

Abstract
Initiation of DNA replication of the papillomavirus genome is a multi-step process involving the sequential loading of viral E1 protein subunits onto the origin of replication. Here we have captured structural snapshots of two sequential steps in the assembly process. Initially, an E1 dimer binds to adjacent major grooves on one face of the double helix; a second dimer then binds to another face of the helix. Each E1 monomer has two DNA-binding modules: a DNA-binding loop, which binds to one DNA strand and a DNA-binding helix, which binds to the opposite strand. The nature of DNA binding suggests a mechanism for the transition between double- and single-stranded DNA binding that is implicit in the progression to a functional helicase.
Keywords: crystal structure/DNA complexes/helicase assembly/papillomavirus/replication–initiation
Introduction
Initiation of DNA replication requires proteins that recognize the origin of replication (ori), melt the DNA duplex, possess helicase activity and recruit other replication factors. For some viral replicons, such as those of papillomavirus and SV40, a single protein—E1 in the former case, T antigen in the latter—carries out all these activities (for reviews see Fanning and Knippers, 1992; Sverdrup and Myers, 1997). Most likely, different oligomeric forms of the two proteins are responsible for the different activities and the sequential assembly of T antigen and E1 complexes ensures an ordered transition between these different activities. Ultimately, E1 and T antigen form a hexameric ring helicase on each strand, which serve as replicative DNA helicases that unwind the DNA in front of the replication fork (Figure 1; Stahl et al., 1986; Borowiec et al., 1990; Sedman and Stenlund, 1998; Fouts et al., 1999).
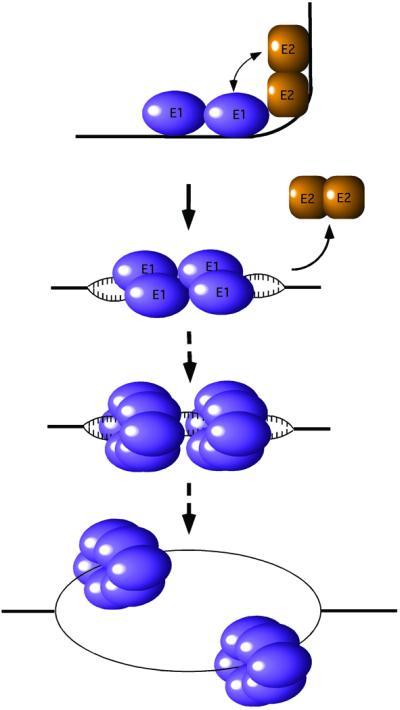
Fig. 1. Schematic figure showing the assembly and conversion of the E1 protein from a site-specific DNA-binding protein to a hexameric replicative DNA helicase. E1 binds site-specifically together with the E2 protein to the ori, forming an E12–E22–ori complex. As a result of the interaction, a sharp bend is induced in the DNA (Gillitzer et al., 2000). In an ATP-dependent process, E2 is displaced and the two additional E1 molecules are bound. Initial melting of the template occurs on both sides of the E1 binding sites and eventually a hexameric E1 helicase is assembled on each strand.
SV40 and papillomaviruses are both small DNA tumor viruses that belong to the papovavirus group and have been extensively studied as model systems for DNA replication in mammalian cells. In addition, papillomaviruses cause benign and malignant lesions in humans (zur Hausen, 1991). Infection of the genital tract by these viruses is the most common sexually transmitted disease (Ho et al., 1998) and infection by certain high-risk types is a prerequisite for invasive cervical carcinoma (Walboomers et al., 1999). Interference with the multi-step process of sequential loading of viral E1 protein subunits onto the ori constitutes a good target for small molecule intervention and disruption of the viral life cycle.
E1 is a multi-functional protein that binds DNA specifically, melts the DNA duplex and also functions as a 3′ to 5′ helicase (Seo et al., 1993b; Yang et al., 1993; Sedman et al., 1997). E1 is a 70 kDa polypeptide, which is monomeric in solution and has a well-defined DNA-binding domain and a C-terminal ATPase/helicase domain. E1 also interacts with DNA polymerase α (Park et al., 1994; Masterson et al., 1998; Conger et al., 1999) and replication protein A (RPA) (Han et al., 1999). E1 binds via its DNA-binding domain (DBD) to an 18 bp nearly-palindromic sequence in the ori, which contains multiple E1 binding sites (Figure 2A; Holt and Wilson, 1995; Mendoza et al., 1995; Chen and Stenlund, 1998).

Fig. 2. DNA oligonucleotides used in this study. (A) Numbering schemes for the oligonucleotides. The wild-type origin sequence is shown on top and the two dsDNA oligonucleotides used for crystallization are shown below. High-affinity E1-binding sites 2 and 4 are colored blue and moderate-affinity sites 1 and 3 are colored yellow. Bases deviating from the wild-type sequence are shown in red. A numbering scheme for the positions of bases in a generic E1 binding site is shown at the bottom. (B) Top: electron density map calculated utilizing density-modified phases (CNS, solvent-flipping and NCS-averaging) after the initial placement of the protein portion of the dimer complex structure (AMoRe). The maps (1.0σ) show clear duplex structures that overlay on the final refined DNA coordinates. Bottom: final 2Fo–Fc map contoured at 1σ. (C) Same as (B) but for the tetramer complex structure.
Initial loading of E1 onto the ori occurs through cooperative binding of E1 and the viral transcription factor E2 (Yang et al., 1991; Seo et al., 1993a; Lusky et al., 1994). This results in an E12–E22–ori complex in which E1 is specifically bound to one pair of E1-binding sites (Chen and Stenlund, 1998, 2001). The initial binding of an E1 dimer together with E2 serves to recognize the ori with high specificity (Sedman and Stenlund, 1995). Subsequently, two additional E1 molecules bind a second pair of binding sites that partially overlap the first pair and E2 is displaced in an ATP-dependent manner (Sanders and Stenlund, 1998). This produces an E1–ori complex with four E1 molecules bound to the overlapping sites (G.Chen and A.Stenlund, unpublished data). Subsequently, additional E1 molecules may bind to the ori. As these larger complexes are generated, ori melting can be detected by permanganate sensitivity (Yang et al., 1993; Gillette et al., 1994; Sanders and Stenlund, 1998). The exact correlation between the number of E1 molecules and ori melting is not clear; however, at least four molecules must be present to detect melting by permanganate sensitivity. Thus, the transition from the initial dimer complex to the larger complexes corresponds to a transition from E1’s origin– recognition function to its DNA–distortion function. Here we describe the crystal structures of the dimeric E1– DBD–DNA complex corresponding to the ori recogni tion function, as well as of a tetrameric E1–DBD–DNA complex that corresponds to the ori distortion function. The mode of DNA binding employed by E1 partitions the two individual DNA strands onto distinct binding surfaces of the protein. This suggests how E1 ultimately progresses to a hexameric helicase where the two strands are fully separated and each strand is encircled by a hexameric ring.
Results and discussion
Crystallization and structure determination
A pair of ATTGTT sequences (E1–2 and E1–4) were previously identified by biochemical and mutational analyses as the highest affinity binding sites for E1 from bovine papillomavirus (BPV; Chen and Stenlund, 1998, 2001). Two secondary binding sites, each with a single nucleotide difference from the highest affinity sequence, have also been identified (E1–1 and E1–3; Chen and Stenlund, 1998, 2001). Two different oligonucleotides containing E1-binding sites (E1BS) were designed to generate complexes corresponding to the two steps in the assembly pathway described above. These oligonucleotide sequences are shown in Figure 2A, along with the wild-type ori sequence. One complex is a dimer of E1–DBD bound to sites 2 and 4, representing the first loading event. The second complex is a tetramer of E1–DBD—i.e. two dimers bound to the four E1BS sites, representing the second step in the assembly process (see Materials and methods). Both crystal structures were determined by molecular replacement using the crystal structure of the unbound E1–DBD (Enemark et al., 2000) as a search model. After solvent flipping and non-crystallographic symmetry (NCS) averaging, the resulting electron density maps had a clearly traceable DNA duplex in both cases (Figure 2B). The dimer complex structure (E1159–307)2 (DNAd) was refined to free and working R values of 31.7 and 28.1%, respectively; the tetramer complex, (E1159–303)4(DNAt), was refined to free and working R values of 28.4 and 26.3%, respectively (Table I).
Table I. Data collection and refinement statistics.
| Dimer | Tetramer | |
|---|---|---|
| Data reduction | ||
| resolution limits (Å) | 50–3.05 (3.16–3.05) | 50–3.20 (3.31–3.20) |
| No. of reflections | 72 557 (6981) | 125 960 (12 242) |
| No. of unique reflections | 20 851 (2091) | 35 038 (3474) |
| completeness (%) | 99.8 (99.6) | 99.7 (99.3) |
| Rsym | 0.108 (0.478) | 0.195 (0.584) |
| <I> | 1581.5 (299.7) | 382.2 (116.9) |
| <σ(I)> | 155.3 (183.5) | 41.8 (47.8) |
| Refinement | ||
| reflections used | 50–3.05, all data | 50–3.2, all data |
| No. of atoms | 4798 (3513 P; 1282 D; 3 W) | 2761 (2328 P; 428 D; 5 W) |
| R factor/No. of reflections | 0.2805/18 273 | 0.2634 /30 253 |
| Rfree/No. of reflections | 0.3172/842 | 0.2844/1612 |
| Ramachandran plot core/allowed (%) | 83.8/16.2 | 86.1/13.9 |
| bond length r.m.s.d. (Å) | 0.003 | 0.007 |
| bond angle r.m.s.d. (°) | 0.93 | 1.4 |
Structural overview
The (E1159–307)2(DNAd) structure (the first complex) consists of a pair of 2-fold-related E1159–307 proteins occupying consecutive major grooves of the DNAd duplex (Figure 3A). As expected, the proteins are bound to the highest affinity sites, E1–2 and E1–4. Although strict 2-fold symmetry within the complex would require a base pair to sit directly on the dyad axis, the complex does not appear to deviate much from 2-fold symmetry. Hence, binding by each E1–DBD molecule to its site is indistinguishable in this complex. The 3 bp situated between the binding sites are not contacted at all by either E1 monomer. The crystal of the tetramer complex (the second complex) consists of two unique (E1159–303)4(DNAt) complexes related by a strong NCS translation approximately along the crystallographic a-axis. The tetramer is composed of a perfectly palindromic oligonucleotide with two pairs of (E1159–303)2 dimers (comparable with those described above) occupying the anticipated 2-fold-related binding site pairs (E1–2, E1–4 and E1–1, E1–3; Figure 3B).

Fig. 3. Crystal structures of two intermediates in the assembly of E1–DBD subunits on the ori. (A) Structure of the E1–DBD dimer bound to sites 2 and 4 determined from the [(E1-DBD159–307)2(DNAd)] crystal structure. The binding site nucleotides identified by mutagenesis (Sedman et al., 1997; Chen and Stenlund, 1998) are colored blue and phosphate contacts identified by ethylation interference (Sanders and Stenlund, 1998) are colored red. (B) Structure of the E1–DBD tetramer bound to E1 binding sites 1–4 determined from the [(E1-DBD159–303)4(DNAt)] crystal structure viewed perpendicular and parallel to the DNA helical axis. The four monomers are individually colored; one dimer is colored cyan and purple, the other dimer pink and green and the two sets of binding sites are colored blue (E1–2 and E1–4) and yellow (E1–1 and E1–3).
Intersubunit interactions
Similar to the full-length protein, E1–DBD is monomeric in solution. In both complexes, pairs of E1–DBD monomers interact through their α3 helices (200–210) to form dimers (numbering scheme as in Enemark et al., 2000). We have shown previously that this region is the dimerization interface by disruption of E1–DBD dimers in [(E1–DBD)2DNA] and [(E1–DBD)2(E2–DBD)2(DNA)] complexes in helix α3 mutants (V202R, A206R; Enemark et al., 2000). This α3 interaction was observed in all four native E1–DBD crystal structures solved, one published (Enemark et al., 2000) and three unpublished; E.J.Enemark and L.Joshua-Tor, belonging to distinct crystal forms. However, this interaction is not orientationally rigid and seems to be a flexible hinge point. As a result, the DNA-binding surfaces are closer together in the DNA complex structures than in the native structure (T187Cα–T187Cα: dimer, 37.67 Å; tetramer, 37.99 Å; native, 43.07 Å). The specific dimeric interactions in any of these structures are not appreciably different.
In the origin recognition complex, E1 binds as a dimer to sites 2 and 4 together with E2. This species is converted to a tetrameric E1-only species. Interestingly, in the E1–DBD tetrameric complex, no significant ‘inter-dimer’ interactions are observed, although K186 (chain A) appears to interact with N189Oδ1 (3.2 Å) and T188O (3.7 Å) of chain B. However, the final electron density map does not allow a definitive assignment of all side chain conformations of K186 in the tetramer. The direction of this invariant side chain is intriguing, as it suggests that although K186 has been demonstrated to be critical for DNA binding, this residue also has an important role in the assembly of higher E1 oligomers and progression to functional hexamers.
E1 has two DNA-binding modules that bind the two individual strands
Each E1–DBD monomer in the two complexes utilizes two DBD-binding modules: a DNA-binding loop (DBL, R180–N189) and a DNA-binding helix with a portion of the loop N-terminal to it (DBH, T239–N248). These modules comprise a new DNA-binding motif. Interestingly, each of these modules contacts only one of the two DNA strands (Figure 4). Binding to the six-nucleotide E1BS (ATTGTT) is accomplished by recognition of two consecutive sequence triplets of opposing strands (Figure 2A). The structures presented here demonstrate that the individual sequence triplets are bound by separate elements of the protein, with the DBL binding to the first sequence element (ATT) and the DBH binding to the second (AAC; Figure 4). This partitioning of the strands onto distinct protein regions represents a novel DNA binding mode. In addition, the strand of contact for the two DNA-binding modules in the dimer is reversed from one E1–DBD monomer to the other. The tetramer structure confirms the proposed manner in which multiple proteins bind to partially overlapping sites (Chen and Stenlund, 2001; G.Chen and A.Stenlund, unpublished results). For the overlapping site sequence ATTGTTGTT (starting with base 4 of DNAt), the first protein contacts ATT of strand 1 via the DBL and AAC of strand 2 (base paired with the first GTT) via the DBH. The second protein contacts the first GTT of strand 1 via the DBL and AAC of strand 2 (paired with the second GTT) via the DBH. Thus, on the upstream side of the ori, both DBLs (of the proteins on sites 3 and 4) bind to the top strand and on the downstream side of the ori, both DBLs (of the proteins on sites 1 and 2) bind to the bottom strand. Therefore, we suggest that one hexameric helicase is assembled around one strand from the proteins at sites 1 and 2 on one side of the ori and the other hexameric helicase is assembled around the opposite strand from proteins at sites 3 and 4 on the other side of the ori.

Fig. 4. Protein–DNA contacts for a single binding site. (A) Close-up view of the protein–DNA contacts between one monomer and a single binding site. The DBL contacts one strand of the DNA, shown in pink and the DBH contacts the other strand, shown in gray. Hydrophilic interactions, all involving DNA phosphates, are indicated in blue and van der Waals interactions are indicated in green. Each base of the first three binding-site nucleotides makes van der Waals contacts with an element of the DBL. T-BS2, the only invariant base of the binding site, is extensively contacted at its methyl group. (B) A schematic representation of the protein–DNA contacts. Also shown are stabilizing interactions of DNA contact residues with other residues of the protein (black).
Binding site recognition
Although there is some variation in sequence among the binding sites, the protein–DNA contacts are identical for all molecules in the two structures and consist predominantly of hydrophilic interactions between the protein and phosphate oxygens of the DNA (Figure 4). These contacts are consistent with ethylation interference data for full-length E1 (Sanders and Stenlund, 1998), with additional contacts that are observed in the crystal structures. The predominance of generic DNA contacts is also consistent with the relatively low sequence speci ficity of the E1–DBD. Interactions of DNA with the DBH are less extensive than with the DBL. DNA binding by the DBH is mediated by side chain atoms that interact exclusively with DNA backbone atoms (T239–A14O2P; K241Nζ–A14O1P,O2P; T245Oγ–A13O1P,O5′; N248Nδ2– A130O2P).
All direct protein–base interactions are van der Waals interactions between DBL side chain atoms and the first three nucleotides of the E1BS. The most significant interactions involve the methyl group of the thymidine at position 2 of the binding site, T-BS2 (DNAd–T5; DNAt–T5, T8) and several side chain atoms: T187 (Cα 3.9 and Oγ 3.6 Å; Figure 4), N184 (Cγ 3.8 Å) and K186 (Cβ 4.0 Å; average values). This thymidine is an invariant base of the binding site; introducing any other base at this position nearly abolishes E1 binding (Figure 5). Moreover, removal of the methyl group by substitution with uridine (Figure 5) also abolishes binding to the site. A thymidine at this position is also invariant in the 20 papillomavirus origin sequences we examined (data not shown). A direct protein–base van der Waals interaction is also observed between the methyl group of the following thymidine, T-BS3 (DNAd–T6; DNAt–T6, T9) and K186Cβ (4.0 Å). A final protein:base van der Waals interaction is observed between T187 Oγ and adenine (or guanine) N7 and C8 at position 1 of the binding site (A-BS1; N7 4.2 and C8 3.9 Å). Other van der Waals interactions in the complex do not directly involve the bases; the aromatic side chain of the fully conserved F182 is packed against the sugar phosphate backbone of the nucleotide at position –1 of the binding site, while being sandwiched between the Cβ/Cγ of T187 and the alkyl ring of P266. Thus, all base contacts appear to be with the first three bases of the binding site. No direct hydrogen bonding interactions with the bases were observed.

Fig. 5. Summary of effects of mutations at all six positions of the E1 binding site. The bases at the six positions were changed individually to the three alternative bases and binding was measured in a gel-shift assay as described (Chen and Stenlund, 2001). At position 2, T was also mutated to U to assess the importance of the extensively contacted methyl group.
Two lysines in the DBL, K183 and K186, are highly conserved residues that appear to be important for DNA binding based on mutagenesis studies (Gonzalez et al., 2000). These residues were expected to have explicit DNA-binding properties in the crystal structures. However, a specific extended conformation for K183 is observed for only one of the dimer subunits. K183 is not in contact with any DNA atoms and cannot be envisioned to reach any base atoms. It could interact with a phosphate if the DNA adopted a larger bend. Explicit electron density for the ends of K186 side chains of the various monomers in the complexes is not observed in the final electron density map. However, other possible conformations (directed into the major groove) could generate base-stabilizing hydrogen bonds with T-BS2, T-BS3 and G-BS4. The lack of electron density for these residues in either complex implies that they are not in a fixed conformation or strict contact. Nonetheless, K186 may be in dynamic contact with more than one nucleotide before transitioning to the dimer–dimer interaction described earlier. K183, on the other hand, serves to neutralize the charge of the DNA backbone. It should be noted that although the resolution of these complexes was not sufficient to place water molecules with a high degree of confidence, there are no real candidates for water-mediated base contacts in these structures, though water-mediated contacts with the backbone are still possible.
All of the specific base contacts are thus van der Waals interactions with the DBL: two bases with minor contacts and one (T-BS2) with multiple contacts. Major contact with a single base was consistent with the previous mutational analysis of the E1 binding site (Figure 5; Chen and Stenlund, 2001) demonstrating that only one position of the hexanucleotide recognition sequence (position 2) has a strict requirement for a particular base. The observation that only three bases interact directly with the E1–DBD is intriguing and seemingly fails to account for the sequence selectivity observed at the remaining three positions of the hexanucleotide sequence. However, the sequence requirements at the other positions are more modest and may reflect structural requirements. In fact, a thymidine at position 1 of the binding site cannot be accommodated for steric reasons, in agreement with the mutational analysis (Figure 5). Interestingly, some of the severe mutations in the E1BS (e.g. G4T) can be rescued by a mutation at another position (e.g. T3G) in the site (data not shown), consistent with some mutations having a structural effect, such as the lack of deformability in a T-stretch. Moreover, in the tetramer, the hexanucleotide binding site is constrained by the partial overlap between the binding sites. The second part of the binding site for E1–1 and E1–4 (positions 4, 5 and 6) is also the first part (positions 1, 2 and 3) for E1–2 and E1–3 in the tetramer. Therefore, positions 1, 2 and 3 that contact the loop and have stronger sequence requirements now coincide with the latter part of sites E1–1 and E1–4.
Thus, sequence specificity appears to derive from a combination of base contacts and overall complementarity of the DNA molecule with the DNA-binding surface of the protein. A major element of DNA binding, the DBL, is an extended loop that does not possess regular secondary structure. However, this loop is well-ordered, with a consistent structure that is retained in the absence and presence of DNA [e.g. the root mean square deviation (r.m.s.d.) between the unbound protein and the dimeric complex is 0.34 Å for all Cα’s of the DBL; 0.39 Å for all main-chain atoms of the DBL; r.m.s.d. between two unbound DBLs is 0.37 Å]. In fact, two bromide ions in the unbound structure occupy practically the same positions as two of the phosphates in both complexes (DNAd, P3 and P4; DNAt, P3, P4 and P6, P7). Mutations that disrupt the integrity of the loop structure, such as D185A, result in loss of DNA-binding activity (Gonzalez et al., 2000). Two of the DBL–DNA contacts are hydrogen bonds between phosphate oxygen atoms and main-chain amide protons, which further supports the role of shape complementarity in addition to direct base readout for recognition. DBL side chains hydrogen bonded to phosphate oxygens are stabilized in their respective conformation by interactions with other side chain atoms that are also observed in the unbound E1–DBD structure (Figure 4A; Enemark et al., 2000). On the other hand, the binding elements of the DBH do not have well-defined structure-fixing interactions, other than residing on a well-defined secondary structure element. However, an important interaction may be the sandwiching of the K241 side chain between the F237 side chain, T245 Cγ and T239CO to lock this otherwise flexible side chain into position. The structural consistency of the DBL in all structures suggests that it is pre-organized in a DNA-binding conformation and poised for binding. As a result, the entropic cost of locking bonds into specific conformations for DNA binding has already been paid. Such a structure would be expected to display greater DNA-binding affinity than a comparable structure that requires more extensive conformational fixing.
Ordered overhangs
An interesting aspect of the tetramer structure is that although the ends of the DNA fragment used in this study have 3-base 5′ overhangs on either side (ATA), these bases are well ordered at both ends of the DNA duplex. The phosphate between A3 and A4 is contacted by the fully conserved R180 through Nε (2.65 Å), a contact present in the dimer complex as well and the order of the unpaired A3 is not surprising. The remaining two overhanging nucleotides, A1 and T2, are not observed to contact anything in the crystal lattice. Without direct interactions to fix their positions, these nucleotides were expected to be disordered. The significance of this finding is not clear; however, it should be noted that base opening by full-length E1 can be detected by permanganate sensitivity assays at positions corresponding to the first two base pairs of the oligonucleotide (A1 and T2; Sanders and Stenlund, 2000).
DNA deformation
Comparison of the DNA structures in the two complexes is of particular interest because the transition between the dimer and tetramer is likely to represent the first step towards melting. The DNA in each complex possesses an overall B-DNA structure. The bases are generally parallel and regularly spaced and the sugar phosphate backbones display prominent major and minor grooves. However, the DNA in both structures has an overall S-shape, with the center portion of the duplexes straight and bends in opposite directions on either side. This is significantly more pronounced in the tetramer (Figure 6). Helical analysis has revealed other consistent deviations from regular B-DNA in the two complexes, as well as a progression in these distortions from the dimer to the tetramer. Specifically, the DNA displays changes in groove widths, expansion of the helical diameter, changes in helical twist and other distortions, which are described below.

Fig. 6. DNA deformation in the two E1–DBD–DNA complexes. (A) Superposition of the DNA from the tetramer complex in blue and pink onto canonical form B-DNA. (B) Selected DNA global interbase-pair helical parameters for the dimer (blue) and tetramer (red) structures calculated by the program Curves (Lavery and Sklenar, 1989). Periodicity is observed in the rise, twist and slide parameters, with local maxima occurring at YR steps. These properties are most pronounced at the central YR step of the tetramer structure. Additionally, the central YR step of the tetramer structure displays an unusually negative roll. The corresponding parameters for canonical B-form DNA are shown in green.
The groove parameters for both structures display comparable relative trends in the vicinity of the E1–DBD binding sites. The major groove at the 5′-end of the E1BS is generally wider and the minor groove at the 3′-end is compressed relative to idealized B-DNA. An abrupt shift in major groove depth is present in the region of T-BS2, the invariant thymidine of the binding site. These groove parameters provide a quantitative basis for the ‘shape complementarity’ component in ori sequence recognition. A DNA sequence that is less amenable to adopting the observed groove widths and depths would not be bound as effectively by the protein.
The helical diameter of the dimer is larger than that of idealized B-DNA and this increase is also more pronounced in the tetramer (B-DNA, 18.5 Å; dimer, 19.3 Å; tetramer, 19.7 Å). Without additional structural changes, a direct consequence of a helical diameter increase would be to increase the distance between the complementary bases, perhaps removing the hydrogen bonding between them altogether. In the observed structures, the presence of consistently negative x-displacement parameters, a characteristic of a more A-DNA-like conformation, has allowed the maintenance of all base pairing hydrogen bonds. These displacements move the individual base pairs towards the minor groove relative to idealized B-DNA and allow the complementary bases to maintain a consistent separation upon widening of the helical diameter.
The twist at each base step is generally lower than that of idealized B-DNA, except at the pyrimidine–purine (YR) steps where the twist is considerably larger (37–45°). The largest twist is at the central base pair of the ori in the tetramer complex DNA (Figure 6). In fact, an intriguing feature of the E1BS is the regular and completely 2-fold symmetrical placement of the kinkable YR dinucleotide steps TG, CA, TA (Dickerson, 1998), around the center of the binding site: ATAATTGTTGTTAACAATAATCAC. Interestingly, none of these are CG steps, the most rigid YR step (Dickerson, 1998). The five central steps are at 3 base intervals, and the outer two are spaced at 4 base steps. The correlation of helical twist with this particular distribution of YR steps generates an obvious ‘small– small–big’ pattern in the plot of helical twist angles (Figure 6). This effect is also more pronounced in the tetramer than in the dimer.
Overall, distortions of each DNA structure from idealized B-DNA appear to occur continuously along the duplex, rather than in locally abrupt deviations. Exceptions are a large positive slide, a large negative roll and a large helical twist and rise in the global interbase-pair parameters at the central base step of the tetramer structure (Figure 6). In the dimer, there is a significant change in the slide between base pairs 9 and 10 and 12 and 13, with respect to the slide for the rest of the base steps in the ori. These base steps are at the immediate 3′-end of each E1-binding site towards the center of the ori. In the tetramer, with the binding of an additional dimer to sites E1–1 and E1–3, we observe a single, much more pronounced, slide at the center of the ori (Figure 6). The 3′-ends of E1–2 and E1–3 correspond to an identical base step and this appears to amplify the positive slide. Therefore, it appears that the positioning of the binding sites and thus the placement of the individual proteins on the ori, produces the increased distortion at the center of the ori. In addition, there is a considerable buckling (∼18°) of the GC base pair 5′ to the invariant T in the central E1 binding sites (E1–2 and E1–3) in the tetramer, which is much smaller in the dimer.
It appears that progressive addition of E1–DBD molecules to the ori induces a progressive increase in helical diameter with an associated progression in negative x-displacement. Progressively increased helical twists at the YR steps and progressively decreased helical twists at the other steps are also present. All of these distortions impart destabilizing forces upon the structural integrity of the duplex. The duplex structure has weathered the degree of forces in these complexes, but distortions of this type cannot be added indefinitely and eventually they would create structural failure in the form of melting.
Conclusion and mechanistic implications
The sequential loading and assembly of viral E1 subunits onto the ori constitute the first steps in initiation of replication of the papillomavirus genome. Here we have captured structural snapshots of two sequential steps in the assembly process. Loading of E1 dimers on the origin occurs initially at adjacent major grooves on one face of the double helix. Subsequently, a second dimer is loaded onto another face of the helix rotated 95° and translated 3 bp from the first dimer. One feature apparent in these early stages of E1 assembly is that binding of E1–DBD molecules on the ori causes some distortions in the DNA and these distortions become more pronounced in the progression from the dimer to the tetramer.
It is likely that the manner by which E1, in its various forms, recognizes and binds DNA reflects its different activities; some requiring binding to double-stranded DNA (dsDNA) and some requiring binding to single-stranded DNA (ssDNA). In addition to origin recognition and binding, which allows the helicase to be tethered to dsDNA, E1 eventually assembles into the replicative hexameric helicase around ssDNA. Full-length E1 requires ssDNA binding for oligomerization (Titolo et al., 2000). On the other hand, ssDNA binding is not required for oligomerization in the absence of the DBD (Titolo et al., 2000). Consistent with a transition from dsDNA binding to ssDNA binding, DNA binding by the E1–DBD shows some unusual features. Binding occurs through two modules that bind separately to the two strands of the DNA. The DBL, a long loop that is pre-organized for binding, makes the majority of the contacts, including the base-specific contacts, whereas the DBH makes only backbone contacts. This feature may have important implications for E1 function. Binding of the two individual strands by separate modules allows a simple transition from dsDNA to ssDNA binding, such that the protein maintains binding to one strand and dissociates from the other. Additional subunit–subunit contacts formed in the assembly of the hexameric ring would then compensate for lost protein–DNA interactions. A more common recognition motif, such as ‘helix in the groove’, in which the same structural element contacts both strands on either side of the groove, would not allow such a facile transition. Since interactions between DNA and the DBH are less extensive than interactions with the DBL, the DBL is a more likely candidate for the binding module that retains binding. Moreover, in the tetramer, the two DBLs of the upstream monomers of each dimer bind to the top strand, while the DBLs of the downstream monomers of each dimer bind to the bottom strand. Therefore, we suggest that one hexameric helicase assembles around the top strand on one side of the ori from the proteins at sites 3 and 4 and the other hexameric helicase assembles around the bottom strand on the other side of the ori from the proteins at sites 1 and 2. As the hexameric helicases travel in opposite directions around their respective strands, the correct 3′ to 5′ polarity would occur (Figure 7). Full understanding of the assembly process awaits structural characterization of additional steps. Interfering with the assembly process should prove useful in designing anti-viral therapies against this important group of viruses.
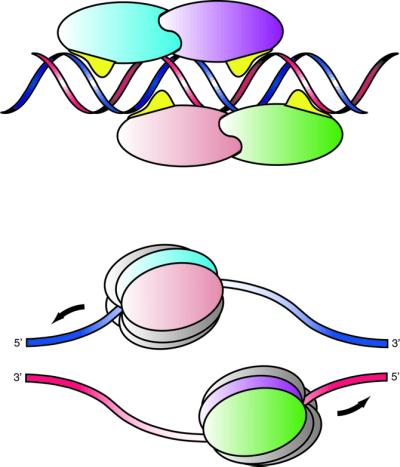
Fig. 7. Schematic diagram of the proposed role of the DBL in strand selection. A cartoon depicting a model for the assembly of two hexameric helicases around single strands at the ori. The two DBLs (in yellow) of the upstream monomers of each dimer bind to the top (blue) strand, while the DBLs of the downstream monomers of each dimer bind to the bottom strand (shown in red). One hexameric helicase assembles from the upstream monomers at sites 3 and 4 (pink and blue) around the top strand and the other hexameric helicase assembles from the downstream monomers at sites 1 and 2 (green and purple). This arrangement results in the correct 3′ to 5′ polarity for the helicases.
Materials and methods
Protein expression and purification
Two different fragments containing the E1–DBD (E1159–303 and E1159–307) of BPV E1 were expressed and purified as N-terminal glutathione S-transferase fusion proteins with an intermediate thrombin cleavage site and purified as described previously (Enemark et al., 2000). A stock solution of each protein was prepared at 10 mg/ml, 25 mM HEPES pH 7.5, 100 mM NaCl and 200 mM dithiothreitol.
Oligonucleotides
Mutations were introduced into the oligonucleotide used for the tetramer in order to generate a perfect palindrome and thus generate 2-fold symmetry, which would facilitate the structure determination. The effect of one of these mutations in the binding site would be to decrease affinity while the effect of the other would be to increase affinity, both on the order of a 2-fold change (see Figure 5). Oligonucleotides were purchased as trityl on commercial synthesis products (Operon) and were purified by reverse phase chromatography on a Biocad SPRINT system, followed by desalting with a Sep–Pak C18 column and cation exchange to Na+ on a Sephadex column. Duplexes were generated by mixing stoichiometric amounts of the two strands, heating to 60°C and slowly cooling to room temperature.
Crystallization and diffraction data measurement
(E1159–307)2(DNAd). Solutions of the protein and the oligonucleotide were combined and used immediately for crystallization trials. Block crystals suitable for X-ray analysis were grown by the hanging drop method, utilizing a 1:1 mixture of the DNA complex and a well solution consisting of 20 mM MgSO4, 2.5 mM spermine tetrahydrochloride and 5% ethylene glycol. Spontaneous growth under these conditions was rare but was readily promoted by seeding.
A crystal was removed from the mother liquor with a fiber loop, rapidly passed through a 33% ethylene glycol:well solution and immediately frozen in a cold nitrogen stream at 100 K. The diffraction limit correlated with the orientation of the crystallographic c-axis. Reflections with a large ‘c-axis contribution’ displayed unusually large intensities in the vicinity of 3.24 Å resolution. The crystals were monoclinic C2 (a = 149.123, b = 110.698, c = 75.227, β = 116.869) with 1.5 dimers/asymmetric unit—one on the crystallographic 2-fold.
(E1159–303)4(DNAt). Direct combination of solutions of the protein and the oligonucleotide resulted in immediate precipitation; therefore, the protein solution was mixed 1:1 with 1 M Ca(CF3COO)2 and subsequently the oligonucleotide solution was added. The resulting solution showed no tendency to form a precipitate. A drop of the solution was equilibrated by the hanging drop method against a distilled water well solution. Large block crystals grew readily and spontaneously. The majority of these crystals were visibly flawed or twinned on a crystallographic axis.
A single (untwinned) crystal suitable for X-ray analysis was removed from the mother liquor with a fiber loop, cryoprotected and frozen as described above. The diffraction limit correlated with the orientation of the crystallographic c-axis. Reflections with a large ‘c-axis contribution’ displayed unusually large intensities in the vicinity of 3.24 Å resolution. The crystals are monoclinic P21 (a = 84.245, b = 103.539, c = 124.878, β = 99.471)—pseudo orthorhombic (C2221) following a [0.5 0 0/ –0.5 0 –2/0 1 0] matrix (a = 42.092, b = 246.029, c = 103.524). This transform treats the two NCS operators (see below) as crystallographic.
Data for both complexes were collected at beamline X26C at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory (BNL). Data were processed with the program HKL (Otwinowski and Minor, 1997). Further details on data collection statistics and refinement are shown in Table I.
Structure determination and refinement
Dimer. The structure was solved by molecular replacement with the program AMoRe (Navaza and Saludjian, 1997) using the coordinates of the crystal structure of E1–DBD159–303 (Enemark et al., 2000) as a search model and a resolution range of 10.0–4.0 Å. Four molecules were initially placed, but the fourth overlapped with a previously placed molecule and was discarded. The remaining three molecules were extremely compelling, displaying two intermolecular contacts identical to those observed in the unbound E1–DBD structure.
Model phases were calculated from the placed protein atoms and used as the starting phases for the density modification routine of CNS (Brünger et al., 1998) utilizing NCS-averaging and solvent flipping with a 70% solvent fraction. The resulting calculated electron density map had a clearly traceable DNA duplex. An idealized B-DNA form of E1BS modified to be perfectly palindromic (excluding a thymine at the central position) was placed in one strand of the density and the individual nucleotides were moved individually to optimally fill the appropriate electron density. The application of crystallographic and non-crystallographic symmetry upon a single protein and a single oligonucleotide strand generates two distinct [(E1–DBD)2(E1BS)1] complexes.
Crystallographic refinement was performed with the program CNS by initially employing strict NCS, a single E1–DBD molecule and one strand of a palindromic sequence. Group temperature factors were refined. The dimer crystal consisted of 1.5 unique (E1159–307)2(DNAd) complexes. Since the oligonucleotide was not perfectly palindromic, it could not possess rigorous 2-fold symmetry. A half-complex (dimer A) sat upon a crystallographic 2-fold with a static disordered oligonucleotide. A full complex (dimer B) sat on a general crystallographic position with a DNA duplex that also appeared to be 2-fold disordered. Each end of dimer A is bluntly packed against one end of dimer B. The remaining end of dimer B has no direct packing interaction. The differences in crystallographic packing of the oligonucleotide ends prevent equivalency between the NCS-related portions and could not be adequately modeled by strict NCS and so the structure was ultimately refined with restrained NCS. The two dimers did not display any appreciable differences in the protein–DNA interface.
Tetramer. The tetramer crystal structure was solved by molecular replacement with the program AMoRe (Navaza and Saludjian, 1997) using the coordinates of the crystal structure of E1159–303 (Enemark et al., 2000) as a search model and a resolution range of 12.0–3.7 Å. Two sets of four molecular orientations were placed, related by the NCS translation. The placed molecules displayed two intermolecular contacts identical to those observed in E1–DBD and the dimer structure (above). Model phases were calculated from the placed protein atoms and used as the starting phases for the density modification routine of CNS (Brünger et al., 1998) utilizing NCS-averaging and solvent flipping with a 66% solvent fraction. The resulting calculated electron density map had a clearly traceable DNA duplex. A single oligonucleotide strand of the dimer structure was placed in one strand of the density and the individual nucleotides were moved individually to optimally fill the appropriate electron density. The application of non-crystallographic symmetry upon two proteins and a single oligonucleotide strand generated two distinct [(E1–DBD)4(E1BS)1] complexes related by strong NCS translation approximately along the crystallographic a-axis. The tetramer is composed of a perfectly palindromic oligonucleotide with two pairs of (E1159–303)2 dimers occupying the anticipated 2-fold-related binding site pairs. The structure was refined with strict NCS translational symmetry and with a strict 2-fold rotational symmetry (the 2-fold of the DNA palindrome) as well as group temperature factors.
Mutagenesis and binding studies
Mutations in the E1 binding site and binding assays were performed as described previously (Chen and Stenlund, 2001).
DNA helix analysis
DNA helix analysis was carried out with the programs Curves (Lavery and Sklenar, 1989), 3DNA (Lu et al., 2000) and Freehelix (Dickerson, 1998).
Figures
Figures 2B, 3, 4A and 6A were prepared with Bobscript (Kraulis, 1991; Esnouf, 1997) and Raster3D (Bacon and Anderson, 1988; Merritt and Murphy, 1994).
Coordinates
Coordinates have been submitted to the Protein Data Bank, under the accession codes 1KSY (dimer) and 1KSX (tetramer).
Acknowledgments
Acknowledgements
We thank Dr Dieter Schneider for help and support with data collection at beamline X26C and Drs Alexander Gann and Adrian Krainer for critical reading of the manuscript. The NSLS at BNL is funded by the US Department of Energy Office of Basic Energy Sciences, Division of Material Sciences and Division of Chemical Sciences under contract number DE-AC02-98CH10886. This work was supported by a National Cancer Institute training grant to E.J.E., National Institutes of Health grant (CA13106) to A.S. and grants from the American Cancer Society (RPG-99-208-01-MBC), the National Institutes of Health (AI46724) and the Louis Morin Charitable Trust to L.J.
References
- Bacon D.J. and Anderson,W.F. (1988) A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graph., 6, 219–220. [Google Scholar]
- Borowiec J.A., Dean,F.B., Bullock,P.A. and Hurwitz,J. (1990) Binding and unwinding—how T-antigen engages the SV40 origin of DNA replication. Cell, 60, 181–184. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromoleclar structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chen G. and Stenlund,A. (1998) Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J. Virol., 72, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. and Stenlund,A. (2001) The E1 initiator recognizes multiple overlapping sites in the papillomavirus origin of DNA replication. J. Virol., 75, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conger K.L., Liu,J.S., Kuo,S.R., Chow,L.T. and Wang,T.S. (1999) Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase α/primase. J. Biol. Chem., 274, 2696–2705. [DOI] [PubMed] [Google Scholar]
- Dickerson R.E. (1998) DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res., 26, 1906–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enemark E.J., Chen,G.Y., Vaughn,D.E., Stenlund,A. and Joshua-Tor,L. (2000) Crystal structure of the DNA-binding domain of the replication initiation protein E1 from papillomavirus. Mol. Cell, 6, 149–158. [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 132–134. [DOI] [PubMed] [Google Scholar]
- Fanning E. and Knippers,R. (1992) Structure and function of simian virus 40 large tumor antigen. Annu. Rev. Biochem., 61, 55–85. [DOI] [PubMed] [Google Scholar]
- Fouts E.T., Yu,X., Egelman,E.H. and Botchan,M.R. (1999) Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J. Biol. Chem., 274, 4447–4458. [DOI] [PubMed] [Google Scholar]
- Gillette T.G., Lusky,M. and Borowiec,J.A. (1994) Induction of structural changes in the bovine papillomavirus type 1 origin of replication by the viral E1 and E2 proteins. Proc. Natl Acad. Sci. USA, 91, 8846–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillitzer E., Chen,G. and Stenlund,A. (2000) Separate domains in E1 and E2 proteins serve architectural and productive roles for cooperative DNA binding. EMBO J., 19, 3069–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A., Bazaldua-Hernandez,C., West,M., Wotek,K. and Wilson,V.G. (2000) Identification of a short, hydrophilic amino acid sequence critical for origin recognition by the bovine papillomavirus E1 protein. J. Virol., 74, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Loo,Y.M., Militello,K.T. and Melendy,T. (1999) Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J. Virol., 73, 4899–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho G.Y.F., Bierman,R., Beardsley,L., Chang,C.J. and Burk,R.D. (1998) Natural history of cervicovaginal papilloma virus infection in young women. N. Engl. J. Med., 338, 423–428. [DOI] [PubMed] [Google Scholar]
- Holt S.E. and Wilson,V.G. (1995) Mutational analysis of the 18-base-pair inverted repeat element at the bovine papillomavirus origin of replication: identification of critical sequences for E1 binding and in vivo replication. J. Virol., 69, 6525–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst., 24, 946–950. [Google Scholar]
- Lavery R. and Sklenar,H. (1989) Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn., 6, 655–667. [DOI] [PubMed] [Google Scholar]
- Lu X.-J., Shakked,Z. and Olson,W. (2000) A-DNA conformational motifs in ligand-bound double helices. J. Mol. Biol., 300, 819–840. [DOI] [PubMed] [Google Scholar]
- Lusky M., Hurwitz,J. and Seo,Y.S. (1994) The bovine papillomavirus E2 protein modulates the assembly but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proc. Natl Acad. Sci. USA, 91, 8895–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson P.J., Stanley,M.A., Lewis,A.P. and Romanos,M.A. (1998) A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J. Virol., 72, 7407–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza R., Gandhi,L. and Botchan,M. (1995) E1 recognition sequences in the bovine papillomavirus type 1 origin of DNA replication: interaction between half sites of the inverted repeats. J. Virol., 69, 3789–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E.A. and Murphy,M.E.P. (1994) Raster3D version 2.0—A program for photorealistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Navaza J. and Saludjian,P. (1997) AMoRe: an automated molecular replacement program package. Methods Enzymol., 276, 581–594. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Park P., Copeland,W., Yang,L., Wang,T., Botchan,M. and Mohr,I. (1994) The cellular DNA polymerase α-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc. Natl Acad. Sci. USA, 91, 8700–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.M. and Stenlund,A. (1998) Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalyzed by a transcription factor. EMBO J., 17, 7044–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.M. and Stenlund,A. (2000) Transcription factor-dependent loading of the E1 initiator reveals modular assembly of the papillomavirus origin melting complex. J. Biol. Chem., 275, 3522–3534. [DOI] [PubMed] [Google Scholar]
- Sedman J. and Stenlund,A. (1995) Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator-specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J., 14, 6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman J. and Stenlund,A. (1998) The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J. Virol., 72, 6893–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman T., Sedman,J. and Stenlund,A. (1997) Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J. Virol., 71, 2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.S., Muller,F., Lusky,M., Gibbs,E., Kim,H.Y., Phillips,B. and Hurwitz,J. (1993a) Bovine papillomavirus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc. Natl Acad. Sci. USA, 90, 2865–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.S., Muller,F., Lusky,M. and Hurwitz,J. (1993b) Bovine papillomavirus (BPV) encoded E1 protein contains multiple activities required for BPV DNA replication. Proc. Natl Acad. Sci. USA, 90, 702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl H., Droge,P. and Knippers,R. (1986) DNA helicase activity of SV40 large tumor antigen. EMBO J., 5, 1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sverdrup F. and Myers,G. (1997) The E1 proteins. In Bernard,H.-U. (ed.), Human Papillomaviruses 1997. Los Alamos National Laboratory, Los Alamos, NM.
- Titolo S., Pelletier,A., Pulichino,A.-M., Brault,K., Wardrop,E., White, P.W., Cordingley,M.G. and Archambault,J. (2000) Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the viral origin. J. Virol., 74, 7349–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers J.M. et al. (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol., 189, 12–19. [DOI] [PubMed] [Google Scholar]
- Yang L., Li,R., Mohr,I., Clark,R. and Botchan,M.R. (1991) Activation of BPV-1 replication in vitro by the transcription factor E2. Nature, 353, 628–633. [DOI] [PubMed] [Google Scholar]
- Yang L., Mohr,I., Fouts,E., Lim,D.A., Nohaile,M. and Botchan,M. (1993) The E1 protein of the papillomavirus BPV-1 is an ATP dependent DNA helicase. Proc. Natl Acad. Sci. USA, 90, 5086–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. (1991) Viruses in human cancers. Science, 254, 1167–1173. [DOI] [PubMed] [Google Scholar]