

Abstract
MuB assembles into a polymer on DNA in the presence of ATP and is directly involved in the selection of an appropriate site on the Escherichia coli chromosome for the insertion of the bacteriophage Mu genome. We have developed an assay using fluorescently tagged proteins to monitor the polymeric state of MuB via fluorescence resonance energy transfer. We show that polymer assembly is initiated by the formation of an ATP–MuB complex. MuB then self-associates into a protomer before binding to DNA. Upon binding to DNA, a dramatic increase in energy transfer is observed, suggesting a conformational change within MuB. Polymer disassembly is much slower than assembly and is greatly stimulated by the MuA transposase. Additionally, MuB is readily exchanged between polymers, and ATP hydrolysis is directly coupled to polymer disassembly. Our data support a model in which a combination of rapid polymer assembly, MuA-mediated disassembly, followed by rapid reassembly of the polymer allows MuB to sample multiple DNA targets until an appropriate site is located for the insertion of the bacteriophage genome.
Keywords: DNA transposition/fluorescence resonance energy transfer/green fluorescent protein/MuB
Introduction
Transposons are mobile genetic elements that transfer DNA from one site within a genome to another site (Mizuuchi, 1992; Craig, 1997). These reactions are mediated by a transposase protein and often are subject to highly complex regulatory pathways. Phage Mu has served as a model system for the study of transposons and other mobile genetic elements and, as such, has generated several important paradigms which have increased the understanding of both DNA transposition and the interactions involved in complex macromolecular assemblies (Mizuuchi, 1992; Chaconas et al., 1996; Craig, 1997).
The Mu transposition reaction, up to and including DNA strand transfer, requires the two phage-encoded proteins MuA and MuB, the host protein cofactors HU and IHF, a donor DNA molecule containing MuA-binding sites and a non-specific target DNA. Transposition proceeds through a series of increasingly stable nucleoprotein complexes called transpososomes (Craigie and Mizuuchi, 1987; Surette et al., 1987; Baker and Mizuuchi, 1992; Mizuuchi et al., 1992). The first step in assembly is the binding of MuA transposase to the consensus sites flanking the donor DNA. Following the initial binding event, a transient three-site synaptic complex is assembled containing both donor ends and the enhancer-like internal activation sequence (IAS) element (Watson and Chaconas, 1996). In the presence of divalent metal, HU and IHF, this complex is converted to the stable synaptic complex (SSC), in which only the donor ends are associated via a tetramer of MuA (Mizuuchi et al., 1992). The SSC is converted to the cleaved donor complex (CDC) by MuA-catalyzed cleavage of the donor DNA ends, and the exposed hydroxyls are used as nucleophiles for strand transfer into a MuB-bound target DNA molecule to form the strand transfer complex (STC).
MuB is a 312 amino acid protein with weak ATPase activity and an ATP-dependent DNA-binding activity (Adzuma and Mizuuchi, 1991; Mizuuchi, 1992). MuB can be divided into two domains by limited proteolysis (Teplow et al., 1988). The 25 kDa N-terminal domain contains the bipartite nucleotide-binding motif Walker A and Walker B boxes (Teplow et al., 1988). Site directed mutagenesis of amino acids within either of the Walker box motifs results in a loss of ATPase activity (Yamauchi and Baker, 1998). Although the structure of the N-terminal domain remains unknown, the C-terminal domain has been solved recently by NMR spectroscopy (Hung et al., 2000). This 8 kDa region is a four-helix bundle displaying structural similarity to the N-terminal region of the Escherichia coli replication helicase DnaB. In the absence of the C-terminal domain, the isolated N-terminal region is able to bind nucleotide, but is incapable of hydrolyzing ATP or stimulating strand transfer and has significantly reduced DNA-binding activity (Teplow et al., 1988).
Transposition is enhanced greatly in the presence of MuB and ATP (Chaconas et al., 1985; Maxwell et al., 1987). In the presence of ATP, MuB polymerizes on DNA to form the transposition target complex, which can then be used as an efficient substrate for MuA-catalyzed insertion of the donor DNA. Thus, the preferred target for donor DNA insertion is a DNA-bound MuB complex and not simply naked DNA. MuB also influences nearly every other step of the transposition reaction (Maxwell et al., 1987; Baker et al., 1991; Mizuuchi, 1992). MuB bound to target DNA interacts with the SSC to stimulate the formation of strand transfer products. MuB stimulates the formation of the CDC and, in reactions containing mutated donor DNA ends, MuB is required for CDC formation (Surette and Chaconas, 1991). When one of the two MuA protomers involved in cleavage of the scissile bonds contains a mutated active site, MuB is required to allow catalysis by the other MuA protomer to proceed (Williams et al., 1999). Additionally, MuB can be chemically cross-linked to all of the transpososome intermediates present during the course of the reaction (Naigamwalla and Chaconas, 1997).
Target immunity, the ability to discriminate between proximal and distal DNA sites relative to the transposon, is a property common to many transposition systems (reviewed in Mizuuchi, 1992; Craig, 1997). Mu target immunity is a consequence of the dynamic behavior of MuB and its response to ATP, DNA and MuA (Adzuma and Mizuuchi, 1988, 1989, 1991). In the absence of MuB, most transposition occurs near or within the donor DNA, which can result in the destruction of the transposon. To avoid this, MuA stimulates the ATP hydrolysis activity of MuB, thereby promoting the dissociation of MuB from DNA to which MuA is bound. This results in the preferential accumulation of MuB on DNA that does not contain nearby MuA-binding sites. Although seemingly simple, many of the details of the target immunity mechansim remain unknown, and a clear understanding of these processes will require detailed knowledge of the steps involved in the assembly and disassembly of the MuB polymer.
Here we present the development of a fluorescence resonance energy transfer (FRET) assay to monitor the polymeric state of MuB and its response to nucleotide, DNA and MuA. Additionally, we provide a kinetic analysis that has allowed us to dissect the steps of the assembly and disassembly pathways of the MuB target complex. These studies show that assembly of the target complex is initiated by the binding of MuB to ATP. MuB then self-associates to form a MuB protomer complex prior to binding DNA. The MuB protomer can then bind to DNA, and our results suggest that DNA binding is coupled to a conformational change within the protomer. Finally, analysis of the target complex disassembly pathway revealed that subunits of MuB are readily exchanged between polymers and that MuB hydrolyzes only one molecule of ATP prior to dissociating from the DNA.
Results
EGFP–MuB has biochemical properties similar to wild-type MuB
We have constructed several fluorescent variants of MuB by directly fusing the protein to either EGFP, ECFP or EYFP (enhanced green, cyan or yellow fluorescent protein, respectively). The fusion proteins were tested initially for their ability to stimulate in vitro transposition (Figure 1). Strand transfer assays performed in the absence of MuB resulted in the accumulation of the nicked form of the donor DNA and a small amount of intramolecular strand transfer products. However, in the presence of wild-type MuB or N-terminal labeled MuB (EGFP–MuB), most of the reaction products were a result of intermolecular strand transfer. The total amount of intermolecular product produced in reactions containing EGFP–MuB was somewhat reduced compared with wild-type MuB, with a concomitant increase observed in the production of intra molecular products (compare wt MuB and EGFP–MuB lanes). In contrast to EGFP–MuB, the C-terminal EGFP-labeled MuB (MuB–EGFP) was unable to stimulate intermolecular strand transfer (Figure 1). Only stimulation of intramolecular strand transfer was observed. This protein did display nucleotide-dependent DNA binding as detected by gel filtration (data not shown). However, even when pre-bound to the target DNA in the presence of ATP (or ATPγS), MuB–EGFP was unable to support intermolecular strand transfer (data not shown).

Fig. 1. EGFP–MuB stimulates intermolecular transposition. Strand transfer reactions were assembled containing 10 ng/µl each φX174 (target DNA) and pMK586 (donor DNA), 25 mM Tris pH 8, 100 ng/µl BSA, 150 mM NaCl, 10 mM MgCl2, 20% glycerol, 1 mM DTT and 4 ng/µl Hu. EGFP–MuB or wild-type MuB were added to a final concentration of 200 nM. Reactions were initiated with the addition of MuA to 50 nM, incubated 1 h at 30°C and terminated with the addition of SDS/EDTA loading dye. Products were resolved with 0.8% HGT–agarose in 1× TAE buffer and stained with ethidium bromide. Intermolecular and intramolecular strand transfer products are indicated. The nicked and supercoiled forms of the target and donor DNAs are indicated as O and SC, respectively. The nicked DNA species represents the accumulation of CDC that did not react with a target DNA molecule.
The ability of MuB to hydrolyze ATP is directly related to its role in establishing target immunity prior to donor DNA insertion (Adzuma and Mizuuchi, 1988). The fluorescent proteins were therefore assayed for the ability to hydrolyze ATP. The Km for ATP hydrolysis was ∼4-fold higher for the EGFP fusion proteins, while their kcat values were nearly identical to those of wild-type MuB (Table I). We also examined the ATPase activity of the fluorescent derivatives of MuB in the presence of DNA and/or MuA. We found that EGFP–MuB responded similarly to wild-type MuB (data not shown). At low ATP concentration (20 µM), DNA had a stimulatory effect on the EGFP–MuB ATPase activity. Conversely, at a higher ATP concentration (500 µM), DNA had an inhibitory effect on the ATPase activity. These properties parallel the known response of wild-type MuB to DNA at varying concentrations of ATP (Adzuma and Mizuuchi, 1991).
Table I. The ATPase activity of the EGFP fusion proteins is similar to that of wild-type MuB.
| kcat (ATP/MuB per min) | Km (µM) | |
|---|---|---|
| Wild-type MuB | 4.1 | 80 |
| EGFP–MuB | 3.9 | 320 |
| MuB–EGFP | 2.5 | 400 |
ATPase assays were assembled containing 0.5, 20, 320, 1280 or 5120 µM ATP total (each reaction containing 0.5 µM [α-32P]ATP). Buffer conditions were the same as with the strand transfer assays, with the omission of DNA and MuA. Reactions were initiated with the addition of MuB proteins to a final concentration of 1.5 µM and incubated at 30°C. Aliquots were removed at 5, 10, 15 and 30 min and terminated with the addition of EDTA to a final concentration of 50 mM. Reaction products were resolved with PEI TLC plates in 0.75 M KH2PO4 and quantitated by phosphoimaging.
The ability of the fluorescently labeled MuB to support target immunity was tested by pre-incubating EGFP–MuB with ATP, an immune plasmid (pMK589) and a non-immune plasmid (φX174) in the presence or absence of MuA (Figure 2). The plasmid pMK589 contains sequences corresponding to the left and right ends of the Mu genome; however, these ends are not in the proper orientation to support transpososome formation (Adzuma and Mizuuchi, 1988). MuA can therefore bind to the immune plasmid without catalyzing transposition. In the absence of MuA, MuB is predicted to become evenly distributed between the two target plasmids. In the presence of MuA, MuB should accumulate preferentially onto the non-immune plasmid. Following pre-incubation with or without MuA, the reactions were chased by the addition of a pre-formed CDC. Target immunity was revealed as the absence of intermolecular strand transfer products with the immune target (pMK598) and the presence of products resulting from donor insertion into the non-immune φX174 target (Figure 2, compare wt MuB lanes with and without the MuA pre-incubation). Both wild-type MuB and EGFP– MuB were able to support target immunity (Figure 2). The fact that EGFP–MuB was able to stimulate strand transfer, hydrolyze ATP and support target immunity suggested that most or all of the structural and biochemical properties of wild-type MuB were retained in this fluorescent derivative of MuB.
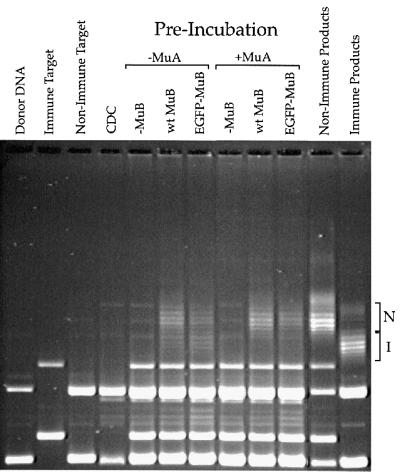
Fig. 2. EGFP–MuB supports target DNA immunity. Duplicate strand transfer reactions were assembled containing 200 nM ATP and both a non-immune target DNA (φX174) and an immune target DNA (pMK589, containing a normal Mu left end and a Mu right end in inverted orientation). EGFP–MuB or wild-type MuB was added to a final concentration of 200 nM. MuA was added to 40 nM to one set of reactions, and all reactions were pre-incubated at 30°C for 30 min. Following the pre-incubation, pre-formed CDC was added along with 2 mM ATPγS and incubations continued for an additional 5 min. Reactions were then terminated with SDS/EDTA buffer and products resolved with 0.7% HGT–agarose. The non-immune (N) and immune (I) strand transfer products are also shown.
Target complex assembly can be detected using FRET
MuB forms a large polymeric structure when bound to DNA and, under some conditions, MuB can oligomerize in the absence of DNA (Adzuma and Mizuuchi, 1991; Mizuuchi, 1992; Y.Wang, R.Ghirlando and K.Mizuuchi, unpublished observations). It therefore seemed likely that the association of MuB monomers into a polymeric structure could be detected using FRET. Using ECFP– MuB and EYFP–MuB as a fluorescence donor–acceptor pair (Miyawaki and Tsien, 2000), we determined that FRET occurred between MuB proteins bound to DNA. ATPγS, an analog of ATP that MuB cannot hydrolyze (Adzuma and Mizuuchi, 1991), was used as the nucleotide cofactor to drive all of the MuB into the polymeric state and to limit the redistribution of MuB during the experiment. ECFP–MuB or EYFP–MuB complexes were formed with ATPγS and DNA, the reaction mixes were excited at 425 nm and the emission spectra of each complex were measured (Figure 3). The emission spectra for the ECFP–MuB and EYFP–MuB complexes were the same as those for the free proteins in the absence of nucleotide and DNA (data not shown). The two separate complexes were then mixed together and the emission spectra immediately measured. The resulting spectrum was identical to the sum of the two spectra for the individual ECFP–MuB and EYFP–MuB complexes, indicating that under these conditions, no energy transfer was detectable (Figure 3). However, when a mixed complex containing both ECFP–MuB and EYFP–MuB was formed in the presence of DNA and ATPγS, a dramatic increase in EYFP–MuB emission was observed along with a concomitant decrease in the emission from ECFP–MuB, corresponding to ∼40% energy transfer efficiency (Figure 3). These results showed that an efficient energy transfer interaction occurred between MuB monomers bound to the same DNA and indicated a very intimate association between monomers within the target complex.

Fig. 3. Target complex formation can be detected with fluorescence energy transfer between ECFP–MuB and EYFP–MuB. MuB–DNA complexes were assembled on ice in reactions containing 2.5 ng/µl φX174, 1 mM ATPγS, 150 mM NaCl, 10 mM MgCl2, 20% glycerol, 1 mM DTT and either 0.2 µM ECFP–MuB (fluorescence donor) only, 0.2 µM EYFP–MuB (fluorescence acceptor) only or 0.1 µM each ECFP–MuB and EYFP–MuB. Reactions were incubated for 5 min at 25°C prior to data collection. Excitation was at 425 nm and emission spectra were collected from 450 to 600 nm. The energy transfer efficiency for the ECFP–MuB/EYFP–MuB complex was 40% (see Materials and methods). All spectra were normalized to account for differences in protein concentration.
The cofactor requirements for complex formation were investigated further using FRET to report the polymeric state of MuB. Energy transfer could be detected with ATPγS, in the presence or absence of DNA, at all MuB concentrations examined (0.2–5 µM; Figure 4). With ATPγS as the nucleotide cofactor, the FRET signal reached a maximum at concentrations of MuB ≥1 µM, indicating that all of the MuB was incorporated into the polymer. ATP alone was also sufficient to drive polymerization of MuB in the absence of DNA, although the FRET efficiency was less than that observed with ATPγS (Figure 4). Because MuB forms polymeric structures in the presence of ATP or ATPγS, in the absence of DNA, it was possible that these complexes represented a precursor of the polymeric DNA-bound form of MuB. Therefore, we refer to MuB complexes formed in the absence of DNA as protomers.

Fig. 4. Nucleotide and DNA requirements for MuB polymer assembly. Reactions were assembled with 2 mM ATP, ATPγS or ADP in the presence or absence of DNA (10 ng/µl φX174) as indicated. Each reaction contained equimolar concentrations of ECFP–MuB and EYFP–MuB, and the total concentration of MuB was 0.2, 0.5, 1.0, 2.0 or 5.0 µM. All data are plotted as the ratio of fluorescence acceptor emission to fluorescence donor emission (Fa/Fd) as a function of MuB concentration.
Efficient energy transfer was detected in the presence of ATP and supercoiled, linear or single-stranded DNA (data not shown). The addition of DNA to reactions containing ATP or ATPγS produced a substantial increase in the FRET signal (Figure 4). This suggested that either more extensive polymerization was occurring on the DNA or a conformational change was occurring within the MuB polymer that led to increased transfer efficiency. Significant energy transfer was not detected in the absence of nucleotide, with DNA only, or with ADP only (Figure 4). Interestingly, at high MuB concentrations (2–5 µM), in the presence of DNA, ADP was able to stimulate some limited MuB polymerization as indicated by FRET (Figure 4). This signal may represent the ADP-bound form of MuB, which is known specifically to stimulate intramolecular strand transfer (Yamauchi and Baker, 1998). In general, these results were consistent with previously described reaction conditions required for assembly of MuB complexes (Adzuma and Mizuuchi, 1991; Y.Wang, R.Ghirlando and K.Mizuuchi, unpublished observations).
Formation of the MuB protomer is kinetically distinct from DNA binding
The ability to detect MuB polymerization using FRET provided an assay to monitor directly the behavior of MuB during the course of a reaction. To dissect the steps involved in the target complex assembly pathway, the kinetics of MuB polymer formation were examined with a stopped-flow fluorimeter. The change in the ratio of donor to acceptor emission (Fa/Fd) as free ECFP–MuB and EYFP–MuB were incorporated into the target complex was monitored as a function of time after the addition of DNA and ATP (Figure 5A). Polymer assembly displayed two distinct phases, a short lag of ∼1–2 s followed by an increase in FRET which reached completion in ∼1 min, and similar kinetics were observed at 0.1, 0.2 and 0.5 µM MuB (data not shown).

Fig. 5. Assembly of the MuB target complex is preceded by the formation of a MuB protomer. (A) Target complex assembly was monitored by rapidly mixing ECFP–MuB, EYFP–MuB and φX174 with ATP. The final concentrations of reaction components in (A) and (B) were as follows: 125 nM ECFP–MuB, 125 nM EYFP–MuB, 10 ng/µl φX174 (15 µM base pairs) and 2 mM ATP plus or minus 100 nM MuA. (B) DNA binding of a pre-formed protomer was monitored by mixing ECFP–MuB, EYFP–MuB and ATP with φX174. (C) Protomer assembly was monitored by mixing ECFP–MuB and EYFP–MuB with ATP. The final concentrations of reaction components in (C) were as follows: 0.25 µM ECFP–MuB, 0.25 µM EYFP–MuB and 2 mM ATP.
The source of the initial lag in target complex assembly was determined by pre-forming MuB protomers in the presence of ATP. The protomers were then mixed rapidly with DNA (Figure 5B). If the lag phase observed during target complex assembly represented the time required to form the MuB protomer, then pre-incubating MuB with ATP should eliminate the lag prior to complex assembly. Consistent with this proposed mechanism, under conditions where the protomer was pre-assembled, no lag phase was observed during target complex assembly. These results indicated that the initial lag phase was occurring before assembly of the MuB protomer (Figure 5B).
To confirm further that the initial lag during complex assembly was occurring prior to DNA binding, we examined the kinetics of protomer assembly by mixing ECFP–MuB and EYFP–MuB with ATP in the absence of DNA (Figure 5C). The FRET signal in the absence of DNA is weaker than the signal obtained from MuB bound to DNA (see Figure 4). To compensate for the reduced signal, the reactions were performed at a higher MuB concentration than those presented for polymer assembly in the presence of DNA (0.5 versus 0.2 µM MuB). Under these conditions, assembly was completed within 5–10 s, and another short lag (∼1 s) was observed prior to the onset of protomer assembly. This lag phase was independent of MuB concentration from 0.5 to 2 µM and could be reduced by increasing the concentration of ATP from 2 to 10 mM (data not shown). These results suggested that the lag represented a step before assembly of the MuB protomer, either the binding of ATP by the MuB monomer or a post-ATP binding conformational change within the MuB monomer. The time required to complete assembly of the MuB protomer was much shorter than that required to complete target complex assembly, even when target complex assembly was initiated with pre-assembled protomers or when the reactions with DNA were performed at higher concentrations of MuB. Therefore, we concluded that target complex assembly was preceded by the formation of a MuB protomer that was then capable of binding DNA. However, we cannot formally rule out the possibility that ATP-bound MuB subunits can also bypass protomer formation and bind directly to DNA.
MuB complexes are assembled and disassembled continually under steady-state conditions
The dynamic behavior of MuB under conditions supporting strand transfer is related directly to its ability to support target site selection (Adzuma and Mizuuchi, 1988, 1989). To assess the behavior of MuB under steady-state conditions, we used FRET to monitor the exchange of MuB between pre-assembled complexes. First, the stability of the MuB protomer was examined by assembling ECFP– MuB or EYFP–MuB protomers in the presence of only ATP (Figure 6A). If the protomers were stable and not undergoing redistribution of monomers, it is predicted that upon mixing no energy transfer would be observed between ECFP–MuB and EYFP–MuB. However, if the protomers were undergoing continual assembly and disassembly, then the fluoresence signals from the donor and acceptor fluorophores should display a concomitant decrease and increase, respectively, at a rate equivalent to the flux of MuB between protomers. Upon mixing, monomers of MuB within the protomer were redistributed rapidly between the ECFP–MuB and the EYFP–MuB complexes, with complete redistribution occurring within ∼15 s (Figure 6A). Redistribution was reduced substantially in the presence of ATPγS, requiring ∼15 min to reach completion (data not shown). From these results, we concluded that the protomeric form(s) of MuB was in rapid equilibrium with free MuB and that the dissociation of MuB subunits from the protomer was stimulated by ATP hydrolysis.

Fig. 6. MuB within the target complex is in a constant state of flux. (A) MuB protomers or (B) polymers were assembled containing only ECFP–MuB or only EYFP–MuB. Protomers were assembled using 2 mM ATP and 0.5 µM ECFP–MuB or EYFP–MuB. Target complexes were assembled onto φX174 (10 ng/µl) in the presence of 2 mM ATP and 250 nM ECFP–MuB or EYFP–MuB. Complexes were mixed together and the exchange of MuB monitored as an increase in Fa/Fd.
To examine the dynamic nature of the MuB target complex, ECFP–MuB and EYFP–MuB polymers were assembled separately onto DNA in the presence of ATP; these complexes were then mixed together. After mixing, there was a slow increase in the ratio of acceptor to donor emission (Fa/Fd), nearing completion only after 20 min (Figure 6B). The change in FRET signal represented the slow dissociation of MuB subunits from the polymers followed by rapid reassembly onto another DNA molecule, the final result being the formation of target complexes containing both ECFP–MuB and EYFP– MuB. This indicated that MuB molecules within the target complex were continually undergoing disassembly and reassembly during the course of the reaction. ATPγS dramatically slowed, but did not completely inhibit the redistribution of MuB in the presence of DNA, with reactions requiring ∼3 h to reach completion (data not shown). Currently, we do not know whether this slow reaction takes place without nucleotide hydrolysis or whether it represents the previously undetected slow hydrolysis of ATPγS. Because DNA binding, protomer assembly and the exchange of MuB subunits between protomers all occurred on relatively rapid time scales, the slow exchange rate observed with ATP should have closely approximated the disassembly rate of the MuB polymer under steady-state conditions. From these results, we concluded that once a MuB polymer had assembled onto a DNA molecule the subunits within that polymer were able to dissociate slowly from the complex. Therefore, even in the absence of MuA, the MuB target complex is undergoing assembly and disassembly continually under steady-state conditions.
During the exchange reactions, the only ADP present is that which was formed by the MuB-catalyzed hydrolysis of ATP. MuB is a relatively weak ATPase, with a kcat of ∼1 molecule/min. Therefore, during these experiments, ATP was always in excess over ADP. This was verified experimentally by assembling a target complex containing both ECFP–MuB and EYFP–MuB and monitoring the FRET signal over time (data not shown). If the ratio of ADP to ATP had approached or exceeded 1:1, then the complex would have disassembled over time as the ADP concentration increased (see below). However, we observed no significant change in the FRET signal for up to 1 h, confirming that the concentration of ADP remained low for the duration of our experiments.
Polymer disassembly is coupled to ATP hydrolysis and is stimulated by MuA
Under steady-state conditions, in the presence of ATP, MuB was clearly capable of dissociating from the target complex and could then be re-incorporated into another polymer on a different molecule of DNA. To monitor the target complex disassembly pathway more directly, we sought to identify conditions under which the disassembly of the MuB polymer could occur with little or no polymer reassembly. Previous work has indicated that ADP has an inhibitory effect on intermolecular transposition; ADP allows MuB-mediated stimulation of intramolecular transposition but does not support intermolecular transposition (Maxwell et al., 1987; Yamauchi and Baker, 1998). In addition, we observed little or no FRET signal in reactions containing ADP, with or without DNA, at MuB concentrations that support transposition. Together, these data suggest that the assembly of a large MuB target complex is inhibited in the presence of ADP. To verify that ADP inhibits polymer assembly, ECFP–MuB, EYFP–MuB and DNA were reacted with ATP and increasing concentrations of ADP (Figure 7A). In reactions performed with equimolar concentrations of ADP and ATP, the extent of polymer assembly, as judged by the magnitude of the FRET signal, was reduced by 50%. When ADP was present in a 5-fold molar excess, polymer assembly was inhibited by 80%. These results directly demonstrated that when ADP is present in excess over ATP, the assembly of MuB into a polymer was inhibited.

Fig. 7. Target complex disassembly is very slow and is stimulated by MuA. (A) The ability of ADP to inhibit polymer assembly was examined by mixing ECFP–MuB, EYFP–MuB and φX174 with ATP in the presence of increasing concentrations of ADP. All reactions (in A, B and C) contained final concentrations of 250 nM each ECFP–MuB, EYFP–MuB, 10 ng/µl φX174 and 1 mM ATP. The ratio of ATP to ADP is indicated at the right-hand side of each of the data curves, and a minus nucleotide control is also indicated. (B) Disassembly of the MuB polymer was observed by mixing a pre-formed complex of ECFP–MuB, EYFP–MuB and φX174 formed in the presence of ATP with a 10-fold excess of ADP. (C) The effect of MuA on the rate of polymer disassembly was assessed by mixing a pre-formed complex of ECFP–MuB EYFP–MuB and φX174 formed in the presence of ATP with MuA. The final concentrations of MuA and ADP were 100 nM and 10 mM, respectively.
Having already shown that polymer disassembly occurred in the presence of ATP under steady-state conditions, we now sought to examine the disassembly pathway directly under conditions where reassembly was limited. Polymer disassembly was examined by pre-assembling a target complex containing ECFP–MuB, EYFP–MuB, ATP and DNA; the pre-assembled target complex was then mixed with a 10-fold excess of ADP (Figure 7B). By initiating target complex disassembly with the addition of a large excess of ADP, we were effectively establishing single-turnover conditions for the hydrolysis of ATP by the DNA-bound MuB. After mixing with ADP, the ratio of acceptor to donor emission decreased over time, indicating that the target complex was being disassembled during the course of the reaction. The reaction reached completion after 20 min, with a half-life of ∼2.5 min. Suprisingly, the kinetics of polymer disassembly under single-turnover conditions in the presence of excess ADP were remarkably similar to those of monomer exchange between polymers under steady-state conditions in the presence of ATP. Based on the similarities between these two experiments, we concluded that under these steady-state conditions MuB was probably catalyzing the hydrolysis of a single ATP, then dissociating from the DNA before being re-incorporated into another polymer.
The interactions between MuA and MuB are critical for establishing and maintaining target immunity. Specific ally, MuA should be able simultaneously to stimulate the disassembly of the MuB target complex while having little or no influence on the rate of assembly. To verify this, the effect of MuA on target complex assembly was examined by mixing ECFP–MuB, EYFP–MuB and MuA with ATP plus DNA. In the presence of MuA, the extent of target complex assembled at steady-state was reduced. However, the assembly kinetics prior to reaching steady-state were very similar for the plus and minus MuA reactions (Figure 5A). To determine how MuA influences disassembly of the MuB polymer, a pre-formed target complex was mixed with ADP and MuA (Figure 7C). In the presence of 100 nM MuA, the disassembly rate was increased dramatically, coming to completion in <2 min and reducing the target complex half-life from ∼2.5 min to ∼20 s. The φX174 plasmid, used in this experiment, contains several pseudo binding sites for MuA. In the absence of a specific DNA competitor, the MuA was bound directly to the φX174 DNA and therefore was reacting in cis with the MuB polymer. The ability of MuA to stimulate polymer disassembly in trans was examined by pre-binding MuA to an oligonucleotide containing a MuA-binding site (Craigie et al., 1984). A pre-formed target complex was mixed with ADP and the oligo-bound MuA. Under these conditions, no significant stimulation of polymer disassembly was observed with 100 nM MuA–oligo complex. These results indicated that MuA must be bound to the same DNA as the MuB polymer to stimulate its disassembly efficiently (data not shown). Polymer disassembly was also examined in the presence of MuA1–615, a truncated version of MuA lacking the C-terminal domain responsible for the interaction with MuB (Baker et al., 1991; Wu and Chaconas, 1994). MuA1–615 had no effect on polymer disassembly (data not shown). This indicated that polymer disassembly in the presence of full-length MuA was due only to specific interactions between MuA and MuB and was not the result of competition between MuA and MuB for DNA-binding sites. Together, these results demonstrated that under conditions closely mimicking those encountered during the establishment of target immunity, MuA stimulated nearly an 8-fold increase in the disassembly rate of the MuB target complex, while having little or no influence on the assembly pathway.
Discussion
Fluorescence-based methods offer the potential for real-time analysis of complex biological systems with exquisite sensitivity unachievable with most other technologies. Here we report the development of a fluorescence-based system for studying the transposition protein MuB. Fusion of EGFP to the N-terminus of MuB yielded a protein that was nearly indistinguishable from wild-type MuB in terms of its capacity to hydrolyze ATP, bind DNA and interact with MuA. EGFP–MuB was able to support both intermolecular transposition and target immunity. Using ECFP–MuB and EYFP–MuB, we have established a FRET assay to monitor the polymeric state of MuB. Because these fluorescent versions of MuB are biochemically similar to wild-type MuB, we infer that the FRET signal is due to the formation of biochemically relevant species. Therefore, use of the fluorescence assay as a tool for monitoring the state of MuB should yield data that qualitatively reflect the structural, mechanistic and biochemical properties of wild-type MuB.
Target complex assembly follows a well ordered pathway and is coupled to conformational changes in the MuB polymer
The target complex assembly pathway can be divided clearly into at least three kinetically distinct events: the initial binding of ATP to the MuB monomer, protomer assembly, followed by the protomer binding to DNA. This proposed pathway for target complex assembly is supported by several of our observations. Prior to protomer formation, there is a short lag phase of ∼1 s. This lag can be reduced by increasing the concentration of ATP, indicating that it probably reflects the initial binding of ATP. After this step, MuB monomers self-associate to form a protomeric complex. The assembly of the MuB protomer is rapid, coming to completion in just 5 s. The final step in target complex assembly is the binding of the MuB protomer to DNA. This step is relatively slow, requiring ∼30 s to reach completion.
When DNA is added to the protomer, the efficiency of energy transfer increases significantly. There are at least two simple explanations for this observation. It is possible that in the presence of DNA, the extent of MuB polymerization is greater than in its absence. While this alone could potentially explain the effect of DNA, we do not favor this interpretation. If MuB polymerization is incomplete in the absence of DNA, higher concentrations of MuB should increase the steady-state level of the MuB polymers. However, even at higher MuB concentrations, a significant difference persists in the presence and absence of DNA; this is especially evident in reactions with ATPγS (Figure 4; data not shown). Therefore, the difference in the FRET signal in the presence and absence of DNA does not appear to reflect entirely differing extents of polymerization. Therefore, we favor the alternative explanation that upon binding DNA there is a conformational change within the MuB polymer that is reflected as increased FRET efficiency. Additionally, during the assembly of the target complex, protomer binding to DNA is rate limiting, suggesting the possibility that an additional step may be required prior to DNA binding, and this may also reflect the existence of a slow conformational change in MuB prior to the completion of target complex assembly. Supporting this notion, reactions performed with ATPγS exhibit a much slower assembly time, requiring ≥3 min to reach completion (data not shown). It should be noted here that there is a significant difference in the FRET efficiency between the MuB protomer in the presence of ATP and that in the presence of ATPγS. It is currently unclear whether this difference reflects the effect of the thiol substitution in ATPγS on the conformation of MuB or is the result of partial hydrolysis of the ATP bound to MuB in the complexes made with ATP. While at this time we cannot determine directly the nature of the conformational changes, we believe that in the presence of ATP and DNA, both more extensive polymerization and a conformational change are likely to play a role in the target complex displaying greater FRET than the MuB protomer.
MuB catalyzes the hydrolysis of a single ATP prior to dissociating from DNA
Following ATP hydrolysis and dissociation of ADP, there are two possible fates for a DNA-bound molecule of MuB: rebinding of another molecule of ATP without the dissociation from the DNA or dissociation from the DNA prior to rebinding another molecule of ATP. Using FRET, we have investigated the disassembly pathway of the MuB target complex under two different sets of conditions: pre-steady-state conditions where the reaction is driven to near completion by the addition of excess ADP, and steady-state conditions in the presence of only ATP. In disassembly reactions initiated with the addition of excess ADP, MuB is unlikely to catalyze multiple rounds of ATP hydrolysis because of the reduced likelihood of encountering another ATP molecule. Disassembly in the presence of excess ADP therefore mimics single-turnover conditions where DNA-bound MuB can only hydrolyze the molecule of ATP to which it is bound at the outset of the reaction. However, during the steady-state reactions, ATP is always in excess over ADP. If MuB were catalyzing multiple rounds of ATP hydrolysis prior to dissociation from the DNA, then the exchange rate of MuB between polymers on different DNA molecules should have been inversely related to the number of rounds of ATP hydrolysis that a molecule of MuB was capable of catalyzing prior to dissociation from the DNA. In this case, we would have expected the exchange rate of MuB between target complexes to be slower than the disassembly rate observed in the excess ADP reactions. However, because the exchange kinetics were very similar to the disassembly kinetics, we conclude that the MuB bound to DNA under steady-state conditions probably catalyzed a single ATP hydrolysis event prior to dissociating from the DNA.
Polymeric nature of MuB
While we repeatedly have referred to the complexes formed by MuB as subunits, protomers or polymers, we have not attempted to define the number of MuB monomers that make up the complexes in this study. These details of the reaction have remained difficult to elucidate, primarily because there appears to be a multitude of oligomeric states available to MuB in the presence of nucleotide cofactor. For instance, with ATP or ATPγS, MuB cannot be resolved as a single species by either sucrose gradient centrifugation or gel filtration (Adzuma and Mizuuchi, 1991; data not shown). Rather than behaving as a single species, the protein complexes display a very broad range of apparent molecular weights. Additionally, under conditions where MuB forms a polymer (ATP or ATPγS plus or minus DNA), monomers of MuB can be chemically cross-linked to one another, and products representing dimers, trimers, tetramers and considerably larger species are detected (Adzuma and Mizuuchi, 1991; our unpublished observations). Because of these current technical limitations, we cannot yet define the true oligomeric state of the MuB complexes present in a reaction.
The 40% energy transfer efficiency observed for the MuB polymer on DNA in the presence of ATPγS is very remarkable. In particular, the simplest scenario is one in which there should exist donor–donor and acceptor– acceptor pairs within the target complex that do not contribute to signal. The Förster distance for randomly oriented ECFP and EYFP is 50 Å (Miyawaki and Tsien, 2000). An energy transfer efficiency of 40% would correspond to a hypothetical distance of just 53 Å between two adjacent molecules of CFP and YFP. However, because structural details are lacking, it is difficult to understand precisely what this means in terms of the MuB polymer. One simple possibility is that MuB is a linear polymer in which a single monomer directly interacts only with its two nearest neighbors. However, this model is difficult to reconcile with our observations because formation of non-productive fluorophore pairs should substantially reduce the FRET efficiency.
Finally, we note that in the presence of ADP, both wild-type MuB and EGFP–MuB are capable of stimulating intramolecular strand transfer (Yamauchi and Baker, 1998; data not shown), even at lower MuB concentrations where no FRET signal was detected. This does not necessarily indicate that MuB is behaving as a monomer during intramolecular strand transfer. It is possible that some oligomeric forms of MuB are not readily detectable by FRET, if for instance the fluorophores are not oriented properly relative to one another. Perhaps more probably, the polymeric state of MuB in the presence of ADP and DNA may not be populated to the extent that it can be detected with FRET at lower MuB concentrations. This notion is supported by the observation that a FRET signal can be detected at elevated concentrations of MuB in the presence of ADP and DNA.
How do MuA and MuB interact to mediate target immunity?
The data presented here indicate that Mu target immunity results from a combination of slow dissociation of MuB from DNA, which is greatly stimulated when MuA is bound to the same DNA as the MuB polymer, followed by the very rapid reassembly of the MuB polymer onto a new DNA site. The combination of these parameters effectively allows MuB to sample multiple DNA molecules (or regions) until one is found that lies outside the influence of MuA. While seemingly simple, many of the details of this mechanism are still unclear. In particular, how does MuA interact with MuB at early stages of the reaction in order to establish target immunity and stimulate transpososome assembly, yet avoid a potentially devastating intramolecular strand transfer event? In the simplest scenario, MuA-stimulated dissociation of MuB from DNA may occur by a simple collisional mechanism whereby one collision of MuA (or the transpososome) stimulates the removal of one monomer of MuB (or other small unit, i.e. dimer, trimer, tetramer, etc.). Proximal DNA sites nearest the transpososome should have less MuB and distal DNA sites should have more MuB. The distribution of MuB on the DNA between the distal and proximal sites should be inversely related to the collisional frequency of the transpososome with the particular DNA site. While this mechanism would yield a gradient of decreasing MuB density on DNA flanking the transpososome, it would not in and of itself provide a mechanism for target immunity. Some bound MuB would still occupy the proximal DNA sites and the effective concentration of this proximal MuB would be the same as that of the distal MuB due to the increased collisional frequency between the transpososome and the proximal sites. This problem could potentially be overcome via a mechanism involving a much larger unit of MuB to activate the transpososome for target capture and subsequent strand transfer. In such a scenario, the small amount of MuB present near the transpososome would be irrelevant if it was not in the appropriate polymeric state. In support of this model, a preliminary result has indicated that ∼100 monomers of MuB are needed to support a single intermolecular strand transfer event (Y.Wang and K.Mizuuchi, unpublished observations).
Although the Mu transposition system has been well characterized, several important questions remain unanswered. The experiments presented here provide the initial framework for a detailed analysis of the behavior of MuB using fluorescence-based technologies, and future experiments using the fluorescent derivatives of MuB should reveal some of the more elusive details of the processes involved in transpositional DNA recombination.
Materials and methods
Cloning and protein expression
MuB GFP fusion proteins were constructed by PCR amplifying the coding region of EGFP (or the variants) from the plasmids pEGFP, pECFP or pEYFP (Clontech). PCR primer pairs were as follows: (i) 5′-GGGAATTCCATATGGTGAGCAAGGGCGAGGAGC-3′ and 5′-GGGAATTCCATATGCTTGTACAGCTCGTCCATGCCG-3′ (for the N-terminal fusion) or (ii) 5′-CGCGGATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ and 5′-CGCGGATCCTTACTTGTACAGCTCGTCCATGCCG-3′ (for the C-terminal fusion). PCR fragments were digested with NdeI or BamHI, gel purified, and cloned into the NdeI or BamHI sites of pET14b-MuBwt (Yamauchi and Baker, 1998) to generate the N- and C-terminal fusion constructs, respectively. Vectors were transformed into E.coli strain HMS174(DE3)pLysS (Stratagene). Cells were grown in LB/ampicillin (100 µg/ml)/chloramphenicol (25 µg/ml) at 37°C to an OD600 of 0.6, then chilled to 16°C. Cells were induced at 16°C with the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Cells were grown for an additional 8–12 h following induction. Lysis was performed essentially as described (Baker et al., 1993) except that 1 M NaCl was included in the lysis buffer.
Purification of GFP fusion proteins
GFP fusion proteins were purified with Ni2+ affinity chromatography. Lysates were loaded onto a chelating Sepharose column (Pharmacia) pre-equilibrated with buffer A (10% glycerol, 50 mM Tris pH 7.6, 1 M NaCl). The columns were washed with 10 vols of buffer A + 10 mM imidazole and proteins eluted with a linear gradient from 10 to 500 mM imidazole in buffer A. The GFP fusion proteins were dialyzed into buffer B [20% glycerol, 30 mM HEPES pH 7.6, 1 mM EDTA, 1 mM dithiothreitol (DTT)] and centrifuged at 15 000 g to remove any insoluble material. Proteins were subsequently loaded onto an SP-Sepharose column (Pharmacia) in buffer B. The column was washed with buffer B containing 75 mM NaCl and proteins eluted with a linear gradient from 75 to 400 mM NaCl. The proteins were dialyzed into buffer B containing 150 mM NaCl for storage. Protein concentrations were determined spectrophotometrically using extinction coefficients of 26 000, 55 000 and 84 000/M/cm at 433, 489 and 514 nm for ECFP–MuB, EGFP–MuB and EYFP–MuB, respectively (Clontech). The spectral properties of the fusion proteins were essentially indistinguishable from the reported values for the free GFP proteins (data not shown).
Fluorescence emission spectra
All steady-state fluorescence measurements were made with a Spex Fluorolog 2 spectrafluorometer (Edison, NJ), equipped with a 450 W xenon arc lamp. Excitation and emission wavelengths are indicated in the figure legends and all data were collected at 25°C. Background fluorescence from unlabeled proteins, DNA and buffer solutions was negligible and therefore not subtracted from the spectral data. The efficiency of energy transfer for the data presented in Figure 3 was calculated by subtracting the integrated intensity from 450 to 500 nm for the reaction containing the separate ECFP–MuB only and EYFP–MuB only complexes, from the integrated intensity over the same range for the mixed ECFP–MuB/EYFP–MuB complex. Similarly, the integrated intensity from 510 to 600 nm for the mixed ECFP–MuB/EYFP–MuB complex was subtracted from the integrated intensity over the same range for the reaction containing the separate ECFP–MuB only and EYFP– MuB only complexes. From this, we calculated a 38% decrease in donor emission and a 42% increase in acceptor emission, yielding an estimated FRET efficiency of ∼40%.
All kinetic measurements were made using a Kinetek stopped-flow spectrafluorometer (Model SF-2001; Kintec Corp.) equipped with dual photomultiplier tubes (PMTs) and a 75 W xenon arc lamp. The PMTs were equipped with optical filters appropriate for either ECFP or EYFP (Omega filter 480AF30 or Omega 510ALP, respectively) and data were plotted as a ratio of the signals from the two channels. Excitation was at 425 nm for all stopped-flow experiments, and all kinetic data curves represent the average of at least three separate experiments.
ATPase assays
ATPase assays were assembled in 25 mM Tris pH 8, 100 ng/µl bovine serum albumin (BSA), 150 mM NaCl, 10 mM MgCl2, 20% glycerol, 1 mM DTT and 0.5 µM [α-32P]ATP (800 Ci/mmol; NEN). ATP concentrations were adjusted using unlabeled ATP. Reactions were terminated with the addition of an equal volume of 10% acetic acid and products resolved on polyethyleneimine (PEI) TLC plates in 0.75 M KH2PO4. Reaction products were quantified using a phosphoimager (Fuji) and data were analyzed by plotting as Eadie–Hofstee plots.
Transposition reactions
Strand transfer reactions were assembled containing 10 ng/µl each of φX174 (target DNA) and pMK586 (donor DNA), 25 mM Tris pH 8, 100 ng/µl BSA, 150 mM NaCl, 10 mM MgCl2, 20% glycerol, 1 mM DTT, 4 ng/µl HU and 200 nM wild-type MuB, EGFP–MuB or MuB-EGFP. Reactions were initiated with the addition of MuA to 50 nM in a final volume of 25 µl and placed at 30°C for 1 h. Reactions were terminated with the addition of 5 µl of loading buffer (0.1% bromophenol blue, 2.5% SDS, 50 mM EDTA and 25% ficoll) and products resolved on 0.8% Seakem HGT–agarose in 1× TAE with buffer recirculation. Products were detected with ethidium bromide staining.
Target immunity assays
Immunity assays were performed essentially as described (Adzuma and Mizuuchi, 1988) with the exception that products were resolved on 0.7% Seakem HGT–agarose in 1× TBE to allow better resolution of strand transfer products.
Acknowledgments
Acknowledgements
We thank Drs Meni Melek, Bob Craigie, Jessica Jones and members of our laboratory for their helpful comments and critical reading of this manuscript. We also wish to thank Tania Baker for the generous gift the His-tagged MuB expression vector. This work was supported in part by the fund from the NIH Intramural Aids Targeted Antiviral Program.
References
- Adzuma K. and Mizuuchi,K. (1988) Target immunity of Mu-transposition reflects a differential distribution of Mu-B-protein. Cell, 53, 257–266. [DOI] [PubMed] [Google Scholar]
- Adzuma K. and Mizuuchi,K. (1989) Interaction of proteins located at a distance along DNA—mechanism of target immunity in the Mu DNA strand-transfer reaction. Cell, 57, 41–47. [DOI] [PubMed] [Google Scholar]
- Adzuma K. and Mizuuchi,K. (1991) Steady-state kinetic-analysis of ATP hydrolysis by the B-protein of bacteriophage-Mu—involvement of protein oligomerization in the ATPase cycle. J. Biol. Chem., 266, 6159–6167. [PubMed] [Google Scholar]
- Baker T.A. and Mizuuchi,K. (1992) DNA-promoted assembly of the active tetramer of the Mu-transposase. Genes Dev., 6, 2221–2232. [DOI] [PubMed] [Google Scholar]
- Baker T.A., Mizuuchi,M. and Mizuuchi,K. (1991) MuB protein allosterically activates strand transfer by the transposase of phage-Mu. Cell, 65, 1003–1013. [DOI] [PubMed] [Google Scholar]
- Baker T.A., Mizuuchi,M., Savilahti,H. and Mizuuchi,K. (1993) Division-of-labor among monomers within the Mu-transposase tetramer. Cell, 74, 723–733. [DOI] [PubMed] [Google Scholar]
- Chaconas G., Giddens,E.B., Miller,J.L. and Gloor,G. (1985) A truncated form of the bacteriophage-Mu-B Protein promotes conservative integration, but not replicative transposition, of Mu-DNA. Cell, 41, 857–865. [DOI] [PubMed] [Google Scholar]
- Chaconas G., Lavoie,B.D. and Watson,M.A. (1996) DNA transposition: jumping gene machine, some assembly required. Curr. Biol., 6, 817–820. [DOI] [PubMed] [Google Scholar]
- Craig N.L. (1997) Target site selection in transposition. Annu. Rev. Biochem., 66, 437–474. [DOI] [PubMed] [Google Scholar]
- Craigie R. and Mizuuchi,K. (1987) Transposition of Mu-DNA—joining of Mu to target DNA can be uncoupled from cleavage at the ends of Mu. Cell, 51, 493–501. [DOI] [PubMed] [Google Scholar]
- Craigie R., Mizuuchi,M. and Mizuuchi,K. (1984) Site-specific recognition of the bacteriophage-Mu ends by the Mu-A protein. Cell, 39, 387–394. [DOI] [PubMed] [Google Scholar]
- Hung L.H., Chaconas,G. and Shaw,G.S. (2000) The solution structure of the C-terminal domain of the Mu B transposition protein. EMBO J., 19, 5625–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell A., Craigie,R. and Mizuuchi,K. (1987) B-protein of bacterio phage Mu is an ATPase that preferentially stimulates intermolecular DNA strand transfer. Proc. Natl Acad. Sci. USA, 84, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A. and Tsien,R.Y. (2000) Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol., 327, 472–500. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. (1992) Transpositional recombination—mechanistic insights from studies of Mu and other elements. Annu. Rev. Biochem., 61, 1011–1051. [DOI] [PubMed] [Google Scholar]
- Mizuuchi M., Baker,T.A. and Mizuuchi,K. (1992) Assembly of the active form of the transposase-Mu DNA complex—a critical control point in Mu-transposition. Cell, 70, 303–311. [DOI] [PubMed] [Google Scholar]
- Naigamwalla D.Z. and Chaconas,G. (1997) A new set of Mu DNA transposition intermediates: alternate pathways of target capture preceding strand transfer. EMBO J., 16, 5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette M.G. and Chaconas,G. (1991) Stimulation of the Mu-DNA strand cleavage and intramolecular strand transfer-reactions by the Mu-B-protein is independent of stable binding of the Mu-B-protein to DNA. J. Biol. Chem., 266, 17306–17313. [PubMed] [Google Scholar]
- Surette M.G., Buch,S.J. and Chaconas,G. (1987) Transpososomes—stable protein DNA complexes involved in the in vitro transposition of bacteriophage-Mu-DNA. Cell, 49, 253–262. [DOI] [PubMed] [Google Scholar]
- Teplow D.B., Nakayama,C., Leung,P.C. and Harshey,R.M. (1988) Structure–function-relationships in the transposition protein-B of bacteriophage-Mu. J. Biol. Chem., 263, 10851–10857. [PubMed] [Google Scholar]
- Watson M.A. and Chaconas,G. (1996) Three-site synapsis during Mu DNA transposition: a critical intermediate preceding engagement of the active site. Cell, 85, 435–445. [DOI] [PubMed] [Google Scholar]
- Williams T.L., Jackson,E.L., Carritte,A. and Baker,T.A. (1999) Organization and dynamics of the Mu transpososome: recombination by communication between two active sites. Genes Dev., 13, 2725–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.G. and Chaconas,G. (1994) Characterization of a region in phage Mu transposase that is involved in interaction with the Mu-B protein. J. Biol. Chem., 269, 28829–28833. [PubMed] [Google Scholar]
- Yamauchi M. and Baker,T.A. (1998) An ATP–ADP switch in MuB controls progression of the Mu transposition pathway. EMBO J., 17, 5509–5518. [DOI] [PMC free article] [PubMed] [Google Scholar]