

Abstract
We investigated whether mitochondria represent morphologically continuous and functionally homogenous entities within single intact cells. Physical continuity of mitochondria was determined by three-dimensional reconstruction of fluorescence from mitochondrially targeted DsRed1 or calcein. The mitochondria of HeLa, PAEC, COS-7, HUVEC, hepatocytes, cortical astrocytes and neuronal cells all displayed heterogeneous distributions and were of varying sizes. There was a denser aggregation of mitochondria in perinuclear positions than in the cell periphery, where individual isolated mitochondria could be seen clearly. Using fluorescence-recovery after photobleaching, we observed that DsRed1 and calcein were highly mobile within the matrix of individual mitochondria, and that mitochondria within a cell were not lumenally continuous. Mitochondria were not electrically coupled, since only individual mitochondria were observed to depolarize following irradiation of TMRE-loaded cells. Functional heterogeneity of mitochondria in single cells was observed with respect to membrane potential, sequestration of hormonally evoked cytosolic calcium signals and timing of permeability transition pore opening in response to tert-butyl hydroperoxide. Our data indicate that mitochondria within individual cells are morphologically heterogeneous and unconnected, allowing them to have distinct functional properties.
Keywords: calcium/FRAP/membrane potential/mitochondria/permeability transition
Introduction
Renewed interest in mitochondria in recent years, sparked by the recognition of their role in processes such as apoptosis and calcium homeostasis, has reopened the debate on how many mitochondria, or populations of mitochondria, exist in a single cell. Do mitochondria exist as multiple isolated organelles, or is the mitochondrion a single network more akin to the endoplasmic reticulum (ER)? Electrical and lumenal continuity across a mitochondrial network would have important consequences for many cellular processes. Electrical continuity, for example, would tend to equalize the potential across the inner membrane of all mitochondrial regions, whether they were respiring or not. Such an electrically coupled network may allow ‘energy transmission’ across the cell from regions of low to regions of high ATP consumption (Skulachev, 2001). A lumenally continuous mitochondrial network would raise the possibility of ‘Ca2+ tunnelling’ via a non-cytoplasmic route similar to the ER (Mogami et al., 1997). Mitochondria are considered as rapid uptake, slow-release buffers of cytosolic Ca2+ (Babcock et al., 1997), with preferential access to Ca2+ at the site of its release (Rizzuto et al., 1993). Ca2+ tunnelling could result in the delivery of Ca2+ to an area of the cell distant from the site of the original signal, and with a significant delay. This would alter the spatio-temporal characteristics of the cytosolic Ca2+ response. Furthermore, Ca2+ tunnelling could promote the synchronization of increases in mitochondrial metabolism (Hajnoczky et al., 1995) or permeability transition (Ichas et al., 1994) across a cell.
Mitochondria play a key role in some of the signalling pathways for apoptosis. Release of pro-apoptotic factors from the inter-membrane space is thought to occur after mitochondrial permeability transition pore (PTP) opening, matrix swelling and outer-membrane rupturing (for a review see Crompton, 1999). Paradoxically, the early stages of apoptosis can involve the depolarization of mitochondria, yet also require ATP, otherwise apoptosis turns to necrosis (Eguchi et al., 1997; Leist et al., 1997). A non-continuous mitochondrial network could allow the possibility that certain mitochondria are involved in pro-apoptotic signalling, whilst others continue to supply ATP during apoptosis.
Several lines of evidence have been presented to suggest that mitochondria are both physically interconnected and functionally homogenous. By triggering mitochondrial depolarization with laser irradiation of fluorescent dye-loaded cells, large networks of electrically connected mitochondria have been demonstrated in COS-7 cells (De Giorgi et al., 2000) and cardiac myocytes (Amchenkova et al., 1988). Reconstruction of electron micrographs revealed mitochondrial networks in rat hepatocytes (Brandt et al., 1974) and a single large mitochondrion in yeast cells (Hoffman and Avers, 1973). More recently, evidence for the existence of a largely interconnected mitochondrial network in HeLa cells was presented using non-confocal, deconvolution imaging and ‘fluorescence-recovery after photobleaching’ (FRAP) (Rizzuto et al., 1998).
In contrast, early electron microscopy revealed populations of mitochondria with different matrix densities within single cells (Simon et al., 1969; Ord, 1979), thought to reflect differences in metabolic states (Ord, 1979). In cardiac (Jahangir et al., 1999) and skeletal muscle (Battersby and Moyes, 1998; Lombardi et al., 2000) cells, two distinct populations of mitochondria are proposed to exist, with differing biochemical and respiratory properties. In pancreatic acinar cells, detailed spatial analysis has revealed mitochondrial populations preferentially sequestering Ca2+ from different sources, and apparently not communicating Ca2+ between one another (Park et al., 2001).
In the present study, we used a combination of approaches to examine the connectivity and functional homogeneity of mitochondria in several cell types. Our analysis indicates that mitochondria exist as separate entities that vary substantially in length and cellular distribution. Furthermore, they are lumenally discontinuous. This physical compartmentalization is accompanied by a functional heterogeneity with respect to membrane potential, Ca2+ sequestration and sensitivity to PTP activation.
Results
Morphological heterogeneity of mitochondria
HeLa cells, porcine aortic endothelial cells (PAEC), COS-7 cells, human umbilical vein endothelial cells (HUVEC), cortical astrocytes and cortical neuronal cells were transfected with DsRed1 targeted to the mitochondrial matrix (mito-DsRed1). In our hands, mito-DsRed1 has a distinct mitochondrial localization, with very little cytoplasmic signal (Collins et al., 2001) and no apparent self-aggregation of the fluorophore. For primary hepatocytes, which could not be transfected, the mitochondria were specifically visualized by loading with calcein (see Materials and methods). Deconvolved confocal images were surface-rendered to generate three-dimensional reconstructions of the mitochondria within the mito-DsRed1-expressing cells (Figure 1). Mitochondria were distributed throughout the cytosol of all cells, with a tendency to be aggregated around the nucleus. Individual, isolated mitochondria could be clearly resolved in the periphery of all cell types. In the perinuclear region, where the mitochondria were more densely packed, it was difficult to discern whether mitochondria were connected or only touching. The morphology of individual mitochondria ranged from small ‘grains’ to larger, sometimes branched, ‘threads’. Mitochondria were often observed under the nucleus, and occasionally also above it (e.g. Figure 1A).

Fig. 1. Surface-rendered three-dimensional reconstructions of cells expressing mito-DsRed1. (A–E) Typical three-dimensional reconstructions of mito-DsRed1 fluorescence in the following cell types: (A) HeLa, (B) COS-7, (C) cortical astrocyte, (D) cortical neuron and (E) HUVEC. (F) A reconstruction of mitochondrial calcein fluorescence in a hepatocyte. The dashed circles indicate the position of nuclei. Scale bars, 5 µm.
The three-dimensional reconstructions shown in Figure 1 were obtained with cells equilibrated at 22°C. This temperature was chosen because it dramatically slows the movement of mitochondria. This is important when taking image stacks, so that the same mitochondria do not appear in different focal planes, which could lead to a false impression of the size and connectivity of the organelles. In single confocal images, we did not see any significant effect of changing temperature from 22 to 37°C on the structure of the mitochondria (data not shown).
Lumenal connectivity between mitochondria was determined using FRAP. In this process, the fluorophore is bleached in a subcellular region using brief high-intensity illumination. Any subsequent recovery of fluorescence in the bleached region occurs due to inward diffusion of unbleached fluorophore molecules. For the cell types that could be transiently transfected (HeLa, PAEC, COS-7, HUVEC, cortical astrocytes and neuronal cells), we utilized mito-DsRed1 for FRAP experiments. To examine the continuity of mitochondria in primary hepatocytes, FRAP experiments were performed using calcein-loaded cells. Since mito-DsRed1 and calcein are localized within the mitochondrial matrix, recovery of fluorescence after local photobleaching would indicate lumenal connectivity within the mitochondrial network.
With all cell types examined, the brief high-intensity illumination caused bleaching of the DsRed1/calcein fluorescence in the irradiated region. Typical responses from mito-DsRed1-expressing HeLa and HUVEC cells are shown in Figure 2A–C. The lack of fluorescence recovery in a single calcein-loaded hepatocyte is illustrated in Figure 2D. In most cases, the area in which the fluorescence was bleached extended a few micrometres beyond the actual region illuminated. This was most probably due to the tunnelling of the fluorophores along the matrix of mitochondria that protruded outside the illumination region. Similar results were obtained using cells at 22 and 37°C (Figure 2A–C). Monitoring the average fluorescence intensity across the entire bleached region indicated that the fluorescence did not recover to >10% of its initial value up to 1 h after irradiation. Occasionally, there was a weak but persistent recovery of fluorescence after ∼20 min. This slow recovery of the fluorescence was more commonly observed at 37°C than at 22°C. We suggest that this recovery was not due to true FRAP within mitochondria (which is fast—see below), but rather movement of unbleached mitochondria into the bleached area or fusion between bleached and unbleached mitochondria. The FRAP procedure did not damage the mitochondria, since the organelles could be subsequently visualized by loading with a membrane-permeant mitochondrial indicator such as tetramethylrhodamine ethyl ester (TMRE) (data not shown).

Fig. 2. Assessment of lumenal continuity between mitochondria using FRAP. A HeLa cell (Ai–iii and Bi–iii) and a HUVEC cell (Ci–iii) expressing mito-DsRed1, and a primary hepatocyte loaded with calcein (Di–iii) were imaged before (Ai–Di), ∼10 s after (Aii–Dii) and 20 min after (Aiii–Diii) photobleaching a subcellular region of each cell type. The white boxes in (Aii–Dii) denote the bleached regions in each cell. The experiments depicted in (A) and (D) were performed at 22°C, and those in (B) and (C) were at 37°C. Scale bars, 5 µm. The results shown in this figure are typical of every trial (n >5) for each cell type and temperature.
The lack of recovery of fluorescence illustrated in Figure 2 was not due to slow diffusion of DsRed1 or calcein within the mitochondrial matrix. To demonstrate this, we employed a similar FRAP protocol to that used above, except that the bleaching was performed in the periphery of cells where individual mitochondria were clearly identifiable. The high-intensity illumination was directed towards the middle of a long (∼22 µm) DsRed1-expressing mitochondrion (Figure 3Ai, arrow). This procedure caused the total loss of DsRed1 fluorescence from the irradiated area, and also decreased the DsRed1 fluorescence along the length of the mitochondrion outside the bleached area (Figure 3Aii). The DsRed1 fluorescence within the long mitochondrion was uniform before the bleach (Figure 3Ai), and had fully equilibrated within 90 s after the bleach (Figure 3Aiii). An analysis of the time-course of the fluorescence recovery revealed that this equilibration had a half-time of ∼15 s (Figure 3Bii). For the few small mitochondria that were completely encompassed within the illumination area, the fluorescence did not recover (e.g. trace a in Figure 3Bii), while those mitochondria outside the illumination region were unaffected (e.g. trace g in Figure 3Bii). The data depicted in Figure 3 were obtained from a HeLa cell, but are typical of responses from the other cell types at both 22 and 37°C (data not shown). Calcein was even more mobile than DsRed1 in the mitochondrial matrix. Equilibration of calcein fluorescence along HUVEC mitochondria at 22°C occurred within a few seconds after photobleaching (data not shown).

Fig. 3. DsRed1 is rapidly diffusible in the mitochondrial matrix. (Ai) A portion of a HeLa cell expressing mito-DsRed1. Both long (denoted by an arrow) and short discrete mitochondria can be seen in this region. The region bounded by the white box in (Aii) was photobleached using a 5 s illumination as described in Materials and methods. The bleached region encompassed the middle portion of the long mitochondria and completely surrounded several smaller mitochondria. The images in (Aii) and (Aiii) show the gradual recovery of fluorescence in the long mitochondrion, and were obtained at times corresponding to 0.5 and 90 s after the photobleach. (B) A quantitation of the fluorescence recovery within the long mitochondrion. The fluorescence was monitored in the regions denoted in (Bi). Areas a, b, c and d were within the bleached zone, but e, f and g were not. The traces in (Bii) illustrate the intensity of mito-DsRed1 fluorescence in these regions before and after the photobleach. Note that regions a and g were not part of the long mitochondrion. Scale bar, 5 µm. The data presented show a typical response observed in more than five cells.
The electrical continuity of mitochondria was assessed by examining the extent of irradiation-induced depolarization of the mitochondrial population. We utilized the previous observations that mitochondrial depolarization can be induced by a combination of high fluorophore concentration and laser irradiation. This is thought to be due to the generation of reactive-oxygen species (ROS), which subsequently trigger PTP opening (Amchenkova et al., 1988; Hüser et al., 1998; Hüser and Blatter, 1999; De Giorgi et al., 2000; Zorov et al., 2000; Buckman and Reynolds, 2001). TMRE is a fluorescent indicator that has been widely used to monitor mitochondrial membrane potential (Δψmit), but it can also lead to the opening of the PTP, probably via ROS production. We therefore used TMRE to both measure Δψmit and to cause spontaneous depolarizations.
Cells were incubated with 1 µM TMRE for 30 min and confocal images were subsequently acquired at 0.2 Hz for periods of ∼10 min. Depolarization events were mapped frame-by-frame by following changes of TMRE fluorescence. For the HeLa cell example depicted in Figure 4Ai–v, analysis of the regions showing changes in TMRE fluorescence indicated that 85 electrically discrete mitochondria were present. In two additional HeLa cells that were analysed with the same frame-by-frame method, we observed 128 and 185 electrically discrete organelles. In keeping with the diverse morphologies of the mitochondria shown in Figure 1, the areas of TMRE fluorescence change were heterogeneous. Some of these electrically discrete mitochondria were quite long (∼45 µM) (Figure 4Avi). The Δψmit often ‘flickered’ several times before the TMRE fluorescence signal was permanently lost. The shapes of the depolarizing regions did not change from flicker to flicker, indicating that the 85 identified regions were truly discrete and the depolarization did not include different portions of an electrically connected network. Similar flickering of Δψmit was observed in all the cell types, and was present at both 22 and 37°C (data not shown).

Fig. 4. Induced depolarization events reveal multiple, electrically discrete mitochondria. (Ai–v) A TMRE-loaded HeLa cell showing random depolarization of discrete mitochondria. The depolarization events were assessed using a frame-by-frame subtraction. This generated a series of images in which the positions of depolarized mitochondria appear from a dark background. These events were pseudocoloured red and superimposed on the original image of the mitochondria as shown. The experiment was actually recorded for 10 min, but only representative frames are shown. The times at which the images were captured relative to the start of the recording are shown. (Avi) Map of individual electrically isolated mitochondria. This was constructed by monitoring the locations of individual depolarization events over time. The red, green and yellow colouration is used to indicate the locations of the electrically isolated mitochondria. The colours do not indicate any relationships between mitochondria. The arrowhead in (Avi) indicates a single 45 µm mitochondria seen to depolarize in (Ai). Scale bar, 5 µm. (B) Depolarizations of mitochondria within a TMRE-loaded primary hepatocyte imaged at 2 Hz. (Bi) The change in fluorescence of individual mitochondria (the locations at which the mitochondrial fluorescence was monitored are depicted by black dots in the inset cell image) plotted over 8 min of laser irradiation. Note that depolarization events only start after a significant period of imaging (∼90 s), confirming that the PTP ‘flickering’ is a laser- and dye-induced phenomenon. (Bii) De piction of the image of the TMRE-loaded cell. The region bounded by the dashed box is shown on an expanded scale in (Biii–viii), and the depolarization events are superimposed with red colouration. (Bix) Map of individual electrically isolated mitochondria constructed by monitoring the locations of individual depolarization events over time. The red, green, cyan and yellow colouration is used to indicate the locations of the electrically isolated mitochondria, and does not indicate any relationships between mitochondria. Scale bar, 5 µm.
Primary hepatocytes were consistently observed to have the shortest and least interconnected mitochondria. This was particularly evident from observations of TMRE- and laser-induced PTP flickering, as depicted in Figure 4B. The flickering of individual mitochondria occurred randomly (Figure 4Bi). Occasionally, several mitochondria depolarized simultaneously, and at other times, the same mitochondria would flicker on their own. Unlike in the other cell types, the regions of depolarization in hepatocytes were observed to be generally <5 µm (Figure 4Bix). The spontaneous depolarization of mitochondria in all cell types was dependent on a critical loading with TMRE, since with low levels of indicator spontaneous Δψmit depolarization was not observed (data not shown).
Functional heterogeneity of mitochondria
Δψmit. The distribution of Δψmit throughout the mitochondrial population of different cell types was monitored with the dual emission potentiometric dye tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1) (Reers et al., 1995; Salvioli et al., 1997). JC-1 accumulates preferentially in polarized mitochondria, existing as green (530 nm emission) fluorescent monomers at low membrane potentials and as orange/red (590 nm emission) fluorescent aggregates at high membrane potentials. In none of the cell types tested did we observe a homogenous red or green JC-1 fluorescence from the mitochondria. Instead, the mitochondria were either green or red, indicating a distinct heterogeneity of Δψmit (Figure 5). The proportions of green- and red-emitting mitochondria were not evenly distributed in cells. The red-fluorescing, highly energized mitochondria were proportionally more prevalent in the periphery of cells (Figure 5Ai). By randomly sampling mitochondria from the periphery and perinuclear regions of HeLa cells, it was observed that 68 ± 5% (± SEM; n = 25 cells) of peripheral mito chondrial were red fluorescing, whilst only 28 ± 4% of perinuclear mitochondria were red. To show that the JC-1 staining was specific for mitochondria, cells were co-loaded with TMRE (e.g. Figure 5Aii). JC-1 and TMRE showed the same cellular localization. In addition, the red fluorescence from both dyes disappeared upon addition of antimycin (Figure 5Aiii), indicating that the TMRE signal and the aggregation of JC-1 were dependent on respiring mitochondria. A predominance of red-fluorescing mitochondria in the periphery of the cell was also seen with JC-1-stained hepatocytes (Figure 5B), HUVECs (Figure 5Ci and ii), COS-7, PAEC and cortical astrocytes and neurons (data not shown). A similar pattern of staining was observed in cells at 22 or 37°C. Figure 5Ci depicts the JC-1 fluorescence of a HUVEC cell loaded with 1 µM JC-1 for 1 h at 22°C. Raising the temperature of the cells to 37°C and equilibrating them for a further 30 min in the continued presence of 1 µM JC-1 did not did dramatically alter the staining pattern: the green-staining mitochondria were still perinuclear, and the red-staining organelles were largely peripheral.

Fig. 5. Heterogeneity in membrane potential revealed by JC-1. (Ai) HeLa cell mitochondria after incubation with JC-1, illustrating the heterogeneity in mitochondria membrane potential within a single cell. To demonstrate co-localization of TMRE with JC-1, the same cells were subsequently co-loaded with TMRE (0.1 µM, 20 min). As illustrated in (Aii), the same organelles are stained. The mitochondria within the cells were then depolarized by addition of antimycin (10 µM) plus oligomycin (2 µM). (B) The peripheral location of red staining mitochondria in a hepatocyte stained with JC-1 at 22°C. (Ci and ii) A section of the same JC-1-loaded HUVEC cell at 22 and 37°C. The HUVEC cell was incubated in JC-1 at 22°C for 1 h before the image in (Ci) was obtained. The cell was then slowly warmed to 37°C (∼10 min) and then incubated for a further 30 min in the continual presence of JC-1. Scale bar, 5 µm.
TMRE partitions across the mitochondrial inner membrane in a manner proportional to Δψmit, and can also be used to monitor differences in Δψmit. We therefore examined the distribution of TMRE within cells. Although confocal imaging allows optical sectioning of cells, out-of-focus fluorescence can enter the image plane and make it appear artificially bright. The amount of out-of-focus light that enters a particular confocal plane is dependent on the intensity of fluorescence in the optical sections above and below it (Fink et al., 1998). Since the density of mitochondria is much greater around the nucleus than in the periphery (Figure 1), TMRE fluorescence from mitochondria outside a confocal plane could make the perinuclear mitochondria appear brighter than the more sparsely distributed peripheral mitochondria. To eliminate this problem and obtain a better indication of the brightness of individual mitochondria, we used image deconvolution (Fink et al., 1998).
Typically, stacks of confocal images (z-stack; 0.2 µm steps) were obtained by scanning through the depth of a TMRE-loaded cell (cells were loaded with 0.1 µM TMRE for 20 min). These image stacks were deconvolved using a blind deconvolution algorithm. Since TMRE partitions between mitochondria and the cytosol in a Δψmit-dependent manner, ratios of fluorescence intensity in mitochondria and their adjacent cytosolic areas were obtained. For HeLa cells, this analysis indicated that TMRE fluorescence intensity was significantly higher (P <0.005; mean ± SEM, 33.8 ± 3.0; n = 58 mitochondria from 10 cells) than in the perinuclear region (mean ± SEM, 22.4 ± 1.8; n = 63 mitochondria from 10 cells). Similar observations were seen with the other cell types (data not shown).
Differences in Δψmit should also be manifest in the rate of TMRE uptake, in that the more polarized mitochondria will load with TMRE faster than those with lower membrane potential. To locate mitochondria and compensate for differences in mitochondrial density, the increase of TMRE fluorescence was ratioed against the signal from mitochondria previously loaded with Mitotracker Green FM. Mitotracker Green FM, unlike other Mitotracker dyes, accumulates in mitochondria regardless of the Δψmit (Haugland, 1999). Cells were therefore initially stained with 1 µM Mitotracker Green FM for 20 min before adding TMRE. Dual-channel images were acquired with a Bio-Rad MRC1024 confocal microscope. It was consistently observed that the normalized TMRE accumulation in HeLa cells proceeded at a greater rate for peripheral mitochondria than in perinuclear mitochondria (mono-exponential rate constants: 0.19 ± 0.04/min for peripheral versus 0.07 ± 0.01/min for perinuclear) (Figure 6).

Fig. 6. Heterogeneity in Δψmit reported by TMRE. The figure illustrates TMRE loading of peripheral (thick black line) and perinuclear (thin grey line) mitochondria pre-loaded with Mitotracker Green FM (1 µM, 25 min). The arrowhead indicates addition of TMRE (0.1 µM). TMRE loading is expressed as a ratio of TMRE fluorescence:Mitotracker Green FM fluorescence. The data in this figure were obtained by monitoring TMRE sequestration in six cells. For each of these cells, TMRE uptake was analysed in 10 randomly chosen perinuclear and peripheral mitochondria. The graph therefore represents the averaged response of 60 mitochondria for both the peripheral and perinuclear traces. The inset image shows the increase in TMRE fluorescence in the nucleus and peripheral cytoplasm.
The peripheral regions of cells have a higher plasma membrane surface to volume ratio than the perinuclear areas. We were concerned that the distinct rates of TMRE sequestration in peripheral and perinuclear mitochondria could simply have reflected different kinetics of TMRE accumulation in the cytosol surrounding the organelles. We therefore examined the rate of TMRE accumulation in the peripheral cytoplasm and nucleus. The time-course of TMRE fluorescence increase was identical for both regions (Figure 6, inset), suggesting that TMRE reaches these parts of the cytoplasm with the same kinetics.
Ca2+ sequestration. Rapid (15 Hz) confocal imaging of histamine-stimulated rhod-2-loaded HeLa cells revealed that mitochondria in the periphery of the cells sequestered more Ca2+ than mitochondria located in the perinuclear region (Figure 7A). Although the onset of mitochondrial Ca2+ accumulation was similar for peripheral and perinuclear mitochondria, the former continued to sequester Ca2+ when their counterparts were beginning to plateau. The difference in Ca2+ sequestration was only apparent for inositol 1,4,5,triphosphate (InsP3)-evoked Ca2+ release. More slowly developing Ca2+ signals did not cause such differences in mitochondrial Ca2+ sequestration. For example, Ca2+ entry triggered by re-addition of Ca2+ to cells pre-treated with thapsigargin or cyclopiazonic acid (CPA) in Ca2+-free medium gave equivalent perinuclear and peripheral mitochondrial Ca2+ uptake (Figure 7B). Treatment with CGP37157, an inhibitor of mitochondrial sodium-Ca2+ exchange, failed to reveal any difference in Ca2+ sequestration by peripheral and perinuclear mitochondria during thapsigargin treatment or Ca2+ entry (Figure 7Biii).
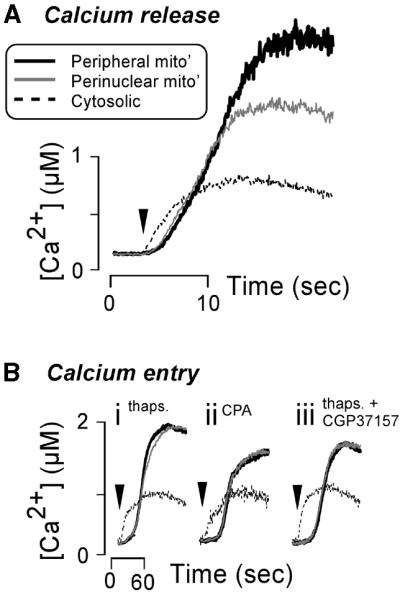
Fig. 7. Perinuclear and peripheral mitochondria sequester agonist- induced Ca2+ signals to different extents. (A) HeLa cells loaded with rhod-2 were treated with 100 µM histamine (arrowhead) to trigger Ca2+ release from the ER. The resultant cytosolic Ca2+ change [thin dashed line in (A)] was monitored by recording rhod-2 fluorescence in the nucleus as described previously (Collins et al., 2001). In addition, Ca2+ increases in the perinuclear mitochondria (thin grey line) and peripheral mitochondria (thick black line) were followed. (B) Depiction of the similarity of Ca2+ sequestration by perinuclear and peripheral mitochondria during Ca2+ entry. Prior to the traces shown in (Bi–iii), the ER Ca2+ pool was depleted by pre-treatment of cells with 2 µM thapsigargin in 4 mM EGTA or 50 µM CPA in 4 mM EGTA. Ca2+ entry was initiated by substituting the EGTA in the extracellular medium with 1.8 mM CaCl2 (arrowheads). The sodium/Ca2+ exchanger was inhibited for the response shown in (Biii) by the addition of 10 µM CGP37157 for 5 min before the replacement of EGTA with CaCl2. For the responses in (A), where the histamine-stimulated Ca2+ signals were rapid, the cells were imaged at 15 Hz. The slower Ca2+ entry signals in (B) were imaged at 0.5 Hz. Traces represent the mean of 10 cells, with >10 individual mitochondria or small clusters of mitochondria being analysed in each cell.
Since the proximity of mitochondria to Ca2+ release sites is thought to be an important determinant of their Ca2+ uptake properties, we examined the co-localization of mitochondria and ER. Cells were transfected with both ER-targeted EGFP and mito-DsRed1. The image in Figure 8A illustrates a deconvolved confocal section of a HeLa cell expressing the two fluorescent proteins. In the perinuclear regions (e.g. region bordered by white box in Figure 8A), the mitochondria were closely apposed to the ER. In contrast, the mitochondria in peripheral regions (e.g. region bordered by dashed box in Figure 8A) were less well associated with the ER. The proximity of ER and mitochondria is more clearly indicated in Figure 8B, where the mitochondrial regions from Figure 8A are shown. The red colouration depicts areas where only the mitochondrial DsRed1 fluorescence was detected, while yellow indicates points where the association between the two organelles was so close that it could not be resolved (i.e. <0.5 µm; green and red fluorescence channels merging). The perinuclear mitochondria have a significant degree of yellow colouration, indicating a close contact with the ER over most of each mitochondrion. The peripheral mitochondria have only a few spots of yellow colouration consistent with a much weaker association at relatively few places. Analysis of the pixels in similarly sized perinuclear and peripheral regions of three different HeLa cells revealed that, on average, the perinuclear mitochondria displayed twice as many yellow pixels compared with those in the periphery (51 ± 5% versus 26 ± 7%; P = 0.044).

Fig. 8. Association between ER and mitochondria in the periphery and perinuclear regions of a cell. (A) Maximum z-projection of a HeLa cell expressing ER-targeted EGFP (shown as green) and mito-DsRed1 (shown as red). The nucleus is out of view to the left of the image. (B) Yellow regions denote where ER and mitochondria co-localize within the resolution of the deconvolved, confocal image. The regions bounded by white boxes in (A) are shown on expanded scales in (Ci) (perinuclear region) and (Di) (peripheral region). The same expanded regions are shown as surface-rendered versions in (Cii) and (Dii), respectively, to show that the perinuclear mitochondria are embedded in ER, whilst the peripheral mitochondria are more remote from the ER. The images are presented as if the cell is viewed from above. The view from underneath the cell shows a similar pattern. Scale bars, 5 µm.
The regions bounded by the white boxes in Figure 8A are shown on expanded scales in Figure 8Ci and Di. The yellow colouration of the perinuclear mitochondrial can be seen clearly, whilst the peripheral mitochondria are mostly red. Figure 8Cii and Dii shows surface representations of the cell sections in Figure 8Ci and Di (i.e. a three-dimensional reconstruction with zero opacity). Essentially, the image in Figure 8Cii illustrates that the perinuclear mitochondria were embedded in the ER as little of the mitochondrial DsRed1 fluorescence is visible between the dense ER GFP signal. In the periphery of the cell, the ER seems to sparsely weave around the mitochondria making regions of en passant points of closer contact (Figure 8Dii).
Permeability transition. The relative sensitivities of mitochondria within a single cell to the permeability transition-inducing factor tert-butyl hydroperoxide (tBuOOH; 100 µM) were determined by imaging calcein loss from mitochondria. In this assay, opening of the PTP causes the redistribution of calcein from the mitochondrial matrix to the cytoplasm. In cells that are simultaneously loaded with cobalt, the fluorescence of the calcein that enters the cytoplasm is rapidly quenched.
Cells were loaded with calcein-AM in the presence of 1 mM CoCl2 for 30 min, then left in 1 mM CoCl2 for another 30 min. Calcein was predominately localized to the mitochondria as shown by co-localization with mito-DsRed1 (Figure 9Ai and ii). A few non-mitochondrial organelles also retained calcein fluorescence (e.g. arrowhead in Figure 9Aiii), but these were a minor component of the total signal. Activation of permeability transition by tBuOOH caused the total loss of calcein fluorescence from individual mitochondria, but over a considerable time window. A few mitochondria released their calcein within ∼30 s, whilst others retained fluorescence until the end of the recording period (20 min after addition of tBuOOH) (Figure 9B).

Fig. 9. Asynchronous PTP activation within a single cell. Location of mito-DsRed1 (Ai) and calcein (Aii) fluorescence in the same cell. (Aiii) Difference image [i.e. (Aii) subtracted from (Ai)], indicating that mito-DsRed1 and calcein co-localized to the same organelles, except in a few small spherical vesicles [e.g. spot marked by arrowhead in panel (Aiii)]. (Bi) Another single calcein-loaded HeLa cell. The circled mitochondria in (Bi) indicate the locations of organelles from which the fluorescence in (Bii) was recorded. PTP activation was initiated by addition of 100 µM tBuOOH [arrowhead in (Bii)]. The drop in fluorescence indicates PTP opening. The traces are expressed as normalized fluorescence calculated as (F – Fmin)/(Fmax – Fmin), except for those mitochondria that did not undergo PTP, which are expressed as F/Fmax (i.e. where there was no Fmin). The numbered mitochondria in (Bi) are those that did not show PTP activation during the experiment. The fluorescence of calcein in these mitochondria is depicted by the correspondingly numbered traces in (Bii). Scale bar, 5 µm.
There were no obvious differences between the peripheral and perinuclear mitochondria of HeLa cells in their response to tBuOOH (Figure 9B). The average times for perinuclear and peripheral mitochondria to show PTP activation were 155 ± 14 s and 169 ± 22 s, respectively, at 20°C, and 181 ± 40 s and 170 ± 31 s, respectively, at 37°C (mean ± SEM; data from 30 peripheral and perinuclear mitochondria in three cells). PTP activation in COS-7 cells displayed a similar sensitivity to tBuOOH. In those cells, the average times for calcein loss from perinuclear and peripheral mitochondria were 145 ± 9 s and 127 ± 10 s, respectively, at 22°C (mean ± SEM; data from 30 peripheral and perinuclear mitochondria in three cells).
Those organelles that retained their fluorescence until the end of the experiment (denoted by numbers in Figure 9Bi and ii) were clearly identified as having mitochondrial morphology rather than the small, spherical morphology of the non-mitochondrial structures, which also loaded with calcein (Figure 9Aiii).
Discussion
Mitochondria are crucial determinants in the life and death of cells, and changes in their morphology and function underlie processes such as apoptosis (e.g. Frank et al., 2001). There are conflicting reports as to how interconnected mitochondria are in intact cells, with suggestions that mitochondria may exist as a single interconnected reticulum akin to the ER (for discussion see Rutter and Rizzuto, 2000).
The data in this study indicate that mitochondria within individual cells are morphologically heterogeneous and physically distinct entities. Similar observations were made for three transformed cell lines (HeLa, PAEC and COS-7 cells) and four primary cell types (HUVEC, hepatocytes, cortical astrocytes and cortical neuronal cells). The lack of lumenal and electrical connectivity allows the mitochondria to function independently with respect to Δψmit, Ca2+ sequestration and PTP activation.
The deconvolved confocal images in Figure 1 clearly reveal the existence of individual mitochondria. In most of the transformed and primary cell types examined in this study, the mitochondria were of extremely variable length and often followed apparently tortuous paths. Furthermore, there were substantial areas of the cytoplasm that were devoid of mitochondria. The striking exception to these observations was hepatocytes (e.g. Figure 4B), where the mitochondria were rarely observed to be longer than 5 µm. The density of mitochondria in hepatocytes was the greatest amongst all the cell types examined (Figure 1F), but the mitochondria were sufficiently isolated from each other that they could be discretely resolved. In contrast to the other cell types where the highest densities of mitochondria were in perinuclear regions, hepatocyte mitochondria were more evenly distributed within the cell.
In the cell types where mitochondria were densely packed in the perinuclear region, it was more difficult to visualize whether mitochondria are connected or simply very close. To determine whether there were individual perinuclear mitochondria, two techniques were employed: FRAP (Figure 2) and TMRE-plus-laser induction of depolarization (Figure 4). The FRAP experiments revealed that the mitochondria in this dense perinuclear region were not lumenally connected. Since the translocation of DsRed1 or calcein within the mitochondrial matrix was rapid (Figure 3), lumenal connectivity would have ensured a significant recovery of the fluorescence signal after photobleaching within a few tens of seconds. However, in all cells tested at either 22 or 37°C, the bleached areas recovered only modestly after 20 min (Figure 2), and were not fully recovered even after 1 h (data not shown).
Our observations contrast with data reported by Rizzuto et al. (1998). In their experiments with HeLa cells expressing mitochondrially targeted GFP, Rizzuto et al. (1998) also found that photobleached areas did not fully recover for several minutes; however, they did see recovery of GFP fluorescence in the bleached area after 30 min. They did not directly measure the mobility of the mitochondrially targeted GFP used in their experiments. However, since the fluorescent markers used in this study and by Rizzuto et al. (1998) are similar in mass, they should have the same mobility in the mitochondrial matrix. We would therefore suggest that the lack of rapid FRAP in the experiments of Rizzuto et al. (1998) supports our conclusion that the mitochondrial are not lumenally contiguous. Our conclusion concurs with that of Ricken et al. (1998), who also did not see recovery of fluorescence after local bleaching of a mitochondrion in HT29 cells. Although these authors also did not measure the mobility of the fluorophore, they used (Fura2) within the matrix.
Inspection of the images presented by Rizzuto et al. (1998) suggests that the morphology of their mitochondria changed significantly over the period of the experiment. This indicates that there was considerable motion or fission/fusion of the mitochondria in the optical section they examined. We would therefore suggest that the movement or fusion of unbleached mitochondria may have caused an apparent increase in fluorescence, but not true FRAP. The limited recovery of DsRed1 fluorescence that we observed after photobleaching was most probably due to movement of unbleached mitochondria or low frequency mitochondrial fusion events. Such fusion events between bleached and unbleached mitochondria would replenish the fluorescence of a few mitochondria within the optical section.
Functional heterogeneity of mitochondria was apparent in several ways. First, monitoring Δψmit indicated that mitochondria were heterogeneous with respect to their energetic status. Although highly energized mitochondria were present throughout cells, there appeared to be proportionally more highly energized mitochondria in the periphery of cells. Such heterogeneity in Δψmit shown by JC-1 has been documented previously (Reers et al., 1991, 1995; Salvioli et al., 1997).
Three separate experimental observations [JC-1 emission (Figure 5), steady-state TMRE distribution and TMRE uptake rate (Figure 6)] all suggest that the peripheral mitochondria have, on average, a higher Δψmit than their perinuclear counterparts. Such differences in Δψmit should have some functional consequences. Indeed, the difference in membrane potential of perinuclear and peripheral mitochondria was substantiated by the observation that peripheral mitochondria accumulate substantially more Ca2+ than those in the perinuclear region (Figure 7A).
To facilitate Ca2+ sequestration, mitochondria are thought to require close association with the source of Ca2+ in order to be exposed to high microdomain concentrations of Ca2+ present at the mouth of a Ca2+ release channel (Rizzuto et al., 1993; Hoth et al., 1997). A plausible prediction from this hypothesis would be that the perinuclear mitochondria, which tend to be embedded in ER (Figure 8), would sequester the most Ca2+ from an agonist-evoked Ca2+ release. However, as for Chinese hamster ovary T cells (Rutter et al., 1996), peripheral mitochondria in HeLa cells were found to sequester more Ca2+ from agonist-evoked cytosolic Ca2+ signals than perinuclear mitochondria (Figure 7A).
Interestingly, there was no difference between peripheral and perinuclear mitochondria in sequestration of Ca2+ entering the cell (Figure 7B). Aside from the source, a major difference between Ca2+ release and entry signals is their kinetics: release is much more rapid than the entry-induced signals. With a more slowly developing signal, Ca2+ efflux from the mitochondria, via the relatively slow Na+/Ca2+ exchanger, may become more significant. However, treatment with the Na+/Ca2+ exchange inhibitor CGP37157 failed to reveal differences in peripheral and perinuclear mitochondria sequestration of Ca2+ influx (Figure 7Biii).
The driving force for the Ca2+ uniporter is the Ca2+ electrochemical gradient across the inner mitochondrial membrane. This gradient is determined by both Δψmit and the Ca2+ concentration difference across the inner mitochondrial membrane (ΔCaimm) (Gunter and Pfeiffer, 1990; Gunter and Gunter, 1994). With a rapidly elevated cytosolic Ca2+ signal, ΔCaimm would be greater than for a more slowly rising signal. In vitro examination of mitochondrial Ca2+ sequestration has shown that the rate of Ca2+ uptake increases more steeply with Δψmit at high extra-mitochondrial Ca2+ concentrations (i.e. high ΔCaimm) compared with lower Ca2+ concentrations (Wingrove et al., 1984). This may explain why sequestration of agonist-evoked Ca2+ transients differed between perinuclear and peripheral mitochondria, whereas the more slowly rising Ca2+ entry signal was not (Figure 7). These data suggest that under some conditions, Δψmit and the nature of the Ca2+ signal are more important determinants of mitochondrial Ca2+ uptake than the proximity of mitochondria to the Ca2+ source.
By monitoring calcein fluorescence from individual mitochondria, opening of the PTP in response to pro-apoptotic factors can be followed (Petronilli et al., 1999). This approach revealed heterogeneity in the sensitivity of individual mitochondria to tBuOOH. Some mitochondria responded almost immediately (∼30 s), others only after 15 min, and some did not show any response within the time scale of the experiment (20 min) (Figure 9). This asynchronous permeability transition is in contrast to studies of cytochrome c release from HeLa cells treated with pro-apoptotic agents (Goldstein et al., 2000). By monitoring GFP–cytochrome c fusion protein release from mitochondria, it was observed that all the mitochondria within a single cell lose their cytochrome c within 5 min of each other, regardless of the strength or type of the pro-apoptotic stimulus (Goldstein et al., 2000). Whilst cytochrome c release is thought to occur subsequent to PTP opening (Heiskanen et al., 1999), there is evidence that cytochrome c loss may occur via another route (Bossy-Wetzel et al., 1998; Goldstein et al., 2000). Consistent with this is the temporary maintenance of Δψmit in some cells after cytochrome c release (Goldstein et al., 2000; Waterhouse et al., 2001). However, it is unclear whether the release of cytochrome c and maintenance of Δψmit occur in the same mitochondria within individual cells. The observation that certain populations of mitochondria may not depolarize at the same time as others in response to a pro-apoptotic stimulus raises the possibility that some mitochondria are involved in signalling during apoptosis, whilst others maintain ATP production during the critical early stages of apoptosis (Leist et al., 1997).
The work presented here demonstrates that the mitochondria do not necessarily form significant electrically or lumenally connected networks. Mitochondria display a wide range of morphologies and are functionally heterogeneous. The morphological and functional heterogeneity of mitochondria raises the possibility that subpopulations of these organelles can carry out diverse processes within different areas of a cell. However, we are not suggesting that the mitochondria in the cells we visualized will always look as they are presented here. Rather, our measurements represent the steady-state situation under typical experimental conditions. Mitochondrial morphology is regulated by the balance of fusion and fission processes. Many factors can tip the balance between these counteracting processes. For example, apoptosis can cause mitochondria to fragment (e.g. Frank et al., 2001), whilst hypoxia can evoke the reverse effect (for discussion see Skulachev, 2001). Other factors can dynamically regulate functional aspects of mitochondria, such as Δψmit and Ca2+ sequestration (e.g. Jouaville et al., 1995, 1999). The plasticity of these organelles plausibly accounts for the discrepant observations of heterogeneity/homogeneity in mitochondrial form and function between different studies. Furthermore, the dynamic nature of mitochondria underscores the conclusion that there is probably no single mitochondrial morphology or functional status, even within an individual cell.
Materials and methods
Cell culture and chemicals
Primary cultured HUVECs were obtained from Clonetics (BioWhittaker Inc, Walkersville, MD), cortical astrocytes and neurons were from Dr L.Prothero (Babraham Institute), hepatocytes were kindly provided by Dr C.W.Taylor (Department of Pharmacology, University of Cambridge, UK), and COS-7 and PAEC cells were from Dr K.Anderson (Babraham Institute). For HeLa, PAEC and COS-7 cells, cell culture was performed as described previously for HeLa cells (Bootman et al., 1994). The primary HUVEC cells were grown as specified in the vendor’s endothelial cell growth medium.
The culture medium was replaced with an extracellular medium (EM) containing (mM): NaCl, 121; KCl, 5.4; MgCl2, 0.8; CaCl2, 1.8; NaHCO3, 6; d-glucose, 5.5; HEPES, 25; pH 7.3. Histamine, thapsigargin, antimycin, oligomycin (mixed isomers) and bromo-A23187 were obtained from Sigma. All fluorescent dyes were obtained from Molecular Probes (Eugene, OR). The experimental steps were carried out at room temperature (22°C) except where indicated. Experiments at 37°C were performed using a thermostated cell chamber.
Statistical significance was analysed using Student’s t-test, with paired or unpaired tests as appropriate.
Transfection
Cells were transfected with mitochondrially targeted DsRed1 (mito-DsRed1) from Clontech (Palo Alto, CA) and ER-targeted EGFP (EGFP with elastase ER-targeting sequence; kindly supplied by Dr E.Joly) with Effectene transfection reagent (Qiagen, Crawley, UK), following the manufacturer’s recommended protocol.
Imaging
Three-dimensional reconstruction. Z-series stacks of mito-DsRed1-expressing cells were acquired with a Bio-Rad MRC1024 LSCM. Subsequent image restoration was achieved with the deconvolution software AutoDeblur (Autoquant, New York) using the ‘power accelerated’ blind deconvolution algorithm. Image analysis and processing was performed with the public domain software ImageJ (NIH; http://rsb.info.nih.gov/ij). Single channel surface-rendered images were processed with ImageJ running the VolumeJ plugin (M.Abràmoff; http://www.isi.uu.nl/people/michael/vr.htm); dual-channel surface-rendered images were processed with Volocity software (Improvision, Coventry, UK).
FRAP. For the FRAP experiments illustrated in Figure 2, cellular areas typically between 25 and 100 µm2 of perinuclear mitochondria were bleached with a Bio-Rad MRC1024 by briefly digitally zooming into the region of interest with an enhanced laser intensity. In all cell types except hepatocytes, FRAP experiments were conducted using cells expressing mito-DsRed1. Since transfection of primary hepatocytes was not possible, the FRAP experiments in these cells utilized mitochondrially localized calcein. Cells were loaded with calcein acetoxymethyl (AM)ester (1 µM) for 30 min in the presence of 1 mM CoCl2 and left to de-esterify the dye in the presence of 1 mM CoCl2 alone. The cobalt quenches the calcein fluorescence except in mitochondria, which do not transport cobalt.
To monitor the mobility of fluorophores in the mitochondrial matrix (Figure 3), DsRed1 was bleached using the digital zoom function of a Noran Oz confocal microscope. The subsequent images showing redistribution of DsRed1 fluorescence were obtained at ∼2 Hz. The laser intensity for bleaching fluorophores during the FRAP experiments was empirically adjusted on a cell by cell basis. Typical intensities for bleaching were in the order of 100 µW at the objective (determined using a Coherent Lasercheck power meter) applied for ∼5 s.
Ca2+mit. To simultaneously monitor Ca2+ concentration in the mitochondria and cytoplasm (Ca2+mit and Ca2+cyt), cells were incubated in 1 µM rhod-2 acetoxymethyl ester for 30 min followed by a 30 min de-esterification period. Rhod-2 fluorescence was converted into Ca2+ concentration using a previously determined calibration (Collins et al., 2001). The affinity of rhod-2 for calcium (i.e. Kd) and dynamic range of the indicator (i.e. maximum Ca2+-bound fluorescence relative to minimum Ca2+-free fluorescence) was the same in perinuclear and peripheral mitochondria (data not shown).
Δψmit. To determine the spatial variation in Δψmit, the dual emission potentiometric dye, JC-1, was imaged. JC-1 aggregates fluoresce red (∼597 nm), whereas monomers fluoresce green (∼539 nm). The red-fluorescing aggregates form in proportion with increasing Δψmit to approximately –240 mV (Reers et al., 1995). The relative distribution of red and green fluorescence was used to indicate the spatial variation in Δψmit. Cells were incubated with 1 µM JC-1 (Molecular Probes). The loading time was determined empirically for each cell, and was typically 15–60 min. Dual-channel images were acquired with a Bio-Rad MRC1024 confocal microscope or an UltraView confocal microscope (Perkin-Elmer Life Sciences, Cambridge, UK).
Concerns have been raised regarding the suitability of JC-1 to monitor changes in Δψmit (Mojet et al., 2000). However, many of these issues relate to use of JC-1 to measure changes in Δψmit rather than resting Δψmit, which is of interest here. Others consider JC-1 to be superior to other carbocyanine- and rhodamine-based dyes for potentiometric measurements (Salvioli et al., 1997; Mathur et al., 2000). It has been proposed that JC-1 loads into non-mitochondrial membranes. This does not seem to be the case in our hands, since TMRE co-localizes with JC-1 and the red fluorescence emission of both dyes is abolished by treatment with anti-mycin (Figure 5). However, after mitochondrial depolarization, JC-1 monomers did redistribute to non-mitochondrial as well as mitochondrial membranes (Figure 5C).
Several previous studies have discussed the possible drawbacks of measuring Δψmit using JC-1 or rhodamine-based potentiometric dyes. JC-1 and rhodamine dyes both can have toxic effects. JC-1 has been suggested to inhibit complex I of the respiratory chain (Mojet et al., 2000), whilst rhodamine derivatives have been shown to inhibit oxygen consumption by isolated mitochondria, with TMRE being the most toxic (Scaduto and Grotyohann, 1999). In situ calibration of 123rhodamine showed it to be insensitive to Δψmit below –140 mV (Ubl et al., 1996) and calibration of tetramethylrhodamine methyl ester was not reported below –165 mV (Scaduto and Grotyohann, 1999). JC-1 has been calibrated to –240 mV (Reers et al., 1995). With JC-1, differences in Δψmit are manifest as absolute changes in emission wavelength due to formation of red-emitting aggregates. In contrast, with rhodamine-based dyes, differences in Δψmit would be indicated by variation in the ratio of mitochondrial to cytosolic emission intensity. Owing to the greater dynamic range of JC-1, it could be considered superior in the measurement of heterogeneous Δψmit in a mitochondrial population. Modest differences in TMRE emission intensity between mitochondria may be difficult to discern, especially when imaging small objects of varying sizes and aggregation and in different focal planes (Fink et al., 1998). This may explain why previous studies using rhodamine-based indicators concluded that Δψmit is uniform throughout the mitochondria within an individual cell (Chen, 1988).
In the present study, evidence for mitochondrial heterogeneity was obtained using both JC-1 and TMRE. First, we observed that the ratio of mitochondrial to cytosolic TMRE intensity was significantly higher with peripheral mitochondria than for perinuclear mitochondria (see Results). However, we have to concede that the interpretation of these data is not without problems due to several issues relating to the dye itself. At high concentrations, i.e. high Δψmit, auto-quenching of TMRE occurs (Duchen et al., 1998). TMRE also triggers permeability transition (Figure 4) and rhodamine-derived dyes have been shown to behave in a non-Nernstian way as they bind to mitochondrial membranes (Scaduto and Grotyohann, 1999). Furthermore, the mitochondrial signal is ∼20–30 times brighter than the cytosolic signal. This makes it difficult to obtain accurate relative measurements of TMRE within the mitochondrial and cytosolic compartments without having saturating mitochondrial fluorescence or rendering the cytosolic signal undetectable. To circumvent these problems, the rate of TMRE uptake by HeLa cell mitochondria was normalized using a mitochondria-specific Δψmit-independent fluorophore (Mitotracker Green FM; Figure 6).
Permeability transition. The opening of the PTP was imaged with calcein (Petronilli et al., 1999). Cells were loaded with calcein-AM as described above for the FRAP experiments involving hepatocytes. When mitochondria undergo PTP, the calcein is lost from the mitochondria and the cytoplasmic cobalt quenches its fluorescence.
Acknowledgments
Acknowledgements
We thank Louise Armstrong-Denby (Improvision, Coventry, UK) for her help in processing the dual-channel, surface-rendered images of ER and mitochondria with Volocity software. This work was supported by the BBSRC. M.D.B. gratefully acknowledges the support of a Royal Society University Research Fellowship.
References
- Amchenkova A.A., Bakeeva,L.E., Chenksov,Y.S., Skulachev,V.P. and Zorov,D.B. (1988) Coupling membranes as energy-transmitting cables. 1. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol., 107, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock D.F., Herrington,J., Goodwin,P.C., Park,Y.B. and Hille,B. (1997) Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol., 136, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby B.J. and Moyes,C.D. (1998) Are there distinct subcellular populations of mitochondria in rainbow trout red muscle? J. Exp. Biol., 201, 2455–2460. [DOI] [PubMed] [Google Scholar]
- Bootman M.D., Cheek,T.R., Moreton,R.B., Bennett,D.L. and Berridge,M.J. (1994) Smoothly graded Ca2+ release from inositol 1,4,5-trisphosphate-sensitive Ca2+ stores. J. Biol. Chem., 269, 24783–24791. [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer,D.D. and Green,D.R. (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J., 17, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J.T., Martin,A.P., Lucas,F.V. and Vorbeck,M.L. (1974) The structure of rat liver mitochondria: a reevaluation. Biochem. Biophys. Res. Commun., 59, 1097–1103. [DOI] [PubMed] [Google Scholar]
- Buckman J.F. and Reynolds,I.J. (2001) Spontaneous changes in mitochondrial membrane potential in cultured neurons. J. Neurosci., 21, 5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.B. (1988) Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol., 4, 155–181. [DOI] [PubMed] [Google Scholar]
- Collins T.J., Lipp,P., Berridge,M.J. and Bootman,M.D. (2001) Mitochondrial Ca2+ uptake depends on the spatial and temporal profile of cytosolic Ca2+ signals. J. Biol. Chem., 276, 26411–26420. [DOI] [PubMed] [Google Scholar]
- Crompton M. (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem. J., 341, 233–249. [PMC free article] [PubMed] [Google Scholar]
- De Giorgi F., Lartigue,L. and Ichas,F. (2000) Electrical coupling and plasticity of the mitochondrial network. Cell Calcium, 28, 365–370. [DOI] [PubMed] [Google Scholar]
- Duchen M.R., Leyssens,A. and Crompton,M. (1998) Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol., 142, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y., Shimizu,S. and Tsujimoto,Y. (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res., 57, 1835–1840. [PubMed] [Google Scholar]
- Fink C., Morgan,F. and Loew,L.M. (1998) Intracellular fluorescent probe concentrations by confocal microscopy. Biophys. J., 75, 1648–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S., Gaume,B., Bergmann-Leitner,E.S., Leitner,W.W., Everett,R.G., Catez,F., Smith,C.L. and Youle,R.J. (2001) The role of Dynamin-Related Protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell, 1, 515–525. [DOI] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse,N.J., Juin,P., Evan,G.I. and Green,D.R. (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nature Cell Biol., 2, 156–162. [DOI] [PubMed] [Google Scholar]
- Gunter K.K. and Gunter,T.E. (1994) Transport of calcium by mitochondria. J. Bioenerg. Biomembr., 26, 471–485. [DOI] [PubMed] [Google Scholar]
- Gunter T.E. and Pfeiffer,D.R. (1990) Mechanisms by which mitochondria transport calcium. Am. J. Physiol., 258, C755–C786. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G., Robbgaspers,L.D., Seitz,M.B. and Thomas,A.P. (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell, 82, 415–424. [DOI] [PubMed] [Google Scholar]
- Haugland R.P. (1999) Handbook of Fluorescent Probes and Research Chemicals. Molecular Probes Inc., Eugene, OR.
- Heiskanen K.M., Bhat,M.B., Wang,H.W., Ma,J.J. and Nieminen,A.L. (1999) Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem., 274, 5654–5658. [DOI] [PubMed] [Google Scholar]
- Hoffman H.P. and Avers,C.J. (1973) Mitochondrion of yeast: ultrastructural evidence for one giant, branched organelle per cell. Science, 181, 749–751. [DOI] [PubMed] [Google Scholar]
- Hoth M., Fanger,C.M. and Lewis,R.S. (1997) Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol., 137, 633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüser J. and Blatter,L.A. (1999) Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J., 343, 311–317. [PMC free article] [PubMed] [Google Scholar]
- Hüser J., Rechemacher,C.E. and Blatter,L.A. (1998) Imaging the permeability pore transition in single mitochondria. Biophys. J., 74, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F., Jouaville,L.S., Sidash,S.S., Mazat,J.P. and Holmuhamedov,E.L. (1994) Mitochondrial calcium spiking—a transduction mechanism-based on calcium-induced permeability transition involved in cell calcium signaling. FEBS Lett., 348, 211–215. [DOI] [PubMed] [Google Scholar]
- Jahangir A., Holmuhamedov,E.L. and Terzic,A. (1999) Two mitochondrial populations in the heart: Are subsarcolemmal mitochondria the primary target of mitochondrial K-ATP channel opener action? Circulation, 100, 1798.10534467 [Google Scholar]
- Jouaville L.S., Ichas,F., Holmuhamedov,E.L., Camacho,P. and Lechleiter,J.D. (1995) Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature, 377, 438–441. [DOI] [PubMed] [Google Scholar]
- Jouaville L.S., Pinton,P., Bastianutto,C., Rutter,G.A. and Rizzuto,R. (1999) Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl Acad. Sci. USA, 96, 13807–13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M., Single,B., Castoldi,A.F., Kuhnle,S. and Nicotera,P. (1997) Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med., 185, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A., Damon,M., Vincent,A., Goglia,F. and Herpin,P. (2000) Characterisation of oxidative phosphorylation in skeletal muscle mitochondria subpopulations in pig: a study using top-down elasticity analysis. FEBS Lett., 475, 84–88. [DOI] [PubMed] [Google Scholar]
- Mathur A., Hong,Y., Kemp,B.K., Barrientos,A.A. and Erusalimsky,J.D. (2000) Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc. Res., 46, 126–138. [DOI] [PubMed] [Google Scholar]
- Mogami H., Nakano,K., Tepikin,A.V. and Petersen,O.H. (1997) Ca2+ flow via tunnels in polarized cells: Recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell, 88, 49–55. [DOI] [PubMed] [Google Scholar]
- Mojet M.H., Jacobson,D., Keelan,J., Vergun,O. and Duchen,M.R. (2000) Monitoring mitochondrial function in single cells. In Tepikin,A.V. (ed.), Calcium Signalling: A Practical Approach. Oxford University Press, Oxford, UK, pp. 79–106.
- Ord M.J. (1979) The effect of chemicals and radiations within the cell: An ultrastructural and micrurgical study using Amoeba proteus as a single-cell model. Int. Rev. Cytol., 61, 229–281. [DOI] [PubMed] [Google Scholar]
- Park M.K., Ashby,M.C., Erdemli,G., Petersen,O.H. and Tepikin,A.V. (2001) Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J., 20, 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V., Miotto,G., Canton,M., Brini,M., Colonna,R., Bernardi,P. and Di Lisa,F. (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J., 76, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reers M., Smith,T.W. and Chen,L.B. (1991) J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane-potential. Biochemistry, 30, 4480–4486. [DOI] [PubMed] [Google Scholar]
- Reers M., Smiley,S.T., Mottolahartshorn,C., Chen,A., Lin,M. and Chen,L.B. (1995) Mitochondrial-membrane potential monitored by JC-1 dye. Methods Enzymol., 260, 406–417. [DOI] [PubMed] [Google Scholar]
- Ricken S., Leipziger,J., Greger,R. and Nitschke,R. (1998). Simultaneous measurements of cytosolic and mitochondrial Ca2+ transients in HT29 cells. J. Biol. Chem., 273, 34961–34969. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini,M., Murgia,M. and Pozzan,T. (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science, 262, 744–747. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton,P., Carrington,W., Fay,F.S., Fogarty,K.E., Lifshitz,L.M., Tuft,R.A. and Pozzan,T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science, 280, 1763–1766. [DOI] [PubMed] [Google Scholar]
- Rutter G.A. and Rizzuto,R. (2000) Regulation of mitochondrial metabolism by ER Ca2+ release: an intimate connection. Trends Biochem. Sci., 25, 215–221. [DOI] [PubMed] [Google Scholar]
- Rutter G.A., Burnett,P., Rizzuto,R., Brini,M., Murgia,M., Pozzan,T., Tavare,J.M. and Denton,R.M. (1996) Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin—significance for the regulation of pyruvate-dehydrogenase activity. Proc. Natl Acad. Sci. USA, 93, 5489–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvioli S., Ardizzoni,A., Franceschi,C. and Cossarizza,A. (1997) JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett., 411, 77–82. [DOI] [PubMed] [Google Scholar]
- Scaduto R.C. and Grotyohann,L.W. (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J., 76, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J., Bhatnagar,P.L. and Milburn,N.S. (1969) An electron microscope study of changes in mitochondria of flight muscle of ageing houseflies (Musca domestica). J. Insect Physiol., 15, 135–140. [DOI] [PubMed] [Google Scholar]
- Skulachev V.P. (2001) Mitochondrial filaments and cluster as intracellular power transmitting cables. Trends Biochem. Sci., 26, 23–29. [DOI] [PubMed] [Google Scholar]
- Ubl J.J., Chatton,J.Y., Chen,S.H. and Stucki,J.W. (1996) A critical evaluation of in situ measurement of mitochondrial electrical potentials in single hepatocytes. Biochim. Biophys. Acta, 1276, 124–132. [DOI] [PubMed] [Google Scholar]
- Waterhouse N.J., Goldstein,J.C., von Ahsen,O., Schuler,M., Newmeyer,D.D. and Green,D.R. (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol., 153, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingrove D.E., Amatruda,J.M. and Gunter,T.E. (1984) Glucagon effects on the membrane-potential and calcium-uptake rate of rat-liver mitochondria. J. Biol. Chem., 259, 9390–9394. [PubMed] [Google Scholar]
- Zorov D.B., Filburn,C.R., Klotz,L.O., Zweier,J.L. and Sollott,S.J. (2000) Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med., 192, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]