

Abstract
The human cytomegalovirus gene products US2 and US11 induce proteasomal degradation of MHC class I heavy chains. We have generated an enhanced green fluorescent protein–class I heavy chain (EGFP–HC) chimeric molecule to study its dislocation and degradation in US2- and US11-expressing cells. The EGFP–HC fusion is stable in control cells, but is degraded rapidly in US2- or US11-expressing cells. Proteasome inhibitors induce in a time-dependent manner the accumulation of EGFP–HC molecules in US2- and US11-expressing cells, as assessed biochemically and by cytofluorimetry of intact cells. Pulse–chase analysis and subcellular fractionation show that EGFP–HC proteins are dislocated from the endoplasmic reticulum and can be recovered as deglycosylated fluorescent intermediates in the cytosol. These results raise the possibility that dislocation of glycoproteins from the ER may not require their full unfolding.
Keywords: dislocation/endoplasmic reticulum/HCMV US2/HCMV US11/proteasomal degradation
Introduction
Misfolded proteins, a consequence of imperfections in protein folding, are unavoidable by-products of protein biosynthesis, regardless of the destination of the translation product (Schubert et al., 2000). Polypeptides that do not acquire their proper conformation or quaternary structure usually fail to reach their target and are destroyed (Bonifacino and Weissman, 1998). The endoplasmic reticulum (ER) is the point of insertion of most secretory and membrane proteins and serves as an important station for quality control (Ellgaard et al., 1999). In the ER, the requirements for protein folding and sensing of the misfolded state are not easily reconciled with the coincident degradation of misfits. The distinctions between a nascent polypeptide that has yet to reach its final conformation, and a protein that has exhausted all attempts at proper folding defy a simple description.
In living cells, folding and degradation of ER-inserted proteins are carefully segregated in space. The ER provides an environment that is conducive to protein folding, usually catalyzed by chaperones that include calnexin and calreticulin as well as oxidoreductases such as PDI and ERp57 (Frand et al., 2000; High et al., 2000). Proteolysis of unwanted polypeptides is segregated from the folding environment by the ER membrane and can occur in the cytosol. This arrangement requires the transfer of misfolded proteins from the ER to the cytosol, a process known as dislocation or retrograde translocation (Wiertz et al., 1996a). Based on genetic experiments in yeast and biochemical analysis in mammalian cells, the dislocation reaction is carried out via the translocon or Sec61 complex (Wiertz et al., 1996b; Pilon et al., 1997; Plemper et al., 1997, 1998; Bebok et al., 1998; Zhou and Schekman, 1999). Some lumenal chaperones, such as calnexin, BIP and PDI, have also been implicated in this reaction (McCracken and Brodsky, 1996; Plemper et al., 1997; Gillece et al., 1999; Nishikawa et al., 2001). In addition, ER membrane proteins as well as cytosolic proteins are involved in the degradation process (Hampton et al., 1996; Hiller et al., 1996; Knop et al., 1996; Biederer et al., 1997; Bordallo et al., 1998; Tiwari and Weissman, 2001; Zhang et al., 2001). However, most of the proteins in the latter category are associated with ubiquitylation events, usually a necessary step for proteasomal degradation (Hirsch and Ploegh, 2000; Pickart, 2000).
Virus-infected cells must avoid detection by the immune system if they are to continue to produce infectious virus (Tortorella et al., 2000). The Herpes viruses are particularly adept at avoiding immune recognition. To achieve temporary invisibility, they have co-opted the mechanisms of protein turnover to selectively eliminate major histocompatibility complex (MHC) class I molecules involved in antigen presentation (Tortorella et al., 2000). The human cytomegalovirus (HCMV)-encoded US2 and US11 proteins accelerate destruction of MHC class I heavy chains (Wiertz et al., 1996a; Huppa and Ploegh, 1997; Tortorella et al., 1998; Shamu et al., 1999). By conferring specificity for class I molecules and by accelerating the rate constant of degradation, the HCMV-encoded US2 and US11 proteins manage to selectively reduce the half-life of the class I heavy chains from hours to minutes (Wiertz et al., 1996a,b). Since the presence of US2 or US11 is sufficient to observe enhanced degradation, these viral glycoproteins must somehow have adopted the machinery used for turnover of ER proteins. Does removal of glycans, when present, precede extraction or is it confined to the cytosol, and is it always catalyzed in the same manner? Is ubiquitin conjugation obligatorily required, and if so, where does this happen, and in what type of linkage is ubiquitin attached (Shamu et al., 1999)? Is reduction of intra- and inter-chain disulfide bonds a prerequisite for dislocation (Tortorella et al., 1998)? Can folded proteins be extracted, or is complete unfolding required? The answers to these questions will require a detailed analysis of a number of substrates susceptible to this mode of degradation.
Here we used an enhanced green fluorescent protein– class I heavy chain fusion (EGFP–HC) as a model protein to study US2- and US11-mediated dislocation and degradation. Characterization of EGFP–HC expressed in control U373-MG cells that do not express US2 or US11 shows that this reporter construct largely behaves like its endogenous counterpart. We show that the chimeric molecules and the endogenous class I heavy chains are dislocated and degraded in a similar manner in the presence of HCMV US2 and US11. Fluorescence microscopy and spectroscopy data are consistent with the possibility that dislocation does not require complete unfolding of the dislocation substrate.
Results
The EGFP–HC chimeric molecule is inserted into the membrane and folds properly
In the course of US2-/US11-induced class I degradation, ER-resident class I molecules are dislocated into the cytosol for degradation by the proteasome. To visualize the dislocation of class I molecules in US2- and US11- expressing cells, we generated a reporter construct comprised of an N-terminal signal sequence, followed by the EGFP moiety fused to the N-terminus of the HLA-A2 heavy chain (EGFP–HC). Because GFP is a stable molecule with a compact tertiary structure, the fluorescent properties of this reporter should be helpful in addressing the unfolding requirements of the EFGP/HC chimera prior to degradation.
For a simple GFP fusion protein, folding is inferred from the emergence of green fluorescence. In the case of the EGFP–HC fusion protein, we may use as an additional criterion the acquisition of the W6/32 antibody epitope, present on properly folded class I molecules only. We first examined the biosynthesis of EGFP–HC in a pulse–chase experiment in U373-MG cells stably transfected with EGFP–HC (U373EGFP–HC) (Figure 1). U373EGFP–HC cells were metabolically labeled for 15 min and chased up to 90 min. The product encoded by the EGFP–HC fusion construct was directed to the ER by the H2-Kb signal sequence fused to the N-terminus of the chimera. The ability of the fusion protein to fold and associate with β2-microglobulin (β2m) was assessed by its reactivity with the monoclonal antibody, W6/32 (Figure 1, lanes 1–6). Both the fusion protein and the endogenous MHC class I molecules acquired the W6/32 epitope, indicative of correct folding and assembly, although the EGFP–HC is less effective at acquiring the W6/32 epitope.
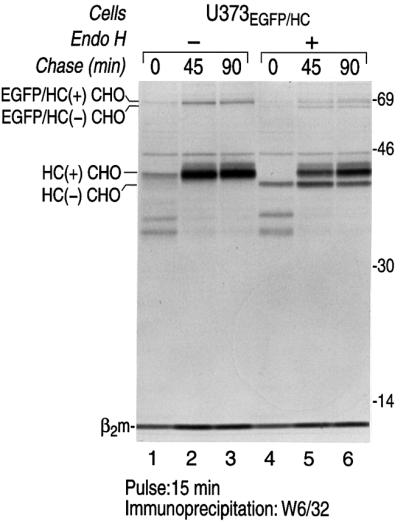
Fig. 1. The EGFP–HC reporter construct is a properly folded EGFP–class I fusion protein. U373EGFP–HC cells were pulse-labeled with [35S]methionine for 15 min and chased up to 90 min. Cells were lysed in NP-40 lysis buffer and immunoprecipitated with W6/32, a mAb that recognizes properly folded class I molecules only. The immunoprecipitates were analyzed by SDS–PAGE (12.5%). The endogenous class I heavy chains (HC) and EGFP–HC associated with β2m were recovered from cell lysates (lanes 1–3). Half of the immunoprecipitates were digested with Endo H (lanes 4–6). The positions of migration of class I molecules that contain an N-linked glycan (+CHO) or lack an N-linked glycan (–CHO) are indicated. Molecular weight markers are indicated on the right in kDa.
Does the EGFP–HC traffic to the cell surface? We first addressed biochemically whether the EGFP–HC molecules exit the ER. We found that a population of EGFP–HC molecules and endogenous class I molecules that were precipitated with W6/32 are resistant to endoglycosidase H (Endo H) treatment (Figure 1, lanes 4–6). Resistance to Endo H is indicative of N-linked glycan modifications that occur in the Golgi. Therefore, the EGFP–HC molecules that associate with β2m can exit the ER although we have not determined whether EGFP–HC travels to the surface as efficiently as its endogenous counterpart. We conclude that a significant population of EGFP–HC molecules is properly folded. For the remainder of our experiments, we focus on the events in the ER and cytosol.
Fluorescence microscopy was used to further analyze the distribution of the fusion protein in intact cells. The localization of the green fluorescent species, EGFP–HC, relative to the red staining pattern derived from W6/32, shows properly folded EGFP–HC at the cell surface and intracellularly, presumably in the ER (Figure 2A). Partial ER localization of EGFP–HC was confirmed by staining with antibodies against ER markers calnexin (Figure 2B) and PDI (Figure 2C). These results establish that the EGFP–HC is found throughout the secretory pathway and at the cell surface.

Fig. 2. Intracellular distribution of EGFP–HC is similar to that of endogenous class I molecules. U373EGFP–HC were grown on chamber slides over night, prior to fixation, immunohistochemistry and confocal laser scanning microscopy. Double-positivity of the green fluorescent EGFP–HC molecule with W6/32 corresponds to properly folded class I complexes that contain the chimeric molecule. These are found at the cell surface (A) and in the ER. ER localization of EGFP–HC is demonstrated by staining with the ER markers calnexin (mAb AF8) (B) and PDI (anti-PDI) (C). For each antibody the green EGFP–HC (left column), the respective second staining in red (middle column) and a merge image (right column) are shown, as indicated for the individual panels.
The EGFP–HC fusion molecule is a substrate for US2- and US11-mediated degradation
Is the EGFP–HC fusion molecule a substrate for the HCMV-encoded US2 and US11 proteins? We generated stable cotransfectants of EGFP–HC and US2 or US11 in U373-MG cells (US2EGFP–HC and US11EGFP–HC). The levels of expression of EGFP–HC, as assessed by pulse labeling, were similar to those in U373EGFP–HC cells. The fate of EGFP–HC in both cell lines was examined by pulse–chase analysis (Figure 3). The EGFP–HC molecules were recovered from SDS-treated cell lysates using a polyclonal anti-GFP serum (αGFP). The class I heavy chains were recovered using a polyclonal anti-heavy chain serum (αHC) that recognizes unfolded class I heavy chains. The immunoprecipitates were then analyzed by SDS–PAGE. Only in US2EGFP–HC and US11EGFP–HC cells did we observe a rapid loss of EGFP–HC and endogenous HC over the chase period (Figure 3A, lanes 4–9). These results are consistent with degradation of class I induced by US2 or US11 (Tortorella et al., 1998). Given the structural information available for the class I–US2 complex, we suggest that the ability of the EGFP–HC fusion to be degraded in a US2-dependent manner is further evidence of a correctly folded state for EGFP–HC (Gewurz et al., 2001a). Even though EGFP–HC and endogenous HC molecules are degraded with similar kinetics in US2EGFP–HC and US11EGFP–HC cells (Figure 3B), different amounts of endogenous HC are recovered at the 0 chase period (Figure 3A, compare lanes 13 and 16). This suggests that the presence of EGFP–HC molecules in US2 cells affects the kinetics of class I degradation, and slows degradation of endogenous class I heavy chains.


Fig. 3. EGFP–HC is degraded in an US2- and US11-dependent manner. (A) US11EGFP–HC and US2EGFP–HC cells were pulse-labeled with [35S]methionine for 15 min and chased up to 90 min. Cells were lysed in 1% SDS, then diluted to 0.07% SDS with NP-40 lysis mix followed by immunoprecipitation with anti-GFP serum (αGFP) (lanes 1–9) and anti-class I heavy chain serum (αHC) (lanes 10–18). The immunoprecipitates were analyzed by SDS–PAGE (12.5%). The positions of migration of the EGFP–HC and endogenous class I heavy chain (HC) polypeptides are indicated. (B) The amount of EGFP–HC and HC polypeptides recovered from U373EGFP–HC cells (black bar), US2EGFP–HC cells (gray bar) and US11EGFP–HC cells (white bar) were quantified by Phosphoimager analysis. The remaining class I molecules recovered at each chase point are given as a percentage of the class I molecules recovered at the 0 chase time.
A pulse–chase analysis of EGFP–HC in US2EGFP–HC and US11EGFP–HC cells was then conducted in the presence of the proteasome inhibitor, ZL3VS. The EGFP–HC and endogenous heavy chains were recovered from cell lysates using αHC serum (Figure 4). A precursor–product relationship between the EGFP–HC molecule and a faster moving polypeptide was observed during the chase period (Figure 4A, lanes 1–6). This intermediate is absent from cells not exposed to proteasome inhibitors (Figure 3). The presence of a biosynthetic intermediate of reduced molecular weight is consistent with the loss of the single N-linked glycan (Figure 4A, lanes 7–9) present in both the endogenous class I heavy chains and the fusion product. Indeed, the intermediate is resistant to digestion with purified N-glycanase. The deglycosylated intermediate is thus the product of an N-glycanase reaction, as confirmed by an isoelectric focusing experiment (Wiertz et al., 1996a; data not shown). The rate of appearance of the deglycosylated EGFP–HC intermediates in pulse–chase experiments was only moderately delayed when compared with the endogenous class I heavy chain intermediates. The presence of a compact folded EGFP moiety is likely to effect the rate of discharge of the fusion protein from the ER. In the presence of the proteasome inhibitor, minor quantities of additional polypeptides were detected over the course of the chase period (* and **, Figure 4A). Subcellular fractionation followed by immunoprecipitation shows that these polypeptides are cytosolic (Figure 4B). The slower migrating polypeptide (*) (Figure 4A) reacts with both αHC and αGFP, while the faster migrating polypeptide (**) (Figure 4A) reacts only with αHC (data not shown). Therefore, these fragments correspond to heavy chain-derived digestion products generated from cytosolic deglycosylated intermediates. At steady state, these products are not detectable (Figure 4C).



Fig. 4. EGFP–HC is dislocated from the ER into the cytosol. (A) US11EGFP–HC and US2EGFP–HC cells were pulse-labeled with [35S]methionine for 15 min and chased up to 90 min in the presence of the proteasome inhibitor, ZL3VS. Cells were lysed in 1% SDS, then diluted to 0.07% SDS with NP-40 lysis mix followed by immunoprecipitation with anti-class I heavy chain serum (αHC). The immunoprecipitates were analyzed by SDS–PAGE (12.5%). The EGFP–HC (lanes 1–9) and endogenous class I molecules (HC) (lanes 10–15) were recovered from US2EGFP–HC and US11EGFP–HC cell lysates. Some class I degradation intermediates (* and **) were present in the US2 and US11 cell lysates. Half of the immunoprecipitates from US11 cells were digested with N-glycanase (PNGase) (lanes 7–9 and 16–18). (B) US11EGFP–HC cells were pulse-labeled with [35S]methionine for 15 min and chased up to 90 min in the presence of the proteasome inhibitor, ZL3VS. Cells were homogenized and subjected to fractionation as described in Materials and methods. The EGFP–HC, endogenous HC (αHC; lanes 1–9) and transferrin receptor (αTfr; lanes 10–18), were recovered from the whole-cell lysate (Whole Cell), from the 100 000 g supernatant (100Kg-sup) and the 100 000 g pellet (100Kg-pellet). The immunoprecipitates were analyzed by SDS–PAGE (12.5%). (C) Steady-state levels of EGFP–HC in U373EGFP–HC and US11EGFP–HC cells. Immunoblots of SDS lysates from equal numbers of untreated (lanes 1 and 2) and ZL3VS-treated (3 h; 50 µM; lanes 3 and 4) cells (0.25 × 106) were performed with a polyclonal anti-GFP serum. U373EGFP–HC cells (lanes 1 and 3) express equal levels of EGFP–HC, independent of the proteasome inhibitor treatment. Inhibition of proteasomal degradation results in accumulation of EGFP–HC in US11EGFP–HC cells (lanes 2 and 4) mostly as the deglycosylated intermediate. Molecular weight markers are indicated on the left in kDa.
In keeping with our earlier observations on the endogenous MHC class I products, the deglycosylated fusion product fractionated with the cytosolic compartment, unlike the transferrin receptor analyzed in the same experiment (Figure 4B). The transferrin receptor was recovered quantitatively in the particulate fraction obtained by differential centrifugation, whereas the deglycosylated MHC class I intermediate remained soluble (Figure 4B, lanes 4–6). We conclude that the EGFP–HC fusion protein behaves like the endogenous class I heavy chain as far as US2 and US11-mediated dislocation and degradation are concerned.
Steady-state expression levels of EGFP–HC in U373EGFP–HC and US11EGFP–HC cells were analyzed by αGFP western blots (Figure 4C). Lysates prepared in SDS from equal numbers of untreated (Figure 4C, lanes 1 and 2) and ZL3VS-treated (Figure 4C, lanes 3 and 4) cells were subjected to SDS–PAGE and immunoblot ting. U373EGFP–HC cells (Figure 4C, lanes 1 and 3) express equivalent levels of EGFP–HC, independent of the presence of the proteasome inhibitor. In contrast, EGFP–HC is barely detectable in untreated US11EGFP–HC cells (Figure 4C, lane 2), but appears as the deglycosylated intermediate when proteasomal degradation is inhibited (Figure 4C, lane 4). No other αGFP-reactive degradation products were detected at steady state, in agreement with the results from pulse–chase experiments.
Time-dependent accumulation of EGFP–HC in US2EGFP–HC and US11EGFP–HC upon inhibition of proteasomal degradation
We used the EGFP–HC reporter in FACS analysis, fluorimetric emission analysis and fluorescence microscopy experiments to explore the dislocation reaction in intact cells. Co-expression of either US2 or US11 with EGFP–HC essentially abolished all fluorescence, a further indication of the ability of the fusion protein to serve as a substrate for US11/US2-mediated degradation (Figure 5A). We quantified the appearance of green fluorescence EGFP–HC in US2EGFP–HC and US11EGFP–HC cells by FACS analysis when proteasome activity is blocked. Treatment of the cells with ZL3VS induces the accumulation of EGFP–HC in a time-dependent manner in US2EGFP–HC and US11EGFP–HC cells, with the half-maximal value attained after ∼1 h (Figure 5A). Untransfected U373-MG cells did not show any fluorescence accumulation (Figure 5A). In the absence of US11 and US2, the levels of EGFP–HC fusion increased only slightly upon proteasomal inhibition (Figure 5A). These results were corroborated using fluorimetric emission analysis of NP-40 lysates from U373-MG, U373EGFP–HC and US11EGFP–HC cells (Figure 5B). The EGFP fluorescence from lysates of US11EGFP–HC cells was significantly greater when cells had been treated with the proteasome inhibitor, ZL3VS. As expected, there was no significant difference in the EGFP fluorescent observed from lysates of ZL3VS treated and non-treated U373EGFP–HC cells.


Fig. 5. Inhibition of proteasomal degradation in US11EGFP–HC and US2EGFP–HC cells induces accumulation of fluorescent EGFP–HC. (A) Flow cytometric quantification of the induction of green reporter fluorescence. Cells were incubated with ZL3VS (50 µM) for the indicated time periods. The mean fluorescence intensity was measured by FACS: untransfected U373 cells (open squares), U373EGFP–HC (closed squares), US11EGFP–HC (open circles) and US2EGFP–HC (open diamonds). The results shown are representative of three experiments. (B) Fluorimetric emission quantification of NP-40 lysates from U373 cells, U373EGFP–HC and US11EGFP–HC cells. Fluorescent units [490 nm(excitation)/515 nm(emission)] of EGFP–HC were measured from cell lysates of cells treated for 5 h with Zl3VS (black bars) or without Zl3VS (white bars). (C) Immunofluorescence of US11EGFP–HC cells. Immunostaining followed by confocal laser scanning microscopy was carried out as described in Materials and methods. For each antibody the green EGFP–HC (left column), the respective second staining in red (middle column) and a merge image (right column) are shown, as indicated for the individual panels. Double-positivity of EGFP–HC with W6/32 shows the localization and the co-localization of properly folded class I complexes and their presence at the cell surface (top row). Almost complete co-localization of EGFP–HC with HC10, the monoclonal antibody reactive with class I independent of its folding, shows that this fluorescence is indeed derived from a green class I fusion protein (middle row). Staining with the αGFP reagent depicts no additive GFP-tagged population that for some reason (unfolding) failed to acquire green fluorescence (anti-GFP; bottom row).
Next, we examined the intracellular localization of ZL3VS-induced EGFP–HC by fluorescence microscopy in fixed cells. Cells grown on glass cover slips were pretreated with ZL3VS for 3–5 h prior to immunohistochemistry. In US11EGFP–HC cells, we observed co-localization of green fluorescence with W6/32 (Figure 5C) or HC10 (Figure 5C) reactive material. Co-localization of EGFP–HC with W6/32 reactive material shows that properly formed class I complexes include the EGFP–HC fusion protein. A small population of W6/32 reactive EGFP–HC can be observed at the cell surface (Figure 5C). However, this co-distribution is substantially less than that seen for EGFP–HC and HC10 (Figure 5C, compare the first two rows). EGFP–HC co-localizes perfectly with HC10, a monoclonal anti-class I reagent that reacts with free (unfolded) class I heavy chains (Stam et al., 1986). Direct examination of EGFP species with anti-GFP serum (Seedorf et al., 1999), does not show a significant population of non-folded, non-fluorescent EGFP–HC molecules (Figure 5C, bottom row). Similar results were obtained when these experiments were performed in US2EGFP–HC cells (data not shown). We conclude that the green fluorescent signal observed in our cells is indeed derived from EGFP–HC chimeric molecules, fully consistent with the result from the biochemical analysis (Figure 3).
Cytosolic EGFP–HC does not form aggresomes
Cells respond to the production of high levels of certain misfolded protein by transport of the aggregated material to a perinuclear inclusion region and the formation of structures referred to as aggresomes (Johnston et al., 1998). Formation of these inclusion bodies was initially described for the cystic fibrosis transmembrane conductance regulator (CFTR) (Johnston et al., 1998), presenilin (Wigley et al., 1999) and a GFP-fusion protein (Garcia-Mata et al., 1999) as a general cellular response to misfolded unassembled proteins. Aggresomes presumably result when cytoplasmic degradation cannot clear the misfolded material in a timely manner. Does inhibition of proteasomal degradation induce such cytosolic structures for EGFP–HC? Aggresomes have been defined by their location close to the centrosome and an attendant redistribution of the intermediate filament protein vimentin into a halo-like cage around the aggregated proteins. Anti-vimentin immunostaining was performed on U373EGFP–HC and US11EGFP–HC cells (Figure 6). Vimentin filaments of U373EGFP–HC cells (Figure 6A) were compared with the ZL3VS-treated US11EGFP–HC cells (Figure 6B). No difference in the distribution of vimentin was apparent. The presence of US11 did not affect the vimentin pattern as compared with control cells, regardless of the inclusion of proteasome inhibitors. Vimentin distribution was equally unaffected by ZL3VS treatment of U373EGFP–HC cells (data not shown). Similar results were observed with US2EGFP–HC cells. Furthermore, some of the perinuclear EGFP–HC must be ER-resident (Figure 2, middle row; Figure 7A). The lack of clearly defined vimentin cages and the broad distribution of the accumulated EGFP–HC in proteasome inhibitor-treated cells argues against the presence of aggresomes in our experimental model (Kopito, 2000).

Fig. 6. Inhibition of proteasomal degradation in US11EGFP–HC cells does not induce the formation of aggresomes. Immunostaining with anti-vimentin mAb V9 and confocal laser scanning microscopy was performed to study the distribution of vimentin filaments in untreated U373EGFP–HC cells (A) and US11EGFP–HC cells treated with ZL3VS (50 µM; 3 h) (B). No significant difference between the vimentin distribution was detected for either cell type. The green EGFP–HC (left column), the anti-vimentin staining in red (middle column) and a merge image (right column) are shown for each panel.


Fig. 7. Direct visualization of EGFP–HC dislocation with intact cells. (A) A significant subpopulation of EGFP–HC is detected in the endoplasmic reticulum. Immunostaining followed by confocal laser scanning microscopy was used to analyze subcellular distribution of the accumulated EGFP–HC. US11EGFP–HC cells were incubated with ZL3VS (50 µM) for 3 h, fixed, and stained. ER localization of EGFP–HC is demonstrated by co-staining with the ER markers concanavalin A (top row), calnexin (anti-calnexin, mAb AF8; middle row) and PDI (anti-PDI; bottom row), as indicated. For each stain the green EGFP–HC (left column), the respective second staining in red (middle column) and a merge image (right column) are shown. Similar results were obtained in four independent experiments. (B) EGFP–HC traffics from the ER to the cytosol. The co-localization program from Bio-Rad was used for evaluation of the confocal laser scanning microscopy. The program depicts colocalization green and red signals from the merge picture (data not shown) and reproduces them as yellow pixels. Single color signals are shown in gray. Stainings were performed as in (A). In the presence of inhibitor, a large proportion of EGFP–HC colocalizes with concanavalin A (top row, left image), calnexin (top row, middle image) and PDI (top row, right image). Following wash-out of the proteasome inhibitor and a 90 min chase in the presence of cycloheximide (CHX) (bottom row), EGFP–HC is no longer detected in concanavalin A- (bottom row, left image), calnexin- (bottom row, middle image) and PDI-positive (bottom row, right image) compartments, yet is readily visible in the cytosol (see also Figure 8). Similar results were obtained in four independent experiments.
Visualization of EGFP–HC dislocation
Direct visualization of the dislocation of proteins in live cells has not been possible so far. We used the EGFP–HC fusion to address two types of question. First, where exactly do cells accumulate EGFP–HC induced by inhibition of proteasomal degradation? Secondly, can we follow the fate of a distinct EGFP–HC subpopulation within a cell? We first examined the intracellular distribution of the EGFP–HC in detail by confocal laser scanning microscopy. US11EGFP–HC cells treated with ZL3VS were stained for the ER markers concanavalin A (Figure 7A, top row), calnexin (Figure 7A, middle row) and PDI (Figure 7A, bottom row) to identify the ER-resident fraction of EGFP–HC. In both US11EGFP–HC and US2EGFP–HC (data not shown) cells, a significant population of EGFP–HC co-localizes with these ER markers (Figure 7A), demonstrating that properly folded fluorescent EGFP–HC molecules are detected in the ER.
We next designed an experiment that would allow the accumulation of EGFP–HC in the ER by blockade of proteasomal proteolysis, followed by removal of inhibition and ensuing dislocation and degradation of EGFP–HC. For this purpose we used the reversible inhibitor ZL3-aldehyde (MG132) (Lee and Goldberg, 1996): proteasomal activity resumes upon removal of the inhibitor. By blocking de novo protein synthesis by the addition of cycloheximide (CHX), the egress of the pre-existing, ER-localized population of EGFP–HC can be followed during the dislocation process without the confounding effect of its de novo synthesis.
In Figure 7B (top row), EGFP–HC molecules colocalized with ER markers in MG132-treated US11EGFP–HC cells are represented as yellow pixels. This yellow signal corresponds to the merged image of the green EGFP–HC and in red the ER staining with concanavalin A, anti-calnexin antibody or anti-PDI antiserum, respectively (Figure 7B). The fraction of immunostaining that does not colocalize is rendered on a gray scale. Examination of US11EGFP–HC cells 1.5 h after the addition of CHX sharply reduces colocalization in cells stained with the respective ER markers (Figure 7B, lower panels). This implies that the initially ER-localized population of EGFP–HC has now left the ER. Likewise, co-localization of W6/32 and EGFP–HC is no longer observed in ER compartments of such treated cells (data not shown). These results demonstrate that a properly folded fluorescent EGFP–HC species can be targeted for dislocation and subsequent degradation in an US11-dependent manner.
To examine the degradation kinetics of fluorescent EGFP–HC molecules, we stopped the degradation of EGFP–HC in US11EGFP–HC cells by blocking proteasomal function with MG132 for 3 h (Figure 8A). The inhibitor was then removed and the mean fluorescence intensity of accumulated EGFP–HC was monitored over a 3 h time period in the presence of the protein synthesis inhibitor CHX. FACS analysis shows that the initial accumulation of green fluorescence is comparable to that seen in cells treated with the irreversible inhibitor ZL3VS (Figure 5A). Within 3 h after the washout of the proteasome inhibitor, this accumulation phase is followed by a decline of signal to background levels (Figure 8A). CHX treatment of cells that did not express US11 failed to show this rapid loss of green fluorescence (Figure 8A). Similar results were obtained from US2EGFP–HC cells (data not shown). Judged from the decay of EGFP signal in FACS analysis, we determined that a period of 1 h after imposition of the CHX block was optimal to obtain samples for subcellular fractionation and fluorimetric emission analysis of the cytosolic fractions. We detected fluorescent EGFP–HC molecules in the cytosol of ZL3VS-treated US11EGFP–HC cells and US2EGFP–HC cells (Figure 8B). After a 1 h CHX chase, there was an increase in recovery of cytosolic fluorescent EGFP–HC molecules. In the absence of proteasome inhibitor, US2EGFP–HC and US11EGFP–HC cells showed very little fluorescence in their cytosolic fractions (data not shown). We conclude that EGFP–HC molecules can exist in the cytosol as fluorescent species and accumulate if dislocation is allowed to proceed in the presence of proteasome inhibitor. EGFP fluorescence implies a properly folded state at least for the EGFP moiety. This raises the question whether EGFP–HC needs to unfold for dislocation to proceed, and if so, whether an unfolded EGFP–HC molecule could re-acquire fluorescent properties.

Fig. 8. Dislocated EGFP–HC is a cytosolic green fluorescent protein and does not, once unfolded, reacquire fluorescence. (A) FACS analysis shows that accumulated EGFP–HC induced by proteasome inhibition can be degraded. U373EGFP–HC (closed squares) and US11EGFP–HC (open circles) were incubated for 3 h with the reversible proteasome inhibitor MG132 (50 µM). After removal of MG132, the accumulated EGFP–HC was chased for an additional 3 h in the presence of cycloheximide (CHX). Similar results were obtained in three independent experiments. (B) Fluorimetric emission quantification of EGFP–HC from cytosolic fractions of U373EGFP–HC, US2EGFP–HC and US11EGFP–HC cells treated with the proteasome inhibitor, ZL3VS (white and black bars). The fluorescence emitted by cytosolic EGFP–HC molecules was examined for US2EGFP–HC and US11EGFP–HC cells after addition of proteasome inhibitor and cycloheximide (CHX) for 1 h (black bars). Cytosolic fractions were prepared as described in Materials and methods. The fluorescence emission of the cytosolic population of EGFP–HC was plotted as a percentage of signal calculated for unfractionated homogenates. (C) Fluorimetric quantification of the EGFP–HC molecules after exposure to the denaturant guanidine hydrochloride (GuHCl). The fluorescence emission of lysates from U373EGFP–HC (open squares), ZL3VS-treated US2EGFP–HC (open diamonds), ZL3VS-treated US11EGFP–HC (asterisks) and U373EGFP (open circles) cells incubated with increasing concentrations of GuHCl were examined. The percentage of EGFP–HC fluorescence intensity was plotted against GuHCl concentration. The mean fluorescence of EGFP–HC at 0 M GuHCl was used as 100% fluorescence. (D) Renaturation of denatured EGFP–HC and EGFP molecules. EGFP–HC molecules from U373EGFP–HC cells and EGFP from U373EGFP cells were denatured with 5.0 M GuHCl (open square and circle, respectively) followed by dilution with PBS to 0.3 M GuHCl (closed square and circle, respectively). The percentage of green fluorescent signal was plotted against GuHCl concentration. The mean green fluorescence signal at 0 M GuHCl was used as 100% fluorescence.
Hysteresis in unfolding of EGFP–HC and EGFP molecules
We addressed the stability of EGFP–HC chimeric molecules by examining in vitro their fluorescent properties in the presence of a denaturant. All fluorescence of EGFP–HC molecules from U373EGFP–HC cells, ZL3VS-treated US2EGFP–HC and ZL3VS-treated US11EGFP–HC cells is extinguished in a guanidine hydrochloride (GuHCl)-concentration dependent manner (Figure 8C). The EGFP–HC molecules are denatured at similar concentrations of GuHCl as free, cytosolic EGFP molecules in U373-MG cells (U373EGFP) (Figure 8C).
Can an unfolded EGFP–HC chimera refold into a fluorescent species? EGFP–HC molecules from U373EGFP–HC cells denatured by addition of 5.0 M GuHCl (Figure 8D; open square) do not re-acquire green fluorescence upon dilution to 0.3 M GuHCl (closed square) (Figure 8D). In contrast, when cytosolically expressed, free EGFP molecules were subjected to the same treatment [5.0 M GuHCl (open circle) to 0.3 M GuHCl (closed circle)], recovery of EGFP fluorescence was essentially complete (Figure 8D). The EGFP moiety itself can thus refold into a fluorescent species after denaturation, in agreement with published data (Battistutta et al., 2000; Fukuda et al., 2000). Taken together, the results imply that the fluorescent EGFP–HC molecules detected in the cytosol do not necessarily arise from an unfolded cytosolic EGFP–HC species that refolds and requires fluorescent properties, but instead may originate from EGFP–HC molecules dislocated in a fluorescent state.
Discussion
To investigate ER dislocation and proteasomal degradation, we use a reporter protein composed of an EGFP-moiety fused at its C-terminus to the HLA-A2 heavy chain (EGFP–HC) preceded by a cleavable N-terminal signal sequence. We show that this protein folds properly, as assessed by the acquisition of the appropriate antibody epitopes and the display of green fluorescence. Furthermore, we demonstrate that the EGFP–HC, like endogenous class I heavy chains, is readily recognized by the US2 and US11 proteins. This interaction leads to degradation of the fusion protein, a reaction accompanied by the accumulation of the diagnostic deglycosylation intermediate when proteasomal proteolysis is inhibited, both in pulse–chase analysis and at steady state. In addition to endogenous class I heavy chains, the only intermediate(s) of the breakdown reaction are the cytosolic deglycosylated forms of the EGFP–HC. Cytochemical and biochemical analysis at steady state failed to reveal the presence of obvious cleavage products or of a free EGFP-moiety that could have confounded further morphological analysis.
Examination of EGFP–HC in US11EGFP–HC and US2EGFP–HC by fluorescence microscopy showed an almost complete lack of fluorescence, unless proteolysis was inhibited by the proteasome inhibitors ZL3VS or MG132. Block of degradation with either proteasomal inhibitor results in the accumulation of the dislocation substrate. This accumulation is not confined only to the cytosol, but extends to the ER as well. We interpret the latter result as the consequence of feedback inhibition: the accumulation of an intermediate in the cytosol affects the reaction(s) immediately upstream, which include(s) the removal from the ER (dislocation) itself. This accumulation is reversible once proteasome inhibitors are removed. When degradation resumes, extraction of fluorescent material formerly arrested in the ER can now occur. This is best visualized when de novo protein synthesis is blocked, and no new material is inserted into the ER. In other words, EGFP–HC is inserted into the ER where it folds, but can still be effectively dislocated thereafter and degraded by proteasomes.
Vimentin filaments do not collapse upon cytosolic accumulation of EGFP–HC in US11EGFP–HC and US2EGFP–HC cells. This finding, in combination with the broad distribution of green fluorescence throughout the cell, argues against the formation of aggresomes for the EGFP–HC substrate, defined as inclusion bodies surrounded by a vimentin cage (Garcia-Mata et al., 1999; Kopito, 2000). Furthermore, EGFP–HC accumulation is reversible and EGFP–HC remains in a cellular compartment that is readily extractable with NP-40 (this work). The formation of aggresomes, as pointed out by Kopito (2000), might be a highly substrate- and cell type-specific process, with certain cell types more prone to aggresome formation than others. Likewise, the type I membrane protein EGFP–HC might behave differently when compared with multi membrane-spanning proteins, such as CFTR or presenilin (12 and eight transmembrane domains, respectively), commonly used to document aggresome formation (Johnston et al., 1998).
How is EGFP–HC dislocated from the ER into the cytosol in a manner consistent with its fluorescent properties in both the ER and the cytosol? As the first of two possible scenarios we propose the following: the ER-resident EGFP–HC molecule is initially denatured by ER chaperones, followed by its dislocation into the cytosol as a completely or partially unfolded molecule. We have observed that the disulfide bonds that stabilize the conformation of class I heavy chains are reduced prior to loss of the single N-linked glycan, brought about by the action of a cytosolic N-glycanase (Tortorella et al., 1998). A catalyst specifically involved in this redox reaction remains to be identified, and perhaps is not even required when a disulfide-bonded protein reaches the cytosol. In the presence of proteasome inhibitors, the EGFP–HC molecule would then refold partially to accumulate as a deglycosylated, fluorescent species. In other words, the folded EGFP–HC molecule would unfold in the ER and at least the EGFP moiety refold in the cytosol. What detracts from this possibility is the fact that the GFP molecule is rather resistant to denaturation. In addition, only in the presence of 6 M GuHCl, but not 6 M urea, is GFP denatured and loses its fluorescent properties (Weber-Ban et al., 1999). Complete unfolding of the EGFP–HC molecule also requires a high concentration of GuHCl (5.0 M) (Figure 8C). A fully unfolded free EGFP molecule can refold in vitro upon the removal of the denaturants, as judged by the re-emergence of fluorescent properties of GFP (Figure 8D) (Battistutta et al., 2000), but the EGFP–HC fusion does not (Figure 8D). The extent to which such unfolding occurs in living cells is not easily assessed. However, the in vitro renaturation of partially or completely unfolded EGFP–HC molecules to a fluorescent species was not observed upon dilution of denaturant (Figure 8D). Clearly, the attachment of the class I moiety affects the folding properties of EGFP in the fusion protein: free EGFP refolds upon removal of denaturant, whereas EGFP fused to the HC does not. We suggest that in living cells the refolding of the unfolded EGFP–HC fusion protein might be likewise compromised. If correct, our data would be consistent with dislocation of EGFP–HC reporter in a conformation that retains at least the fold for fluorescent EGFP. In the presence of the ClpA unfoldase, fluorescent GFP is unfolded in vitro but only if equipped with a degradation signal (Weber-Ban et al., 1999). A 20% decrease in GFP fluorescence was observed in the presence of ClpA. It could be argued that the attached heavy chain moiety is in fact a degradation signal for EGFP–HC that acts in a manner similar to the ssrA sequence used to target GFP to the ClpA unfoldase (Weber-Ban et al., 1999). However, the fact remains that the ubiquitin conjugation apparatus and the proteasome cannot access the EGFP–HC protein until it is exposed to the cytosol.
An additional complication of a model that requires unfolding of the EGFP–HC complex prior to dislocation is the following: the three-dimensional structure of the US2–class I complex has been determined for the lumenal domains (Gewurz et al., 2001a). This complex is quite stable and survives native gel electrophoresis, as well as several chromatography steps used in the purification of the complex (Gewurz et al., 2001b). If unfolding were a prerequisite for dislocation, the unfolding reaction would presumably occur in the same compartment where the complex between US2 and US11 was formed in the first place, the ER. The remarkable efficacy of the entire dislocation process, perhaps not more than 2 min between completion of the polypeptide chain and the dislocation event (Wiertz et al., 1996a,b), raises important questions as to the nature of this presumed unfolding and the catalysts involved, given the stability of the US2–class I complex. In summary, one model consistent with our data postulates unfolding immediately prior to escape of EGFP–HC from the ER, followed by re-acquisition of fluorescent properties of the EGFP moiety upon arrival in the cytosol. The inhibition of the proteasome is essential for this reaction to yield EGFP–HC in detectable amounts.
An alternative model for dislocation of the EGFP–HC molecule through the ER membrane is that it is dislocated as a folded structure, a suggestion not entirely without precedent and perhaps more consistent with the remarkable stability of the US2–class I complex. Nonetheless, we cannot formally exclude the possibility that the unfolded GFP moiety attached to the class I heavy chain may refold upon arrival in the cytosol. However, we note the following examples that may serve as precedents for the transport of folded molecules across a membrane. Unfolding of cytosolic peroxisomal precursor proteins is not required for import into the peroxisome (Subramani et al., 2000). Cytosolic proteins stabilized by crosslinking or by pharmacological means were efficiently translocated into peroxisomes and glycosomes respectively (Walton et al., 1995; Hausler et al., 1996). In fact, multimeric proteins are also transported into the peroxisomal matrix. In yeast, a trimer of chloramphenicol acetyltransferase containing a peroxisomal targeting sequence as well as a dimer of peroxisomal thiolase are imported into peroxisomes as oligomeric complexes (Glover et al., 1994; McNew and Goodman, 1994). Such import sets a precedent for the ability of folded or even multimeric proteins to cross membranes when assisted by the appropriate partners. In view of the shared ancestry of the peroxisomal membrane and the ER (Titorenko and Rachubinski, 2001), the requirement for unfolding, in contrast to the transport of a multimeric protein complex, is an issue that deserves further study in the context of the dislocation reaction. If transport of a multimeric protein complex across the peroxisomal membrane is possible (Smith and Schnell, 2001), there is in principle no reason why such transport should be denied to proteins destined to leave the ER for the cytosol.
The Sec61 complex is believed to be the conduit via which proteins are dislocated from the ER. The inner diameter of the translocon pore has been estimated to fluctuate between 15 Å in its inactive state (Menetret et al., 2000; Beckmann et al., 2001) to 40–60 Å during protein translocation (Johnson and van Waes, 1999). Can GFP pass through the pore of the Sec61 complex as a folded molecule? GFP is a cylinder 42 Å long with a diameter of 24 Å (Ormo et al., 1996). The estimated pore size of the translocon during its active state could therefore allow the passage of the folded GFP molecule. Even if the pore size of the translocon is maximally 60 Å when fully active, further rearrangements of the translocon subunits could produce different results. Relatively modest adjustments in the angle of connexin subunits within the membrane can create a completely closed or an open state for gap junctions (Simon and Goodenough, 1998). In a similar event, re-arrangements of translocon subunits or an adjustment of their stoichiometry initiated from within the ER could modulate pore size.
Materials and methods
Cell lines and antibodies
U373-MG astrocytoma cells transfected with the US2 or US11 cDNA were prepared as described (Jones et al., 1995; Kim et al., 1995), cells were maintained in DME medium as described (Rehm et al., 2001). EGFP–HC (U373EGFP–HC), US2-EGFP–HC (US2EGFP–HC), US11-EGFP–HC (US11EGFP–HC) and EGFP (U373EGFP) cells were maintained in DME medium supplemented with 5% FCS, 5% calf serum and 0.5 mg/ml of Geneticin (Gibco, Fredrick, MD). The αHC serum was generated by immunizing rabbits with the bacterially expressed lumenal fragment of HLA-A2 and HLA-B27 heavy chain (Tortorella et al., 1998). αGFP was generated by immunizing rabbits with the bacterially expressed GFP (Seedorf et al., 1999). mAbs AF8 (Schreiber et al., 1994), HC10 (Stam et al., 1986), W6/32 (Parham et al., 1979) and anti-transferrin receptor (van de Rijn et al., 1983) were used as described. Anti-vimentin mAb V9 (Sigma, St Louis, MO) was used for the characterization of aggresomes (Heath et al., 2001). Polyclonal anti-PDI serum was generated with bacterially expressed human PDI.
Metabolic labeling of cells, pulse–chase analysis, immunoprecipitation and immunoblotting
Cells were detached by trypsin treatment, followed by starvation in methionine-/cysteine-free DME for 45 min at 37°C. Cells were metabolically labeled with 500 µCi/ml of [35S]methionine/cysteine (1200 Ci/mM; NEN-Dupont, Boston, MA) at 37°C for the times indicated. Pulse–chase experiments, cell lysis and immunoprecipitations were performed as described previously (Rehm et al., 2001). The immunoprecipitates were analyzed by SDS–PAGE followed by fluorography (Ploegh, 1995). αGFP immunoblots were performed with SDS lysates of inhibitor-treated or untreated U373EGFP–HC and US11EGFP–HC cells as described (Fiebiger et al., 2001).
cDNA, transfection and Endo H digestion
The EGFP–HC molecule was cloned with the murine class I molecule H2-Kb signal sequence at its N-terminal end and the class I heavy chain HLA-A2 allele (amino acids 25–365) at the C-terminal end. The H2-Kb signal sequence directs the chimeric molecule to the ER. The chimeric construct was inserted into pCDNA 3.1 (Invitrogen, Carlsbad, CA) (Swann et al., 2001). The EGFP–HC construct was transfected into U373-MG, US2 and US11 cells using a liposome-mediated transfection protocol (4 µg of DNA/20 µl of lipofectamine/10 cm dish; Lipofectamine, Gibco, Fredrick, MD). The cytosolic EGFP construct was cloned into pCDNA 3.1 and transfected into U373-MG cells as above. Endo H and N-glycanase (New England Biolabs, Beverly, MA) digestions were performed as described by the manufacturer.
Subcellular fractionation
Subcellular fractionation of metabolically labeled US11EGFP–HC cells was performed as described (Tortorella et al., 1998).
Fluorimetric analysis and folding studies
Whole-cell NP-40 lysates and 100 K supernatants obtained by subcellular fractionation of U373EGFP–HC, US2EGFP–HC, US11EGFP–HC and U373EGFP cells, respectively, were analyzed for the fluorescence emission of EGFP–HC chimeric molecules using a ISS® KS Multifrequency Phase Fluorimeter (ISS® Fluorescence Analytical Instrumentation, Champaign, IL) at 490 nm excitation and 515 nm emission. The lysates were incubated with increasing concentrations of guanidine hydrochloride (GuHCl) for at least 1 h at 25°C. Refolding experiments were performed by the addition of PBS to GuHCl containing samples and incubate at 25°C for 1 h. Results were shown directly as fluorescent units or as a percentage of the signal from unfractionated homogenates. For the folding studies, the fluorescent signal obtained from samples not treated with GuHCl were taken as 100%.
Flow cytometry analysis
Quantitative flow cytometry analysis of accumulation of the green fluorescent reporter construct after the inhibition of proteasomal degradation in living cells was performed by FACS (FACS Calibur; Beckton Dickinson, Mountain View, CA) supported by CellQuest software (Beckton Dickinson). Mean fluorescence intensity correlates to the amount of intracellular EGFP–HC and is monitored under the conditions indicated.
Immunostaining and confocal microscopy
Immunofluorescence experiments were performed essentially as described (Driessen et al., 1999) with minor modifications. Cells were allowed to attach to slides overnight before inhibitor incubation with [ZL3VS (Wiertz et al., 1996b) or the peptide aldehyde inhibitor carboxybenzyl-leucyl-leucyl-leucinal, MG132, (Lee and Goldberg, 1996); 50 µM final from a DMSO stock]. DMSO was used as solvent control. Where indicated, protein de novo synthesis was inhibited by the addition of cycloheximide (CHX) (50 µg/ml, final). After fixation with 3.7% paraformaldehyde for 20 min at room temperature immunohistochemistry was performed in a 0.5% saponin/3% BSA/PBS solution to define the intracellular location of the EGFP–HC fusion protein. Alexa Fluor® 594-conjugated concanavalin A (Molecular Probes, Eugene, OR), anti-calnexin (mAb A8) and anti-PDI (polyclonal serum, rabbit anti-human) staining were performed to define ER structures. mAbs W6/32 and HC10 were used to define localization of properly folded and total MHC class I reactivity, respectively. αGFP serum depicts the EGFP moiety. Anti-mouse Alexa Fluor® 568 (Molecular Probes) and anti-rabbit Cy™3 (Jackson Laboratory, Bar Harbor, ME) were used as the fluorescent probe. Further analysis were performed with a Bio-Rad MRC1024 confocal laser scanning microscope. The merge images were analyzed for the presence of EGFP–HC in the ER with the colocalization program from Bio-Rad (Driessen et al., 1999).
Acknowledgments
Acknowledgements
We thank Rebecca Tirabassi for generating the U373EGFP cell line. We thank Kristina Cunningham (Laboratory of R.J.Collier) for her assistance with the fluorescence measurements. During the course of this study E.F. was supported by a Max Kade Fellowship of the Max Kade Foundation, NY and the Austrian Academy of Sciences. D.T. is a Charles King Trust (Boston, MA) fellow. This study was supported by the NIH grant 5R37-AI33456.
References
- Battistutta R., Negro,A. and Zanotti,G. (2000) Crystal structure and refolding properties of the mutant F99S/M153T/V163A of the green fluorescent protein. Proteins, 41, 429–437. [DOI] [PubMed] [Google Scholar]
- Bebok Z., Mazzochi,C., King,S.A., Hong,J.S. and Sorscher,E.J. (1998) The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61β and a cytosolic, deglycosylated intermediary. J. Biol. Chem., 273, 29873–29878. [DOI] [PubMed] [Google Scholar]
- Beckmann R., Spahn,C.M., Eswar,N., Helmers,J., Penczek,P.A., Sali,A., Frank,J. and Blobel,G. (2001) Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell, 107, 361–372. [DOI] [PubMed] [Google Scholar]
- Biederer T., Volkwein,C. and Sommer,T. (1997) Role of Cue1p in ubiquitination and degradation at the ER surface. Science, 278, 1806–1809. [DOI] [PubMed] [Google Scholar]
- Bonifacino J.S. and Weissman,A.M. (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol., 14, 19–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo J., Plemper,R.K., Finger,A. and Wolf,D.H. (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell., 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen C., Bryant,R.A., Lennon-Dumenil,A.M., Villadangos,J.A., Bryant,P.W., Shi,G.P., Chapman,H.A. and Ploegh,H.L. (1999) Cathepsin S controls the trafficking and maturation of MHC class II molecules in dendritic cells. J. Cell Biol., 147, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Fiebiger E., Meraner,P., Weber,E., Fang,I.F., Stingl,G., Ploegh,H. and Maurer,D. (2001) Cytokines regulate proteolysis in major histocompatibility complex class II-dependent antigen presentation by dendritic cells. J. Exp. Med., 193, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frand A.R., Cuozzo,J.W. and Kaiser,C.A. (2000) Pathways for protein disulphide bond formation. Trends Cell Biol., 10, 203–210. [DOI] [PubMed] [Google Scholar]
- Fukuda H., Arai,M. and Kuwajima,K. (2000) Folding of green fluorescent protein and the cycle3 mutant. Biochemistry, 39, 12025–12032. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R., Bebok,Z., Sorscher,E.J. and Sztul,E.S. (1999) Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol., 146, 1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewurz B.E., Gaudet,R., Tortorella,D., Wang,E.W., Ploegh,H.L. and Wiley,D.C. (2001a) Antigen presentation subverted: Structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc. Natl Acad. Sci. USA, 98, 6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewurz B.E., Wang,E.W., Tortorella,D., Schust,D.J. and Ploegh,H.L. (2001b) Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus- specific manner. J. Virol., 75, 5197–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillece P., Luz,J.M., Lennarz,W.J., de La Cruz,F.J. and Romisch,K. (1999) Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol., 147, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.R., Andrews,D.W., Subramani,S. and Rachubinski,R.A. (1994) Mutagenesis of the amino targeting signal of Saccharomyces cerevisiae 3-ketoacyl-CoA thiolase reveals conserved amino acids required for import into peroxisomes in vivo. J. Biol. Chem., 269, 7558–7563. [PubMed] [Google Scholar]
- Hampton R.Y., Gardner,R.G. and Rine,J. (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell, 7, 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausler T., Stierhof,Y.D., Wirtz,E. and Clayton,C. (1996) Import of a DHFR hybrid protein into glycosomes in vivo is not inhibited by the folate-analogue aminopterin. J. Cell Biol., 132, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath C.M., Windsor,M. and Wileman,T. (2001) Aggresomes resemble sites specialized for virus assembly. J. Cell Biol., 153, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High S., Lecomte,F.J., Russell,S.J., Abell,B.M. and Oliver,J.D. (2000) Glycoprotein folding in the endoplasmic reticulum: a tale of three chaperones? FEBS Lett., 476, 38–41. [DOI] [PubMed] [Google Scholar]
- Hiller M.M., Finger,A., Schweiger,M. and Wolf,D.H. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science, 273, 1725–1728. [DOI] [PubMed] [Google Scholar]
- Hirsch C. and Ploegh,H.L. (2000) Intracellular targeting of the proteasome. Trends Cell Biol., 10, 268–272. [DOI] [PubMed] [Google Scholar]
- Huppa J.B. and Ploegh,H.L. (1997) The α chain of the T cell antigen receptor is degraded in the cytosol. Immunity, 7, 113–122. [DOI] [PubMed] [Google Scholar]
- Johnson A.E. and van Waes,M.A. (1999) The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol., 15, 799–842. [DOI] [PubMed] [Google Scholar]
- Johnston J.A., Ward,C.L. and Kopito,R.R. (1998) Aggresomes: a cellular response to misfolded proteins. J. Cell Biol., 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.R., Hanson,L.K., Sun,L., Slater,J.S., Stenberg,R.M. and Campbell,A.E. (1995) Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol., 69, 4830–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Gatz,C., Hillen,W. and Jones,T.R. (1995) Tetracycline repressor-regulated gene repression in recombinant human cytomegalovirus. J. Virol., 69, 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Finger,A., Braun,T., Hellmuth,K. and Wolf,D.H. (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J., 15, 753–763. [PMC free article] [PubMed] [Google Scholar]
- Kopito R.R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol., 10, 524–530. [DOI] [PubMed] [Google Scholar]
- Lee D.H. and Goldberg,A.L. (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem., 271, 27280–27284. [DOI] [PubMed] [Google Scholar]
- McCracken A.A. and Brodsky,J.L. (1996) Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol., 132, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew J.A. and Goodman,J.M. (1994) An oligomeric protein is imported into peroxisomes in vivo. J. Cell Biol., 127, 1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetret J., Neuhof,A., Morgan,D.G., Plath,K., Radermacher,M., Rapoport,T.A. and Akey,C.W. (2000) The structure of ribosome-channel complexes engaged in protein translocation. Mol. Cell, 6, 1219–1232. [DOI] [PubMed] [Google Scholar]
- Nishikawa S.I., Fewell,S.W., Kato,Y., Brodsky,J.L. and Endo,T. (2001) Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol., 153, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormo M., Cubitt,A.B., Kallio,K., Gross,L.A., Tsien,R.Y. and Remington,S.J. (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science, 273, 1392–1395. [DOI] [PubMed] [Google Scholar]
- Parham P., Barnstable,C.J. and Bodmer,W.F. (1979) Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J. Immunol., 123, 342–349. [PubMed] [Google Scholar]
- Pickart C.M. (2000) Ubiquitin in chains. Trends Biochem. Sci., 25, 544–548. [DOI] [PubMed] [Google Scholar]
- Pilon M., Schekman,R. and Romisch,K. (1997) Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J., 16, 4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper R.K., Bohmler,S., Bordallo,J., Sommer,T. and Wolf,D.H. (1997) Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature, 388, 891–895. [DOI] [PubMed] [Google Scholar]
- Plemper R.K., Egner,R., Kuchler,K. and Wolf,D.H. (1998) Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J. Biol. Chem., 273, 32848–32856. [DOI] [PubMed] [Google Scholar]
- Ploegh H.L. (1995) Current Protocols in Protein Science. John Wiley, New York, NY.
- Rehm A., Stern,P., Ploegh,H.L. and Tortorella,D. (2001) Signal peptide cleavage of a type I membrane protein, HCMV US11, is dependent on its membrane anchor. EMBO J., 20, 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber K.L., Bell,M.P., Huntoon,C.J., Rajagopalan,S., Brenner,M.B. and McKean,D.J. (1994) Class II histocompatibility molecules associate with calnexin during assembly in the endoplasmic reticulum. Int. Immunol., 6, 101–111. [DOI] [PubMed] [Google Scholar]
- Schubert U., Anton,L.C., Gibbs,J., Norbury,C.C., Yewdell,J.W. and Bennink,J.R. (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature, 404, 770–774. [DOI] [PubMed] [Google Scholar]
- Seedorf M., Damelin,M., Kahana,J., Taura,T. and Silver,P.A. (1999) Interactions between a nuclear transporter and a subset of nuclear pore complex proteins depend on Ran GTPase. Mol. Cell. Biol., 19, 1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu C.E., Story,C.M., Rapoport,T.A. and Ploegh,H.L. (1999) The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J. Cell Biol., 147, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A.M. and Goodenough,D.A. (1998) Diverse functions of vertebrate gap junctions. Trends Cell Biol., 8, 477–483. [DOI] [PubMed] [Google Scholar]
- Smith M.D. and Schnell,D.J. (2001) Peroxisomal protein import. The paradigm shifts. Cell, 105, 293–296. [DOI] [PubMed] [Google Scholar]
- Stam N.J., Spits,H. and Ploegh,H.L. (1986) Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol., 137, 2299–2306. [PubMed] [Google Scholar]
- Subramani S., Koller,A. and Snyder,W.B. (2000) Import of peroxisomal matrix and membrane proteins. Annu. Rev. Biochem., 69, 399–418. [DOI] [PubMed] [Google Scholar]
- Swann S.A., Williams,M., Story,C.M., Bobbitt,K.R., Fleis,R. and Collins,K.L. (2001) HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology, 282, 267–277. [DOI] [PubMed] [Google Scholar]
- Titorenko V.I. and Rachubinski,R.A. (2001) The life cycle of the peroxisome. Nature Rev. Mol. Cell. Biol., 2, 357–368. [DOI] [PubMed] [Google Scholar]
- Tiwari S. and Weissman,A.M. (2001) Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J. Biol. Chem., 276, 16193–16200. [DOI] [PubMed] [Google Scholar]
- Tortorella D. et al. (1998) Dislocation of type I membrane proteins from the ER to the cytosol is sensitive to changes in redox potential. J. Cell Biol., 142, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella D., Gewurz,B.E., Furman,M.H., Schust,D.J. and Ploegh,H.L. (2000) Viral subversion of the immune system. Annu. Rev. Immunol., 18, 861–926. [DOI] [PubMed] [Google Scholar]
- van de Rijn M., Geurts van Kessel,A.H., Kroezen,V., van Agthoven,A.J., Verstijnen,K., Terhorst,C. and Hilgers,J. (1983) Localization of a gene controlling the expression of the human transferrin receptor to the region q12 leads to qter of chromosome 3. Cytogenet. Cell Genet., 36, 525–531. [DOI] [PubMed] [Google Scholar]
- Walton P.A., Hill,P.E. and Subramani,S. (1995) Import of stably folded proteins into peroxisomes. Mol. Biol. Cell., 6, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Ban E.U., Reid,B.G., Miranker,A.D. and Horwich,A.L. (1999) Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature, 401, 90–93. [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Jones,T.R., Sun,L., Bogyo,M., Geuze,H.J. and Ploegh,H.L. (1996a) The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell, 84, 769–779. [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Tortorella,D., Bogyo,M., Yu,J., Mothes,W., Jones,T.R., Rapoport,T.A. and Ploegh,H.L. (1996b) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature, 384, 432–438. [DOI] [PubMed] [Google Scholar]
- Wigley W.C., Fabunmi,R.P., Lee,M.G., Marino,C.R., Muallem,S., DeMartino,G.N. and Thomas,P.J. (1999) Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol., 145, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Nijbroek,G., Sullivan,M.L., McCracken,A.A., Watkins,S.C., Michaelis,S. and Brodsky,J.L. (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell., 12, 1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M. and Schekman,R. (1999) The engagement of Sec61p in the ER dislocation process. Mol. Cell, 4, 925–934. [DOI] [PubMed] [Google Scholar]