

Abstract
We investigated whether Epstein–Barr virus (EBV) infection could counteract the antitumor effect of interferon (IFN)-α. EBV-negative subclones isolated from EBV-positive Burkitt’s lymphoma (BL) cell lines Akata, Daudi and Mutu were found to fall into apoptosis after IFN-α treatment. On the other hand, EBV-positive counterparts exhibited striking resistance against IFN-α-induced apoptosis. Transfection of an individual EBV latent gene into EBV-negative BL cells revealed that EBV-encoded poly(A)– RNAs (EBERs) were responsible for IFN resistance. EBERs bound double-stranded (ds) RNA-activated protein kinase (PKR), a key mediator of the antiviral effect of IFN-α, and inhibited its phosphorylation. Transfection of dominant-negative PKR, which was catalytically inactive and could block phosphorylation of endogenous PKR, made EBV-negative BL cells resistant to IFN-α-induced apoptosis. Furthermore, EBERs did not bind mutant PKR, which was catalytically active but lacked dsRNA-binding activity, nor did they inhibit its phosphorylation. These results indicate that EBERs confer resistance to IFN-α-induced apoptosis via binding to PKR and inhibition of its phosphorylation. This is the first report that the virus counteracts IFN-induced apoptosis in virus-associated tumors.
Keywords: apoptosis/Burkitt’s lymphoma/EBV-encoded small RNA/Epstein–Barr virus/interferon
Introduction
Interferon (IFN)-α/β is produced by virus-infected cells and confers cellular resistance against virus infection (Flint et al., 2000). This antiviral effect is mediated by at least two cellular proteins, double-stranded (ds) RNA-activated protein kinase (PKR) and ribonuclease L, and each independently drives cells to a translational block. PKR, a serine/threonine kinase, has two known cellular substrates: α-subunit of translation initiation factor 2 (eIF-2α) (Samuel, 1993) and nuclear factor (NF)-κB inhibitor IκBα (Kumar et al., 1994). Phosphorylation of eIF-2α abrogates translation initiation. The contribution of IκBα to antiviral defense has not been well studied. Ribonuclease L is a nuclease capable of degrading most cellular and viral RNA species. Its concentration increases 10- to 1000-fold after IFN treatment, but the enzyme remains inactive unless activated by dsRNA-activated 2′,5′-oligoadenylate synthetase.
Besides its antiviral effect, IFN has antiproliferative and antitumor effects (Barber, 2000; Borden et al., 2000). IFN has been used in the treatment of several malignancies, including hairy cell leukemia, chronic myeloid leukemia and squamous cell carcinoma. It has been shown to induce G1 phase arrest of cells and cell death by a direct cytotoxic effect in vitro. In some instances, cell death is due to the induction of apoptosis. Although in virus-infected cells, viral dsRNA can serve as a PKR activator, recent studies have identified a cellular protein activator of PKR (termed PACT), which heterodimerizes with PKR and activates it in the absence of dsRNA (Patel and Sen, 1998).
To confound the actions of IFN, viruses have numerous mechanisms (Flint et al., 2000). Influenza virus and vaccinia virus produce proteins that bind and sequester dsRNA, and block activation of PKR and 2-5 A synthetase. Influenza virus and herpes simplex virus induce cellular proteins that bind PKR and inhibit its activity. Adenovirus VA1, non-polyadenylated small RNA, binds PKR and inhibits its activity. All of them release cells from the antiviral state and allow virus production. However, there has been little reported on a viral strategy against the antitumor effect of IFN, although there are many virus-associated malignancies.
Epstein–Barr virus (EBV), a family of human herpesviruses, establishes life-long latent infection in B lymphocytes following primary infection. The virus is associated with various malignancies such as Burkitt’s lymphoma (BL), nasopharyngeal carcinoma and AIDS-associated lymphoma. In these tumor cells, the entire viral genome of ∼170 kbp is maintained as a plasmid, and among ∼80 viral genes, a limited number of genes are expressed (Kieff, 1996; Rickinson and Kieff, 1996). The Akata (Takada, 1984; Takada et al., 1991), Mutu (Gregory et al., 1990) and Daudi (Klein et al., 1968) BL cell lines are unique in that they retain the in vivo phenotype of EBV expression (termed type I latency), which is characterized by expression of EBV-determined nuclear antigen 1 (EBNA1), EBV-encoded poly(A)– RNAs (EBERs) and transcripts from the BamHI A region (BARF0), after long-term culture in vitro. In contrast, most BL cell lines convert EBV expression to that of EBV-immortalized lymphoblastoid cells (LCLs), in which all the latent viral gene products, including six EBNAs and three latent membrane proteins (LMPs), are expressed (Rowe et al., 1987). We have recently isolated EBV-negative subclones from the Akata (Shimizu et al., 1994), Mutu (Kitagawa et al., 2000) and Daudi cell lines. This prompted us to study whether BL cells are susceptible to the antiproliferative effect of IFN and, if so, whether EBV could counteract it. We demonstrate here that EBV-negative subclones from all three BL lines fall into apoptosis after IFN-α treatment, but that EBV-positive counterparts show relative resistance to apoptosis. Furthermore, we show that EBERs are responsible for resistance to IFN-α-induced apoptosis, and that resistance to IFN-α-induced apoptosis in BL cells is mediated by inhibition of PKR activation.
Results
Induction of apoptosis by IFN-α treatment in BL cells and role of EBV in resistance to apoptosis
To examine whether IFN-α induces apoptosis in BL cells, three BL cell lines, Akata, Daudi and Mutu, were chosen as test cells, because they were originally EBV positive, retained BL-type EBV expression that was characterized by expression of a restricted set of EBV latent genes, including EBNA1, EBERs and BARF0, and from which EBV-negative subclones had been isolated (Figure 1A). Therefore, these test cells allowed us to examine not only whether IFN-α induced apoptosis in BL cells, but also whether EBV had any effect on the response of the cells to IFN-α treatment. EBV-positive and -negative Akata, Daudi and Mutu cell clones were incubated in a medium containing 500 U/ml IFN-α, and the appearance of apoptotic cells was assessed at designated times by propidium iodide staining and flow cytometry (Nicoletti et al., 1991). In this assay, apoptotic cells appeared as a broad hypodiploid DNA peak that was easily discriminated from the narrow peak of normal diploid DNA. As shown in Figure 1B and C, EBV-negative Akata, Daudi and Mutu cell clones were prone to fall into apoptosis without addition of IFN-α, and nearly half of the cells underwent apoptosis after 3–4 days of IFN-α treatment. These results clearly demonstrated that IFN-α induced apoptosis in EBV-negative BL cells. On the other hand, the EBV-positive counterparts remained apoptosis-free under ordinary culture conditions, and exhibited striking resistance to induction of apoptosis by IFN-α. Electronic analysis of cellular DNA also revealed the fragmentation of chromatin into nucleosomal-size fragments (ladder) characteristic of apoptosis in IFN-α-treated EBV-negative BL cells, but not in EBV-positive counterparts (Figure 1D). These results revealed that EBV infection conferred resistance to IFN-α-induced apoptosis in BL cells.

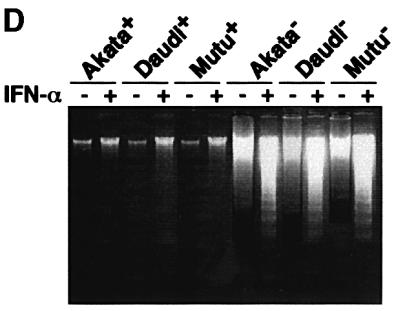
Fig. 1. IFN-α-induced apoptosis in BL-derived Akata, Daudi and Mutu cells. These cell lines were originally 100% EBV-positive, and EBV-negative subclones were isolated by limiting dilution from the parental cultures. (A) PCR analysis of EBV genomes in EBV-positive and -negative cell clones. The EBNA2 region was amplified by 30 cycles of PCR as described previously (Takeda et al., 2000). (B) DNA fluorescence histograms of propidium iodide-stained cells. EBV-positive and -negative cell clones (5 × 104/ml) were incubated in the presence or absence of human IFN-α (500 U/ml) for 60 h, and the frequency of apoptotic cells was determined by flow cytometry. A, apoptotic cells with hypodiploid DNA content. The vertical axis denotes the number of cells counted and the horizontal axis denotes fluorescence intensity. (C) The frequency of apoptotic cells determined by flow cytometry. EBV-positive and -negative cell clones (two clones each; 5 × 104/ml) were incubated in the presence or absence of human IFN-α (500 U/ml) for various times, and the frequency of apoptotic cells was determined. Results are expressed as the means of triplicate wells. (D) DNA laddering. EBV-positive and -negative cell clones (5 × 104/ml) were incubated in the presence or absence of human IFN-α (500 U/ml) for 72 h. DNA from 1 × 106 cells was subjected to 2% agarose gel electrophoresis.
Figure 2 shows the dose responses of apoptosis induction in EBV-positive and -negative Akata cells treated with IFN-α. IFN-α at 600 U/ml gave the maximum induction of apoptosis in EBV-negative Akata cells, while EBV-positive Akata cells exhibited resistance to apoptosis up to 1000 U/ml IFN-α.

Fig. 2. Dose response of apoptosis induction by IFN-α in EBV-positive and -negative Akata cell clones. Cells (5 × 104) were suspended in 1 ml of fresh medium containing various concentrations of IFN-α. After 84 h of incubation, cells were harvested for flow cytometry analysis.
EBERs are responsible for resistance to IFN-α-induced apoptosis
Next we examined which EBV gene was responsible for resistance to IFN-α-induced apoptosis in BL cells. Figure 3A and B shows EBV gene expression in EBV-positive Akata, Daudi and Mutu cell clones. Western blot analysis indicated that they were positive for EBNA1, but negative for other EBNAs and LMP1. RT–PCR analysis indicated that they utilized the Q promoter for EBNA transcription, and were positive for BARF0 and EBER but negative for LMP2B. LMP2A was weakly positive in Akata cell clones, but negative in Daudi and Mutu cell clones. These results indicate that EBNA1, EBERs and BARF0 are commonly expressed in EBV-positive Akata, Daudi and Mutu cell clones.

Fig. 3. EBV expression in EBV-positive and -negative Akata, Daudi and Mutu cell clones. (A) Immunoblot analysis for detection of EBNAs and LMP1. The blots were probed with EBNA-positive human serum (upper blot), an anti-EBNA2 monoclonal antibody (middle blot) and an anti-LMP1 monoclonal antibody (lower blot). Protein samples extracted from 105 cells were loaded per slot. (B) RT–PCR analysis of EBNA promoter usage and EBV latent gene expression. Akata cells were used as a positive control for detection of Qp-initiated EBNA mRNA, and a lymphoblastoid cell line immortalized by Akata EBV (LCL) was used as a positive control for detection of Cp- or Wp-initiated EBNA mRNAs, and EBER, BARF0, LMP2A and LMP2B mRNAs.
Based on these results, an EBV-negative Akata cell clone was transfected with an individual EBV latent gene, and cell clones that stably expressed levels similar to EBV-positive Akata cells were selected and examined for their susceptibility to IFN-α (Figure 4A–C). The results indicated that cell clones transfected with the EBER gene exhibited striking resistance to IFN-α-induced apoptosis (Figure 4D). Similarly, EBV-negative Daudi and Mutu cell clones were transfected with the EBER gene, and cell clones that stably expressed similar levels to EBV-positive counterparts were selected and examined (Figure 5A). The results indicated that cell clones transfected with the EBER gene exhibited striking resistance to IFN-α-induced apoptosis (Figure 5B).

Fig. 4. IFN-α-induced apoptosis in EBV-negative Akata cell clones transfected with an individual EBV latent gene expressed in BL. An EBV-negative Akata cell clone was transfected with an individual EBV gene, and cell clones (two clones each) that stably expressed similar levels to EBV-positive Akata cells were selected and subjected to apoptosis assay. (A) EBNA1 expression. EBNA1 was detected by immunoblotting using EBNA1-positive human serum. (B) BARF0 expression in EBV-negative Akata cell clones transfected with the FLAG epitope-tagged BARF0 gene. BARF0 was detected by immunoblotting with anti-FLAG antibody. (C) EBER expression. EBER and GAPDH expression were determined by RT–PCR. (D) Apoptosis assay. Cells (5 × 104/ml) were incubated in the presence or absence of IFN-α (500 U/ml) for various times. The frequency of apoptotic cells was determined by flow cytometry. Results are expressed as the means of triplicate wells.

Fig. 5. IFN-α-induced apoptosis in EBV-negative Daudi and Mutu cell clones transfected with the EBER gene. EBV-negative Daudi and Mutu cell clones were transfected with the EBER plasmid, and cell clones (two clones each) that stably expressed similar levels to EBV-positive clones were selected and subjected to apoptosis assay. (A) EBER expression. EBER and GAPDH expression were determined by RT–PCR. (B) Apoptosis assay. Cells (5 × 104/ml) were incubated in the presence or absence of IFN-α (500 U/ml) for various times. The frequency of apoptotic cells was determined by flow cytometry. Results are expressed as the means of triplicate wells.
To further confirm that EBERs were responsible for resistance to IFN-α-induced apoptosis, we examined whether an EBV recombinant lacking EBER genes (Kitagawa et al., 2000) could confer resistance to IFN-α. EBER-knockout EBV carrying the neoR gene was infected into EBV-negative Akata cell clones, and EBV-positive cell clones were selected in medium containing G418. These cell clones showed a pattern of EBV expression similar to that of wild-type EBV-infected cell clones, except for the absence of EBER expression (Figure 6A and B). The flow cytometry analysis indicated that EBER-knockout EBV-infected Akata cell clones were susceptible to IFN-α and, like their EBV-negative counterparts, nearly half of the cells underwent apoptosis after 3–4 days of IFN-α treatment (Figure 6C).

Fig. 6. IFN-α-induced apoptosis in EBER-knockout, EBV-infected Akata cells. An EBV-negative Akata cell clone was infected with EBER-positive or -negative EBV, and 100% EBV-positive cell clones (two clones each) were isolated and subjected to analysis. (A) Immunoblot analysis for detection of EBNAs and LMP1. The blots were probed with EBNA-positive human serum (upper blot), an anti-EBNA2 monoclonal antibody (middle blot) and an anti-LMP1 monoclonal antibody (lower blot). Protein samples extracted from 105 cells were loaded per slot. (B) RT–PCR analysis of EBNA promoter usage and EBV latent gene expression. EBV-positive Akata cells were used as a positive control for detection of Qp-initiated EBNA mRNA, and a lymphoblastoid cell line immortalized by Akata EBV (LCL) was used as a positive control for detection of Cp- or Wp-initiated EBNA mRNAs, and BARF0, EBER, LMP2A and LMP2B mRNAs. (C) Apoptosis assay. Cells (5 × 104/ml) were incubated in the presence or absence of IFN-α (500 U/ml) for various times. The frequency of apoptotic cells was determined by flow cytometry. Results are expressed as the means of triplicate wells.
These results clearly demonstrate that the EBERs are responsible for resistance to IFN-α-induced apoptosis.
Inhibition of PKR activation in EBV-infected BL cells
PKR is a key mediator of the antiviral effect of IFN-α, and is also known to bind EBERs in a cell-free system (Clarke et al., 1991). Therefore, we examined whether PKR was involved in resistance to IFN-α-induced apoptosis in EBV-positive and EBER-expressing BL cells.
First, we examined expression of PKR in BL cells by western blot analysis using a polyclonal antibody against PKR. IFN-α treatment upregulated expression of PKR in Akata, Daudi and Mutu cells. However, there was no difference in the level of PKR expression between EBV-positive and -negative BL cells, indicating that EBV infection did not influence PKR expression (Figure 7).
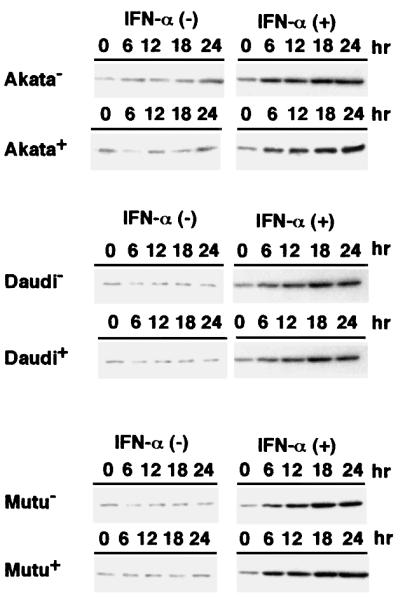
Fig. 7. Induction of PKR expression by IFN-α in EBV-positive and -negative Akata, Daudi and Mutu cell clones. Cells (5 × 104/ml) were incubated in the presence or absence of human IFN-α (500 U/ml) for various times. PKR expression was detected by immunoblotting using a polyclonal antibody to human PKR. Protein samples extracted from 2 × 105 cells were loaded per slot. IFN-α (+), IFN-α treated; IFN-α (–), IFN-α untreated.
It is known that PKR is autophosphorylated to become an active form following IFN-α treatment (Galabru and Hovanessian, 1987). Therefore, we examined whether EBV infection or EBER expression could inhibit phosphorylation of PKR. Since an antibody that could immunoprecipitate PKR or detect the phosphorylated form of PKR was not available, we used FLAG-tagged PKR to investigate the effect of EBV on phosphorylation of PKR. The FLAG-tagged PKR plasmid was transfected into EBV-positive and -negative Akata, Daudi and Mutu cell clones, and cultured for 48 h. FLAG-PKR in cell lysates was immunoprecipitated with anti-FLAG M2 antibody, labeled with [γ-32P]ATP and subjected to SDS–PAGE. Autophosphorylated PKR was visualized by autoradiography. The results indicated that phosphorylation of PKR was substantially inhibited in EBV-positive BL cells (Figure 8A).
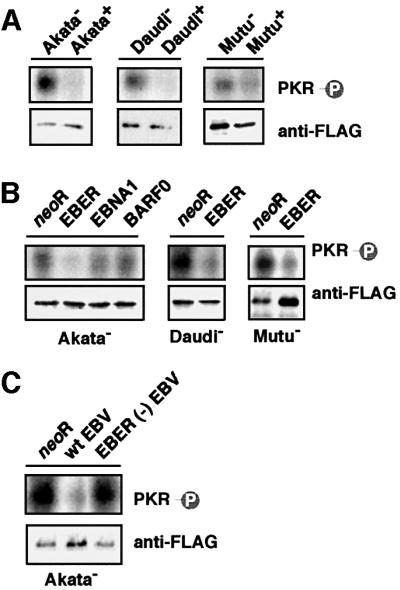
Fig. 8. Effects of EBV infection and EBER expression on phosphorylation of PKR. Cells (5 × 106) were transfected with the FLAG epitope-tagged PKR plasmid by the electroporation method. After 48 h of incubation, FLAG-PKR was immunoprecipitated with anti-FLAG antibody and subjected to in vitro kinase assay. Immunoprecipitated FLAG-PKR was detected by immunoblotting using anti-FLAG antibody (lower panel), and its phosphorylation was visualized by autoradiography (upper panel). (A) Phosphorylation of PKR in EBV-positive and -negative Akata, Daudi and Mutu cell clones. (B) Phosphorylation of PKR in EBV-negative Akata cell clones transfected with an individual EBV latent gene expressed in BL, and in Daudi and Mutu cell clones transfected with the EBER gene. (C) Phosphorylation of PKR in EBV-negative Akata cell clones that were infected with EBER-positive or -negative EBV.
Next, we examined whether EBERs could inhibit phosphorylation of PKR. As shown in Figure 8B, phosphorylation of PKR was substantially inhibited in EBER-transfected cell clones compared with cell clones transfected with neoR, while EBNA1 and BARF0 had no effect. Similarly, phosphorylation of PKR in EBV-negative Akata cell clones was strikingly inhibited by wild-type EBV infection, but not by EBER-knockout EBV infection (Figure 8C). These results clearly demonstrate that EBERs inhibit phosphorylation of PKR in BL cells.
We further examined whether EBER expression could inhibit phosphorylation of two substrates of PKR, eIF-2α (Samuel, 1993) and IκBα (Kumar et al., 1994), by IFN-α treatment. Western blot analysis using monoclonal antibodies against phosphorylated eIF-2α and IκBα indica ted that phosphorylation of both substrates was inhibited in EBV-positive and EBER-expressing BL cell clones (Figure 9A–C).

Fig. 9. Effects of EBV infection and EBER expression on phosphorylation of PKR substrates after IFN-α treatment. Cells (5 × 104/ml) were incubated in the presence of IFN-α (500 U/ml) for 12 h and subjected to immunoblotting for detection of phosphorylated eIF-2α or IκBα. Protein samples extracted from 1 × 105 cells were loaded per slot. (A) Phosphorylation of eIF-2α and IκBα in EBV-positive and -negative Akata, Daudi and Mutu cell clones. (B) Phosphorylation of eIF-2α and IκBα in EBV-negative Akata cell clones transfected with an individual EBV latent gene expressed in BL, and in Daudi and Mutu cell clones transfected with the EBER gene. (C) Phosphorylation of eIF-2α and IκBα in EBV-negative Akata cell clones that were infected with EBER-positive or -negative EBV.
EBERs consist of EBER1 and EBER2. By transient transfection assay, both EBER1 and EBER2 were found to inhibit phosphorylation of PKR in EBV-negative Akata and Daudi cell clones (Figure 10).

Fig. 10. Effect of transient expression of EBER1 or EBER2 on phosphorylation of PKR in EBV-negative Akata and Daudi cell clones. Cells (5 × 106) were co-transfected with a FLAG epitope-tagged PKR plasmid (20 µg) and EBER plasmid (20 µg) by the electroporation method, incubated for 48 h, and subjected to in vitro kinase assay. EBER expression was determined by RT–PCR.
Role of PKR in resistance to IFN-α-induced apoptosis in BL cells
Although it has been shown that EBERs bind to PKR in a cell-free system (Clarke et al., 1991), it is not known whether EBERs bind to PKR in EBER-expressing cells. To clarify this, we transfected EBV-positive Akata, Daudi and Mutu cell clones with the FLAG-tagged PKR plasmid, precipitated with the anti-FLAG antibody, and determined by RT–PCR whether EBERs were co-precipitated along with PKR. The results indicated that EBERs were co-precipitated along with PKR (Figure 11).
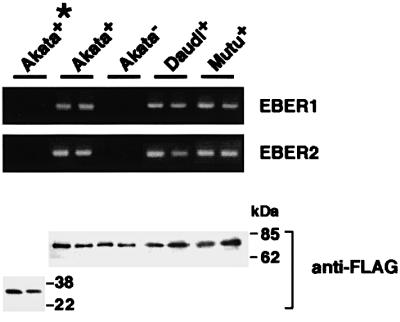
Fig. 11. Binding assay for association of EBER1 and EBER2 with PKR. Cells were transfected with a FLAG-tagged PKR plasmid. As a control, an EBV-positive Akata cell clone was transfected with a FLAG-tagged mutant PKR plasmid (Wu and Kaufman, 1997), which lacked the sequence coding for the dsRNA-binding domain (indicated as Akata+*). After 48 h of transfection, cells were treated with UV irradiation, digested with RNases to remove unbound RNA sequences and subjected to immunoprecipitation with anti-FLAG antibody. RNA was isolated from the immunoprecipitate, and EBER1 and EBER2 were measured by RT–PCR.
Next, we examined whether resistance to IFN-α-induced apoptosis in BL cells was due to inhibition of PKR. A mutant form of PKR (mPKR) that was catalytically inactive and could block phosphorylation of endogenous PKR (Katze et al., 1991) was introduced into EBV-negative Daudi cells, and cell clones that stably expressed mPKR were isolated and examined for their response to IFN-α. The results showed that mPKR expression made EBV-negative Daudi cells resistant to IFN-α-induced apoptosis (Figure 12A and B).

Fig. 12. Effect of dominant-negative PKR (mPKR) on phosphorylation of PKR and IFN-α-induced apoptosis in EBV-negative Daudi cells. HA-tagged mPKR plasmid was transfected into an EBV-negative Daudi cell clone, and cell clones that stably expressed mPKR were selected in the medium containing G418. (A) Expression of transfected HA-tagged mPKR that was detected by immunoblotting using an anti-HA polyclonal antibody. (B) Phosphorylation of wild-type PKR in EBV-negative Daudi cell clones expressing mPKR. Cells (5 × 106) were transfected with the FLAG epitope-tagged wild-type PKR plasmid by the electroporation method. After 48 h of incubation, FLAG-PKR was immunoprecipitated with anti-FLAG antibody and subjected to in vitro kinase assay. Immunoprecipitated FLAG-PKR was detected by immunoblotting using anti-FLAG antibody (lower panel), and its phosphorylation was visualized by autoradiography (upper panel). (C) Apoptosis assay of EBV-negative Daudi cell clones expressing mPKR. Cells (5 × 104/ml) were incubated in the presence or absence of IFN-α (500 U/ml) for various times. The frequency of apoptotic cells was determined by flow cytometry. Results are expressed as the means of triplicate wells.
To clarify further whether EBERs binding to PKR is necessary for inhibition of kinase activity of PKR, we generated two plasmids (K150A and A158D) carrying FLAG-tagged mutant PKR that lacked dsRNA binding activity but had kinase activity (Patel et al., 1996). We transfected these plasmids into EBV-positive Akata cells, and precipitated them with the anti-FLAG antibody. As shown in Figure 13, EBERs were not co-precipitated along with mutant PKR. In vitro kinase assay showed that phosphorylation of mutant PKR was not inhibited in EBER-expressing EBV-positive Akata cells. These results clearly demonstrate that EBERs binding to PKR is necessary for inhibition of PKR activation.

Fig. 13. Effect of EBER expression on kinase activity of mPKR that lacked dsRNA-binding activity. EBV-positive Akata cells were transfected with FLAG-tagged wild-type PKR plasmid or with FLAG-tagged mPKR plasmid (K150A or A158D). For binding assay for association of EBER1 and EBER2 with PKR, after 48 h of transfection cells were treated with UV irradiation, digested with RNases to remove unbound RNA sequences, and subjected to immunoprecipitation with anti-FLAG antibody. RNA was isolated from the immunoprecipitate, and EBER1 and EBER2 were measured by RT–PCR. For in vitro kinase assay, after 48 h of transfection FLAG-PKR was immunoprecipitated from the cells with anti-FLAG antibody and subjected to in vitro kinase assay. Immunoprecipitated FLAG-PKR was detected by immunoblotting using anti-FLAG antibody (lower panel), and its phosphorylation was visualized by autoradiography (upper panel).
Finally, we examined the role of interleukin-10 (IL-10) in resistance to IFN-α-induced apoptosis, since this cytokine was induced by EBER expression in BL cells (Kitagawa et al., 2000) and was reported to inhibit apoptosis in other systems (Alas et al., 2001). We carried out two experiments. First, we examined whether addition of recombinant IL-10 in the culture could block IFN-α-induced apoptosis in EBV-negative Akata cells. Secondly, we examined whether an anti-IL-10 antibody could abrogate resistance to IFN-α-induced apoptosis in EBV-positive Akata cells. Both results indicated that IL-10 was not related to IFN resistance (Figure 14).

Fig. 14. Role of IL-10 in resistance to IFN-α-induced apoptosis. (A) EBV-negative Akata cells (5 × 104/ml) were incubated in RPMI 1640 medium containing human IFN-α (500 U/ml) in the presence or absence of recombinant IL-10 (100 pg/ml; Endogen) for 4 days. (B) EBV-positive Akata cells (5 × 104/ml) were incubated in RPMI 1640 medium containing human IFN-α (500 U/ml) in the presence or absence of purified rat anti-human IL-10 antibody (50 ng/ml; PharMingen) for 4 days. The frequency of apoptotic cells was determined by flow cytometry. Results are expressed as the means of triplicate wells.
Discussion
This is the first report showing that a virus product confers resistance against the IFN-α-induced apoptosis in virus-associated tumor cells. The human T cell lymphotropic virus type I (HTLV-I)-encoded transactivator protein tax has been reported to confer resistance to IFN-triggered apoptosis in HTLV-I-transformed cells (El-Sabban et al., 2000). However, tax is not expressed in in vivo tumor cells of adult T cell leukemia (ATL). Although the non-structural 5A (NS5A) protein of hepatitis C virus (HCV) has been shown to bind and inhibit PKR, it is not known whether NS5A confers resistance to IFN-induced apoptosis in HCV-related carcinoma cells (Gale et al., 1999). All three BL cell lines examined in this study retain the in vivo phenotype of EBV expression, thus suggesting that EBV plays a role in IFN resistance in the in vivo tumor situation of BL, and also in other EBV-associated malignancies, because EBERs were expressed in all EBV-associated malignancies.
EBERs, consisting of EBER1 and EBER2, are non-polyadenylated RNAs transcribed by the RNA polymerase III system. They are 166 and 172 nucleotides long, respectively, and are the most abundant EBV RNAs in latently infected cells (Sharp et al., 1993). EBERs have been reported to bind some cellular proteins: La (Lerner et al., 1981), EAP/L22 (Toczyski and Steiz, 1991; Toczyski et al., 1994) and PKR (Clarke et al., 1991). Among them, the association of EBERs with PKR has been most intensively studied. In vitro assays have demonstrated that they can bind PKR and inhibit its activation, and block phosphorylation of eIF-2α, thus resulting in the blockage of inhibition of protein synthesis by eIF-2α (Sharp et al., 1993). In a previous study, we demonstrated that EBERs conferred resistance to induction of apoptosis by various stimuli such as cycloheximide, glucocorticoid and hypoxic stress, although we did not examine the relationship between resistance to apoptosis and PKR in that study (Komano et al., 1999). After our report, similar results were also reported by Yamamoto et al. (2000), who showed that EBER expression in B lymphoma cells caused inhibition of phosphorylation of PKR in an EBV-negative B lymphoma cell line, BJAB. However, their report did not show whether the resistance to apoptosis in BJAB cells is conferred by inhibition of phosphorylation of PKR. In the present study we demonstrated that transfection of dominant-negative PKR, which was catalytically inactive and could block phosphorylation of endogeneous PKR, made EBV-negative BL cells resistant to IFN-α-induced apoptosis. Furthermore, it was shown that EBERs did not bind mutant PKR, which was catalytically active but lacked dsRNA-binding activity, nor did they inhibit its phosphorylation. These results indicate that EBERs confer resistance to IFN-α-induced apoptosis via binding to PKR and inhibition of its phosphorylation.
Concerning the anti-apoptotic effect of EBERs, Ruf et al. (2000) reported that EBERs failed to support Akata cell survival following serum deprivation. Different experimental conditions, including stimuli for inducing apoptosis, may account for the difference between our results and those of Ruf et al. (2000).
Even though it is clear that IFN-α can exert antitumor activity in various malignancies, surprisingly little is understood about the exact mechanism of its antitumor action. Its cytoreductive effects can be divided into growth inhibition and cytotoxicity. The latter includes induction of apoptosis. IFN-α has been reported to induce FAS expression in basal cell carcinoma cells and myeloma cells, thus committing the cells to apoptosis (Buechner et al., 1997; Spets et al., 1998). However, in the BL cell lines studied here, we could not see any difference in the level of FAS expression before and after IFN treatment (data not shown). Subclones of BL-derived Daudi lines are highly sensitive to the antiproliferative effects of IFN-α and have been routinely used to explore the mechanisms of the antitumor effect of IFN. IFN-α stimulates the Jak–STAT signaling pathway, thereby inducing several changes in proteins associated with the cell cycle, and results in G1 arrest in Daudi cells (Grimley et al., 1998). On the other hand, the mechanism of apoptosis induction in Daudi cells is poorly understood. From that viewpoint, the present finding is particularly important, because this is the first report showing that IFN-α-induced apoptosis in tumor cells is mediated via PKR phosphorylation.
It has been reported that IFN inhibits the proliferation of EBV-infected B lymphocytes and prevents the eventual outgrowth of EBV-transformed cells into LCLs when added at the time of infection (Lotz et al., 1985). On the other hand, EBV-transformed LCLs are insensitive to the antiproliferative effects of exogenously added IFN. Aman and von Gabain (1990) reported that EBNA2 expression correlates well with IFN resistance and that the presence of the EBNA2 and EBNA5 (also called EBNA-LP) sequences is sufficient to mediate resistance to IFN. Furthermore, LMP1 has been reported to induce antiapoptotic protein bcl-2 (Henderson et al., 1991). Although Swaminathan et al. (1992) reported that EBV lacking EBER could still transform B cells in vitro and that the absence of EBER expression did not affect the response of cells to IFN treatment, it is possible that EBNA2 and/or LMP1 conferred resistance to IFN-induced growth inhibition in LCLs transformed with EBER-negative EBV recombinants.
In virus-associated malignancies, it is very important for viruses to compromise host immunity. EBV establishes latent infection in B lymphocytes. These cells are potentially oncogenic and develop to lymphomas under conditions of severe immune deficiency such as AIDS and after transplantation (Rickinson and Kieff, 1996). On the other hand, in other EBV-associated malignancies, including BL and gastric carcinoma, a decrease in the number of EBV genes expressed is a strategy for the virus to survive in immunocompetent hosts. We have recently found that EBERs induce expression of human IL-10 (Kitagawa et al., 2000), which is known to suppress T-helper 1 (TH1) cytotoxic T lymphocytes. These findings, together with the present study, demonstrate ingenious strategies of EBV to escape both IFN-mediated non-specific immunity and TH1-mediated specific immunity through the action of EBERs. Although the present study deals with BL, EBERs should exhibit these activities in other EBV-associated malignancies, since EBERs are expressed in all EBV-associated malignancies known thus far.
Materials and methods
Cell lines and culture
Akata (Takada et al., 1991), Daudi (Klein et al., 1968) and Mutu (Gregory et al., 1990) are type I BL cell lines. We isolated EBV-negative Akata, Daudi and Mutu cell clones from the parental cultures by the limiting dilution method (Shimizu et al., 1994; Kitagawa et al., 2000). Cells were maintained in RPMI 1640 medium (Sigma) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL), penicillin (40 U/ml) and streptomycin (50 µg/ml) at 37°C in a 5% CO2 incubator.
Plasmids, transfection and cell cloning
PKR plasmid carries the simian virus 40 (SV40) promoter-driven FLAG-tagged PKR. Two mutant PKR plasmids, K150A (Lys150→Ala) and A158D (Ala158→Asp), lacking dsRNA-binding activity (Patel et al., 1996) were generated by oligonucleotide-directed mutagenesis using the following oligonucleotides: K150A, 5′-GGTTCTACTGCACAGGAAGCA-3′ and A158D, 5′-CAATTGGCCGATAAACTTGC-3′. The plasmid for the catalytically inactive mutant form (Lys296→Arg) of PKR (mPKR) (Katze et al., 1991) carries SV40-driven influenza virus-encoded hemagglutinin (HA)-tagged mPKR and the neomycin resistance (neoR) gene driven by the SV40 promoter. EBER1 and EBER2 open reading frames are located at 6628–6796 bp and at 6958–7129 bp, respectively, on the EcoRI K fragment of Akata EBV DNA, which corresponds to the EcoRI J fragment of B95–8 EBV DNA (Baer et al., 1984). Since plasmids that contained a single copy of EBER could not induce levels of EBER expression in transfected cells equivalent to those in EBV-infected cells, we used EBER plasmids that contained 10 tandem repeats of the EBER1 and EBER2 subfragments (6297–7325 bp) from the EcoRI K fragment of Akata EBV DNA and neoR (Komano et al., 1999). The EBER1 plasmid and the EBER2 plasmid contained 10 tandem repeats of the EBER1 subfragment (6288–6797 bp) and EBER2 subfragment (6800–7317 bp), respectively, from the EcoRI K fragment of Akata EBV DNA and neoR driven by the SV40 promoter (Kitagawa et al., 2000). The pEBO plasmid carries the SV40 promoter-driven neoR, the EBNA1 gene and ori-P sequence of B95–8 origin. The BARF0 plasmid carries the SV40 promoter-driven neoR and the FLAG-tagged BARF0 gene derived from Akata EBV DNA.
Each of the plasmids was introduced into cells by the electroporation method. For isolation of stable transfectants, transfected cells were cultured for 2 days and transferred to 96-well, flat-bottom plates at 5000–10 000 cells/well in complete culture medium containing an appropriate concentration of G418 (Gibco-BRL) for selection (700 µg/ml for Akata, 1.1 mg/ml for Daudi and 1.5 mg/ml for Mutu). Cultures were fed every 5 days by replacement of half of the medium until colonies emerged.
Generation of recombinant EBV and EBV infection
As described previously (Shimizu et al., 1996), the EBER-positive EBV recombinant carries neoR inserted into the EBV thymidine kinase gene, which is non-essential for infection and replication. An EBER-knockout EBV was generated by replacing the EBER coding region (6609–7270 bp) with neoR (Kitagawa et al., 2000). Preparation of the virus solution was as described previously (Shimizu et al., 1996). For EBV infection, 5 × 106 cells were suspended in 2 ml of diluted EBV solution (1:2–10) for 60 min at room temperature with continuous gentle mixing. Then they were washed and cultured for 2 days. Selection and cloning procedures are described above.
Apoptosis assay
Apoptotic cells were measured by the appearance of cells with sub-G1 DNA content (Nicoletti et al., 1991) and DNA laddering. Apoptotic cells appeared as a broad hypodiploid DNA peak that was easily discriminated from the narrow peak of normal diploid cells. Cells (5 × 104) were suspended in 1 ml of culture medium containing 500 U/ml recombinant human IFN-α (Pepro Tech EC) and incubated for up to 96 h. At the designated time, cells were harvested and fixed in 400 µl of phosphate-buffered saline (PBS) containing 50% ethanol. Then cells were suspended in 300 µl of PBS containing 50 µg/ml RNase A and incubated for 30 min at 37°C. After low-speed centrifugation at 3500 r.p.m. for 3 min, the cell pellet was suspended in 300 µl of PBS containing 1 mg/ml propidium iodide, and the percentage of apoptotic cells with hypodiploid DNA was measured by using a FACScalibur (Becton Dickinson). For the DNA laddering assay, 106 cells were lysed in 100 µl of lysis buffer containing 10 mM EDTA, 10 mM Tris–HCl (pH 8.0) and 0.5% Triton X-100 and then kept on ice for 10 min. Supernatants were collected by centrifugation of the sample at 16 000 r.p.m. for 5 min. Supernatant was then treated with RNase A (200 µg/ml) at 37°C for 1 h and with proteinase K (200 µg/ml) (Takara) at 50°C for 30 min, and nucleic acids were precipitated in 50% 2-propanol overnight. Finally, the pellet was resolved in 20 µl of water.
In vitro kinase assay
Measurement of kinase activity of FLAG-PKR was carried out by the method of Laurent et al. (1985) with a slight modification. Briefly, the FLAG-PKR expression plasmid was transfected into cells by the electroporation method. At 48 h post-transfection, cells were lysed in high-salt buffer I [20 mM Tris–HCl pH 7.5, 50 mM KCl, 400 mM NaCl, 1 mM EDTA, 1 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 100-fold diluted phosphatase inhibitor cocktail I (Sigma), 1% Triton X-100 and 20% glycerol]. Cell extracts corresponding to 3 × 106 cells were pre-incubated with 20 µl of protein G–Sepharose (Amersham Pharmacia) in 1 ml of high-salt buffer I for 1 h at 4°C and incubated with the M2 monoclonal antibody to FLAG (Sigma) for 2 h at 4°C, followed by incubation with 20 µl of protein G–Sepharose for 1 h at 4°C. Immunoprecipitants were washed in high-salt buffer I three times, twice in buffer II (10 mM Tris–HCl pH 7.5, 100 mM KCl, 0.1 mM EDTA, 1 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM PMSF, 400-fold diluted phosphatase inhibitor cocktail I and 20% glycerol), and then once in kinase reaction buffer (20 mM Tris–HCl pH 7.5, 0.01 mM EDTA, 50 mM KCl, 2 mM MgCl2, 2 mM MnCl2, 0.1 mM PMSF and 20% glycerol). Following the last wash, immunoprecipitants were resuspended in 20 µl of kinase reaction buffer containing 2 µM [γ-32P]ATP (3000 Ci/mmol) (NEN), and incubated for 15 min at 30°C. The reaction was stopped by addition of 10 µl of 4-fold concentrated sample buffer (500 mM Tris–HCl pH 6.8, 10% SDS, 4% 2-mercaptoethanol, 0.4% bromophenol blue and 40% glycerol). The samples were analyzed in 10% SDS–polyacrylamide gels. FLAG-PKR expression was determined by immunoblot analysis with the M2 antibody, and its phosphorylation was visualized by autoradiography.
Immunoblot analysis
Cells were lysed in SDS–PAGE loading buffer, sonicated and boiled for 5 min. Cell lysate equal to 105 cells was separated in 10% polyacrylamide gels and transferred to a nitrocellulose membrane (Schleicher & Schuells). After blocking with 5% non-fat dry milk in Tris-buffered saline (TBS-M pH 7.6), the membrane was incubated for 2 h at room temperature with appropriate antibodies diluted in TBS-M. For detection of EBNAs, the membrane was incubated with human serum, washed three times with TBS-M containing 0.1% Tween 20 (TBS-TM), and then serially reacted for 30 min each with biotinylated rabbit anti-human IgG (Dako) diluted 1:500 in TBS-M and alkaline phosphatase-conjugated streptoavidin (Amersham Pharmacia) diluted 1:3000 in TBS-M with washing between each reaction. Antibodies used are PE2 for EBNA2 (a gift from E.Kieff, Harvard Medical School, Boston, MA), CS1-4 for LMP1 (Dako), M2 for FLAG-tagged proteins (Upstate Technology), polyclonal antibodies for PKR (Santa Cruz), HA-tagged proteins (Santa Cruz), eIF-2α and IκBα (Cell Signaling Technology), and phosphorylation site-specific polyclonal antibodies for eIF-2α (Ser51) and IκBα (Ser32 and Ser36) (Biosource International). Antibody reaction and washing were carried out in TBS and TBS-T solutions, respectively. The second antibody reaction was done with horseradish peroxidase-conjugated sheep antibodies to Ig (Amersham Pharmacia) diluted 1:3000 in TBS-M. After the second antibody reaction, the filters were washed five times with TBS-T, immersed in enhanced chemiluminescence solution (Amersham Pharmacia) as specified by the manufacturer, and subjected to autoradiography.
RT–PCR
RT–PCR analysis was carried out to investigate the expression of EBV latent genes and the utilization of EBNA promoters (Qp, Cp and Wp) as described previously (Imai et al., 1998). Total cellular RNA was isolated by guanidium isothiocyanate–phenol extraction using TRIzol reagent (Gibco-BRL) according to the manufacturer’s protocol. Extracted RNA was heated for 10 min at 70°C and then rapidly cooled on ice. cDNA synthesis was performed for 60 min at 37°C with Molony murine leukemia virus RTase (Gibco-BRL) using 100 pmol of random hexamer (Takara) followed by 10 min of heating at 94°C to inactivate RTase. The cDNA samples were then subjected to 30 cycles of PCR in a thermal cycler. Each cycle consisted of denaturation for 30 s at 94°C, annealing for 30 s at 45–55°C and extension for 1 min at 72°C. The reaction mixture contained buffers and reagents as described, with 20 pmol of each primer and cDNA (equivalent to 5 × 104 cells/tube) in a volume of 50 µl. Five microliters of the PCR product were electrophoresed on a 2% agarose gel, blotted onto nylon membranes (Hybond N+; Amersham Pharmacia), and specific amplified DNA was detected by ECL 3′-oligolabeling and detection systems (Amersham Pharmacia). The quality of RNA was checked by parallel amplification of GAPDH mRNA.
Primers used for detection of EBERs were 5′-AGGACCTACGCTGCCCTAGA-3′ (upstream) and 5′-CCCTAGAAATGGTGCCAATG-3′ (downstream) for EBER1 and 5′-AGGACAGCCGTTGCCCTAGTGGTTTCG-3′ (upstream) and 5′-AAAAACAGCGGACAAGCCGAATACC-3′ (downstream) for EBER2. PCR was 12 cycles consisting of denaturation for 30 s at 94°C, annealing for 30 s at 55°C and extension for 1 min at 72°C. One-fifth of the PCR product was subjected to 2% agarose gel electrophoresis and was visualized by ethidium bromide staining.
UV cross-linking and immunoprecipitation
Binding of EBER to PKR was analyzed by UV cross-linking and immunoprecipitation of PKR–RNA complexes. A FLAG-tagged PKR plasmid was transfected into EBV-positive and -negative BL cell clones by the electroporation method. As a control, an EBV-positive Akata cell clone was transfected with a FLAG-tagged mutant PKR plasmid (Wu and Kaufman, 1997), which lacked the sequence coding for the dsRNA-binding domain. At 48 h after transfection, cells (107) were washed with PBS, suspended in 2 ml PBS and transferred to a pre-chilled 10 cm tissue culture dish. UV irradiation was performed for 5 min with 8 W germicidal lamp at a 4 cm distance (GS Gene Linker™; Bio-Rad). N-laurylsarcosine was then added to cell suspension at a final concentration of 0.5% (w/v). Thereafter, cells were washed with PBS and suspended in 200 µl of immunoprecipitation buffer (20 mM Tris–HCl pH 7.4, 0.15 M NaCl, 5 mM EDTA, 4 µg/ml each leupeptin and pepstatin, and 1 mM PMSF], and lysed by sonication for 15 s. The sample was clarified by centrifugation and treated with 20 U of RNases T1 and 10 µg of RNases A for 30 min at 37°C to remove unbound RNA sequences, and incubated with the anti-FLAG M2 antibody for 4 h at 4°C and with protein G–Sepharose for 1 h at 4°C. The immunoprecipitate was treated with proteinase K, and subjected to phenol extraction and ethanol precipitation for RNA isolation. Three-tenths of the RNA sample was used as a template for cDNA synthesis, and half each of that was used for PCR for EBER1 and EBER2, respectively. One-fifth of the PCR product was subjected to agarose gel electrophoresis.
Acknowledgments
Acknowledgements
This work was supported by grants-in-aid from the Ministry of Education, Science, Sports, Culture, and Technology, Japan.
References
- Alas S., Emmanouilides,C. and Bonavida,B. (2001) Inhibition of interleukin 10 by rituximab results in down-regulation of bcl-2 and sensitization of B-cell non-Hodgkin’s lymphoma to apoptosis. Clin. Cancer Res., 7, 709–723. [PubMed] [Google Scholar]
- Aman P. and von Gabain,A. (1990) An Epstein–Barr virus immortalization associated gene segment interferes specifically with the IFN-induced anti-proliferative response in human B-lymphoid cell lines. EMBO J., 9, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer R. et al. (1984) DNA sequence and expression of the B95–8 Epstein–Barr virus genome. Nature, 310, 207–211. [DOI] [PubMed] [Google Scholar]
- Barber G.N. (2000) The interferons and cell death: guardians of the cell or accomplices of apoptosis. Semin. Cancer Biol., 10, 103–111. [DOI] [PubMed] [Google Scholar]
- Borden E.C., Lindner,D., Dreicer,R., Hussein,M. and Peereboom,D. (2000) Second-generation interferons for cancer: clinical targets. Semin. Cancer Biol., 10, 125–144. [DOI] [PubMed] [Google Scholar]
- Buechner S.A., Wernli,M., Harr,T., Hahn,S., Itin,P. and Erb,P. (1997) Regression of basal cell carcinoma by intralesional interferon-α treatment is mediated by CD95 (Apo-1/Fas)-CD95 ligand-induced suicide. J. Clin. Invest., 100, 2691–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P.A., Schwemmle,M., Schickinger,J., Hilse,K. and Clemens,M.J. (1991) Binding of Epstein–Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucleic Acids Res., 19, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sabban M.E. et al. (2000) Arsenic-interferon-α-triggered apoptosis in HTLV-I transformed cells is associated with tax down-regulation and reversal of NF-κB activation. Blood, 96, 2849–2855. [PubMed] [Google Scholar]
- Flint S.J., Enquist,L.W., Krug,R.M., Racaniello,V.R. and Skalka,A.M. (2000) Principles of Virology. ASM Press, Washington, DC.
- Galabru J. and Hovanessian,A. (1987) Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem., 262, 15538–15544. [PubMed] [Google Scholar]
- Gale M., Kwieciszewski,B., Dossett,M., Nakao,H. and Katze,M.G. (1999) Antiapoptotic and oncogeneic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol., 73, 6506–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory C.D., Rowe,M. and Rickinson,A.B. (1990) Different Epstein–Barr virus–B cell interactions in phenotypically distinct clones of a Burkitt’s lymphoma cell line. J. Gen. Virol., 71, 1481–1495. [DOI] [PubMed] [Google Scholar]
- Grimley P.M. et al. (1998) Prolonged STAT1 activation related to the growth arrest of malignant lymphoma cells by interferon-α. Blood, 91, 3017–3027. [PubMed] [Google Scholar]
- Henderson S., Rowe,M., Gregory,C., Croom-Carter,D., Wang,F., Longnecker,R., Kieff,E. and Rickinson,A. (1991) Induction of bcl-2 expression by Epstein–Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell, 65, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Imai S., Nishikawa,J. and Takada,K. (1998) Cell-to-cell contact as an efficient mode of Epstein–Barr virus infection of diverse human epithelial cells. J. Virol., 72, 4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze M.G., Wambach,M., Wong,M.L., Garfinkel,M., Meurs,E., Chong,K., Williams,B.R., Hovanessian,A.G. and Barber,G.N. (1991) Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol. Cell. Biol., 11, 5497–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieff E. (1996) Epstein–Barr virus and its replication. In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 3rd edn. Lippincott-Raven, Philadelphia, PA, pp. 2343–2396.
- Kitagawa N. et al. (2000) Epstein–Barr virus-encoded poly(A)– RNA supports Burkitt’s lymphoma growth through interleukin-10 induction. EMBO J., 19, 6742–6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E., Klein,G., Nadkarni,J.S., Nadkarni,J.J., Wigzell,H. and Clifford,P. (1968) Surface IgM-κ specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer Res., 28, 1300–1310. [PubMed] [Google Scholar]
- Komano J., Maruo,S., Kurozumi,K., Oda,T. and Takada,K. (1999) Oncogenic role of Epstein–Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol., 73, 9827–9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Haque,J., Lacoste,J., Hiscott,J. and Williams,B.R. (1994) Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc. Natl Acad. Sci. USA, 91, 6288–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent A.G., Krust,B., Galabru,J., Svab,J. and Hovanessian,A.G. (1985) Monoclonal antibodies to an interferon-induced Mr 68,000 protein and their use for the detection of double-stranded RNA-dependent protein kinase in human cells. Proc. Natl Acad. Sci. USA, 82, 4341–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner M.R.,Andrews,N.C., Miller,G. and Steitz,J.A. (1981) Two small RNAs encoded by Epstein–Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl Acad. Sci. USA, 78, 805–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz M., Tsoukas,C.D., Fong,S., Carson,D.A. and Vaughan,J.H. (1985) Regulation of Epstein–Barr virus infection by recombinant interferons. Selected sensitivity to interferon-γ. Eur. J. Immunol., 15, 520–525. [DOI] [PubMed] [Google Scholar]
- Margolskee R.F., Kavathas,P. and Berg,P. (1988) Epstein–Barr virus shuttle vector for stable episomal replication of cDNA expression libraries in human cells. Mol. Cell. Biol., 8, 2837–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti I., Migliorati,G., Pagliacci,M.C., Grignani,F. and Riccardi,C. (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods, 139, 271–279. [DOI] [PubMed] [Google Scholar]
- Patel R.C. and Sen,G.C. (1998) PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J., 17, 4379–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.C., Stanton,P. and Sen,G.C. (1996) Specific muations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J. Biol. Chem., 271, 25657–25663. [DOI] [PubMed] [Google Scholar]
- Rickinson A.B. and Kieff,E. (1996) Epstein–Barr virus. In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 3rd edn. Lippincott-Raven, Philadelphia, PA, pp. 2397–2446.
- Rowe M., Rowe,D.T., Gregory,C.D., Young,L.S., Farrell,P.J., Rupani,H. and Rickinson,A.B. (1987) Differences in B cell growth phenotype reflect novel patterns of Epstein–Barr virus latent gene expression in Burkitt’s lymphoma cells. EMBO J., 6, 2743–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf I.K., Rhyne,P.W., Yang,C., Cleveland,J.L. and Sample,J.T. (2000) Epstein–Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J. Virol., 74, 10223–10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C.E. (1993) The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem., 268, 7603–7606. [PubMed] [Google Scholar]
- Sharp T.V., Schwemmle,M., Jeffrey,I., Laing,K., Mellor,H., Proud,C.G., Hilse,K. and Clemens,M.J. (1993) Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein–Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res., 21, 4483–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N., Tanabe-Tochikura,A., Kuroiwa,Y. and Takada,K. (1994) Isolation of Epstein–Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J. Virol., 68, 6069–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N., Yoshiyama,H. and Takada,K. (1996) Clonal propagation of Epstein–Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Virol., 70, 7260–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spets H., Georgii-Hemming,P., Siljason,J., Nilsson,K. and Jernberg-Wiklund,H. (1998) Fas/APO-1 (CD95)-mediated apoptosis is activated by interferon-γ and interferon-α in interleukin-6 (IL-6)-dependent and IL-6-independent multiple myeloma cell lines. Blood, 92, 2914–2923. [PubMed] [Google Scholar]
- Swaminathan S., Huneycutt,B.S., Reiss,C.S. and Kieff,E. (1992) Epstein–Barr virus-encoded small RNAs (EBERs) do not modulate interferon effects in infected lymphocytes. J. Virol., 66, 5133–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K. (1984) Cross-linking of cell surface immunoglobulins induces Epstein–Barr virus in Burkitt lymphoma lines. Int. J. Cancer, 33, 27–32. [DOI] [PubMed] [Google Scholar]
- Takada K., Horinouchi,K., Ono,Y., Aya,T., Osato,T., Takahashi,M. and Hayasaka,S. (1991) An Epstein–Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes, 5, 147–156. [DOI] [PubMed] [Google Scholar]
- Takeda T., Mizugaki,Y., Matsubara,L., Imai,S., Koike,T. and Takada,K. (2000) Lytic Epstein–Barr virus infection in the synovial tissue of patients rheumatoid arthritis. Arthritis Rheum., 43, 1218–1225. [DOI] [PubMed] [Google Scholar]
- Toczyski D.P. and Steitz,J.A. (1991) EAP, a highly conserved cellular protein associated with Epstein–Barr virus small RNAs (EBERs). EMBO J., 10, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski D.P., Matera,A.G., Ward,D.C. and Steitz,J.A. (1994) The Epstein–Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc. Natl Acad. Sci. USA, 91, 3463–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. and Kaufman,R.J. (1997) A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem., 272, 1291–1296. [DOI] [PubMed] [Google Scholar]
- Yamamoto N., Takizawa,T., Iwanaga,Y., Shimizu,N. and Yamamoto,N. (2000) Malignant transformation of B lymphoma cell line BJAB by Epstein–Barr virus-encoded small RNAs. FEBS Lett., 484, 153–158. [DOI] [PubMed] [Google Scholar]