMBO J., 18, 7029–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver G.P., Putnam,C.D., Denton,M.L. and Pikaard,C.S. (1994) The RNA polymerase I transcription factor UBF is a sequence-tolerant HMG-box protein that can recognize structured nucleic acids. Nucleic Acids Res., 22, 2651–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G., Delaglio,F. and Bax,A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR, 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Cramer P., Varrot,A., Barillas-Mury,C., Kafatos,F.C. and Muller,C.W. (1999) Structure of the specificity domain of the Dorsal homologue Gambif1 bound to DNA. Structure Fold. Des., 7, 841–852. [DOI] [PubMed] [Google Scholar]
- Dallas P.B., Pacchione,S., Wilsker,D., Bowrin,V., Kobayashi,R. and Moran,E. (2000) The human SWI–SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol. Cell. Biol., 20, 3137–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Doyle H.J., Kraut,R. and Levine,M. (1989) Spatial regulation of zerknüllt: a dorsal–ventral patterning gene in Drosophila. Genes Dev., 3, 1518–1533. [DOI] [PubMed] [Google Scholar]
- Fattaey A.R. et al. (1993) Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene, 8, 3149–3156. [PubMed] [Google Scholar]
- Fraenkel E., Rould,M.A., Chambers,K.A. and Pabo,C.O. (1998) Engrailed homeodomain–DNA complex at 2.2 Å resolution: a detailed view of the interface and comparison with other engrailed structures. J. Mol. Biol., 284, 351–361. [DOI] [PubMed] [Google Scholar]
- Gregory S.L., Kortschak,R.D., Kalionis,B. and Saint,R. (1996) Characterization of the dead ringer gene identifies a novel, highly conserved family of sequence-specific DNA-binding proteins. Mol. Cell. Biol., 16, 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S. and Bax,A. (1992a) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc., 114, 6291–6293. [Google Scholar]
- Grzesiek S. and Bax,A. (1992b) Improved 3D triple-resonance NMR techniques applied to a 31-kDa protein. J. Magn. Reson., 96, 432–440. [Google Scholar]
- Grzesiek S. and Bax,A. (1993) The origin and removal of artifacts in 3D HCACO spectra of proteins uniformly enriched with 13C. J. Magn. Reson. B, 102, 103–106. [Google Scholar]
- Grzesiek S., Anglister,J. and Bax,A. (1993a) Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B, 101, 114–119. [Google Scholar]
- Grzesiek S., Vuister,G.W. and Bax,A. (1993b) A simple and sensitive experiment for measurement of Jcc couplings between backbone carbonyl and methyl carbons in isotopically enriched proteins. J. Biomol. NMR, 3, 487–493. [DOI] [PubMed] [Google Scholar]
- Hansen M.R., Mueller,L. and Pardi,A. (1998) Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nature Struct. Biol., 5, 1065–1074. [DOI] [PubMed] [Google Scholar]
- Herrscher R.F., Kaplan,M.H., Lelsz,D.L., Das,C., Scheuermann,R. and Tucker,P.W. (1995) The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev., 9, 3067–3082. [DOI] [PubMed] [Google Scholar]
- Hu J.S. and Bax,A. (1997) Determination of φ and χ1 angles in proteins from 13C–13C three-bond J couplings measured by three-dimensional heteronuclear NMR. How planar is the peptide bond? J. Am. Chem. Soc., 119, 6360–6368. [Google Scholar]
- Hu J.S., Grzesiek,S. and Bax,A. (1997) Two-dimensional NMR methods for determining (chi 1) angles of aromatic residues in proteins from three-bond JC′Cγ and JNCγ couplings. J. Am. Chem. Soc., 119, 1803–1804. [Google Scholar]
- Huang T.H., Oka,T., Asai,T., Okada,T., Merrills,B.W., Gertson,P.N., Whitson,R.H. and Itakura,K. (1996) Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res., 24, 1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura I., Kay,L.E. and Bax,A. (1991) Improved three-dimensional 1H-13C-1H correlation spectroscopy of a 13C-labeled protein using constant-time evolution. J. Biomol. NMR, 1, 299–304. [DOI] [PubMed] [Google Scholar]
- Ip Y.T., Kraut,R., Levine,M. and Rushlow,C.A. (1991) The dorsal morphogen is a sequence-specific DNA-binding protein that interacts with a long-range repression element in Drosophila. Cell, 64, 439–446. [DOI] [PubMed] [Google Scholar]
- Iwahara J. and Clubb,R.T. (1999a) 1H, 13C and 15N resonance assignments of the AT-rich interaction domain from the Dead Ringer protein. J. Biomol. NMR, 15, 85–86. [DOI] [PubMed] [Google Scholar]
- Iwahara J. and Clubb,R.T. (1999b) Solution structure of the DNA binding domain from Dead ringer, a sequence-specific AT-rich interaction domain (ARID). EMBO J., 18, 6084–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J., Wojciak,J.M. and Clubb,R.T. (2001a) An efficient NMR experiment for analyzing sugar-puckering in unlabeled DNA: application to the 26-kDa Dead ringer-DNA complex. J. Magn. Reson., 153, 262–266. [DOI] [PubMed] [Google Scholar]
- Iwahara J., Wojciak,J.M. and Clubb,R.T. (2001b) Improved NMR spectra of a protein–DNA complex through rational mutagenesis and the application of a sensitivity optimized isotope-filtered NOESY experiment. J. Biomol. NMR, 19, 231–241. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. and Blevins,R.A. (1994) NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Kalionis B. and O’Farrell,P.H. (1993) A universal target sequence is bound in vitro by diverse homeodomains. Mech. Dev., 43, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan M.H., Zong,R.T., Herrscher,R.F., Scheuermann,R.H. and Tucker,P.W. (2001) Transcriptional activation by a matrix associating region-binding protein—contextual requirements for the function of Bright. J. Biol. Chem., 276, 21325–21330. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Xu,G.Y., Singer,A.U., Muhandiram,D.R. and Forman-Kay, J.D. (1993) A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. B, 101, 333–337. [Google Scholar]
- Kissinger C.R., Liu,B.S., Martinblanco,E., Kornberg,T.B. and Pabo,C.O. (1990) Crystal structure of an engrailed homeodomain–DNA complex at 2.8-Å resolution—a framework for understanding homeodomain– DNA interactions. Cell, 63, 579–590. [DOI] [PubMed] [Google Scholar]
- Kodandapani R., Pio,F., Ni,C.Z., Piccialli,G., Klemsz,M., McKercher,S., Maki,R.A. and Ely,K.R. (1996) A new pattern for helix–turn–helix recognition revealed by the PU.1 ETS-domain–DNA complex. Nature, 380, 456–460. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter,M. and Wuthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph., 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Kortschak R.D., Reimann,H., Zimmer,M., Eyre,H.J., Saint,R. and Jenne, D.E. (1998) The human dead ringer-bright homolog, DRIL1: cDNA cloning, gene structure and mapping to D19S886, a marker on 19p13.3 that is strictly linked to the Peutz–Jeghers syndrome. Genomics, 51, 288–292. [DOI] [PubMed] [Google Scholar]
- Kortschak R.D., Tucker,P.W. and Saint,R. (2000) ARID proteins come in from the desert. Trends Biochem. Sci., 25, 294–299. [DOI] [PubMed] [Google Scholar]
- Kuboniwa H., Grzesiek,S., Delaglio,F. and Bax,A. (1994) Measurement of H-N-H-α J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR, 4, 871–878. [DOI] [PubMed] [Google Scholar]
- Kuszewski J., Gronenborn,A.M. and Clore,G.M. (1997) Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J. Magn. Reson., 125, 171–177. [DOI] [PubMed] [Google Scholar]
- Kuszewski J., Schwieters,C. and Clore,G.M. (2001) Improving the accuracy of NMR structures of DNA by means of a database potential of mean force describing base–base positional interactions. J. Am. Chem. Soc., 123, 3903–3918. [DOI] [PubMed] [Google Scholar]
- Lankhorst P.P., Haasnoot,C.A., Erkelens,C. and Altona,C. (1984) Carbon-13 NMR in conformational analysis of nucleic acid fragments. 2. A reparametrization of the Karplus equation for vicinal NMR coupling constants in CCOP and HCOP fragments. J. Biomol. Struct. Dyn., 1, 1387–1405. [DOI] [PubMed] [Google Scholar]
- Marion D., Kay,L.E., Sparks,S.W., Torchia,D. and Bax,A. (1989) Three-dimensional heteronuclear NMR of 15N-labeled proteins. J. Am. Chem. Soc., 111, 1515–1517. [Google Scholar]
- Muhandiram D.R., Farrow,N.A., Xu,G.-Y., Smallcombe,S.H. and Kay, L.E. (1993) A gradient 13C NOESY-HSQC experiment for recording 13C-labeled proteins dissolved in H2O. J. Magn. Reson. B, 102, 317–321. [Google Scholar]
- Nie Z.Q., Xue,W.T., Yang,D.F., Zhou,S., Deroo,B.J., Archer,T.K. and Wang,W.D. (2000) A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol. Cell. Biol., 20, 8879–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilges M., Clore,G.M. and Gronenborn,A.M. (1988) Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamic simulated annealing. FEBS Lett., 229, 129–136. [DOI] [PubMed] [Google Scholar]
- Ogura K., Terasawa,H. and Inagaki,F. (1996) An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. J. Biomol. NMR, 8, 492–498. [DOI] [PubMed] [Google Scholar]
- Olejniczak E.T., Xu,R.X. and Fesik,S.W. (1992) A 4D HCCH-TOCSY experiment for assigning the side chain 1H and 13C resonances of proteins. J. Biomol. NMR, 2, 655–659. [DOI] [PubMed] [Google Scholar]
- Omichinski J.G., Pedone,P.V., Felsenfeld,G., Gronenborn,A.M. and Clore,G.M. (1997) The solution structure of a specific GAGA factor–DNA complex reveals a modular binding mode. Nature Struct. Biol., 4, 122–132. [DOI] [PubMed] [Google Scholar]
- Pabo C.O. and Nekludova,L. (2000) Geometric analysis and comparison of protein–DNA interfaces: why is there no simple code for recognition? J. Mol. Biol., 301, 597–624. [DOI] [PubMed] [Google Scholar]
- Pervushin K., Riek,R., Wider,G. and Wuthrich,K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl Acad. Sci. USA, 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann J.L. et al. (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science, 290, 2105–2110. [DOI] [PubMed] [Google Scholar]
- Schneider B. and Berman,H.M. (1995) Hydration of the DNA bases is local. Biophys. J., 69, 2661–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandala T., Kortschak,R.D., Gregory,S. and Saint,R. (1999) The Drosophila dead ringer gene is required for early embryonic patterning through regulation of argos and buttonhead expression. Development, 126, 4341–4349. [DOI] [PubMed] [Google Scholar]
- Tjandra N., Garrett,D.S., Gronenborn,A.M., Bax,A. and Clore,G.M. (1997) Defining long range order in NMR structure determination from the dependence of heteronuclear relaxation times on rotational diffusion anisotropy. Nature Struct. Biol., 4, 443–449. [DOI] [PubMed] [Google Scholar]
- Valentine S.A., Chen,G., Shandala,T., Fernandez,J., Mische,S., Saint,R. and Courey,A.J. (1998) Dorsal-mediated repression requires the formation of a multiprotein repression complex at the ventral silencer. Mol. Cell. Biol., 18, 6584–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez M., Moore,L. and Kennison,J.A. (1999) The trithorax group gene osa encodes an ARID-domain protein that genetically interacts with the Brahma chromatin-remodeling factor to regulate transcription. Development, 126, 733–742. [DOI] [PubMed] [Google Scholar]
- Venter J.C. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Whitson R.H., Huang,T. and Itakura,K. (1999) The novel Mrf-2 DNA-binding domain recognizes a five-base core sequence through major and minor-groove contacts. Biochem. Biophys. Res. Commun., 258, 326–331. [DOI] [PubMed] [Google Scholar]
- Wittekind M. and Mueller,L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α-carbon and β-carbon resonances in proteins. J. Magn. Reson., 101, 201–205. [Google Scholar]
- Wu Z.R., Tjandra,N. and Bax,A. (2001) Measurement of 1H3′-P-31 dipolar couplings in a DNA oligonucleotide by constant-time NOESY difference spectroscopy. J. Biomol. NMR, 19, 367–370. [DOI] [PubMed] [Google Scholar]
- Yuan Y.C., Whitson,R.H., Liu,Q., Itakura,K. and Chen,Y. (1998) A novel DNA-binding motif shares structural homology to DNA replication and repair nucleases and polymerases. Nature Struct. Biol., 5, 959–964. [DOI] [PubMed] [Google Scholar]
- Zhang W. and Gmeiner,W.H. (1996) Improved 3D gd-HCACO and gd-(H)CACO-TOCSY experiments for isotopically enriched proteins dissolved in H2O. J. Biomol. NMR, 7, 247–250. [DOI] [PubMed] [Google Scholar]
- Zhu L., Hu,J., Lin,D., Whitson,R., Itakura,K. and Chen,Y. (2001) Dynamics of the mrf-2 DNA-binding domain free and in complex with DNA. Biochemistry, 40, 9142–9150. [DOI] [PubMed] [Google Scholar]
- Zwahlen C., Legault,P., Vincent,S.J.F., Greenblatt,J., Konrat,R. and Kay,L.E. (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-peptide/boxB RNA complex. J. Am. Chem. Soc., 119, 6711–6721. [Google Scholar]