

Abstract
The endoplasmic reticulum (ER) is recognized as an important site for regulating cell surface expression of membrane proteins. We recently reported that only a fraction of newly synthesized δ opioid receptors could leave the ER and reach the cell surface, the rest being degraded by proteasomes. Here, we demonstrate that membrane-permeable opioid ligands facilitate maturation and ER export of the receptor, thus acting as pharmacological chaperones. We propose that these ligands stabilize the newly synthesized receptor in the native or intermediate state of its folding pathway, possibly by inducing stabilizing conformational constrains within the hydrophobic core of the protein. The receptor precursors that are retained in the ER thus represent fully competent folding intermediates that can be targets for pharmacological intervention aimed at regulating receptor expression and cellular responsiveness. The pharmacological chaperone action is independent of the intrinsic signaling efficacy of the ligand, since both agonists and antagonists were found to promote receptor maturation. This novel property of G protein-coupled receptor ligands may have important implications when considering their effects on cellular responsiveness during therapeutic treatments.
Keywords: endoplasmic reticulum/G protein-coupled receptor/opioids/pharmacological chaperone
Introduction
The endoplasmic reticulum (ER) quality control scrutinizes newly synthesized proteins entering the secretory pathway and assures that only correctly folded proteins and fully assembled protein complexes ultimately reach their site of action within the cell (Ellgaard et al., 1999). High fidelity of the ER quality control is of primary importance for the functional integrity of the cell. Thus, even subtle mutations that would not dramatically affect protein function can lead to ER retention of the mutant protein, thereby preventing its physiologically relevant activity. This is exemplified in many important human diseases (Kim and Arvan, 1998; Aridor and Balch, 1999). For example, mutations that produce minor changes in the cystic fibrosis transmembrane conductance regulator (CFTR), α1-antitrypsin and V2-vasopressin receptor (V2R) have been shown to be the underlying cause for cystic fibrosis and some forms of emphysema and nephrogenic diabetes insipidus, respectively (Kim and Arvan, 1998; Aridor and Balch, 1999; Morello and Bichet, 2001).
Restoration of impaired trafficking of the disease-causing mutant proteins has become an important experimental and therapeutic goal in the past few years. This concept is based on the finding that a protein may be able to adopt a functionally competent conformation even if it is normally retained by the ER quality control. This is demonstrated by the ability of the so-called chemical chaperones, such as glycerol, trimethylamine-N-oxide and dimethyl sulfoxide, to rescue targeting and function of the affected protein (Brown et al., 1996; Sato et al., 1996; Tamarappoo and Verkman, 1998; Zhou et al., 1999; Burrows et al., 2000). In line with these findings are our recent observations on the V2R, a member of the G protein-coupled receptor (GPCR) superfamily. We found that two non-peptidic V2R antagonists were able to functionally rescue several receptor mutants that were normally retained in the ER (Morello et al., 2000a). We hypothesized that these antagonists function as pharmacological chaperones by binding to incompletely folded mutant receptors intracellularly and enhance their release from the ER retention machinery (Morello et al., 2000b).
Recently, it has been proposed that retention and degradation of incorrectly folded proteins entering the secretory pathway may not be restricted to products of mutated genes. Schubert and colleagues proposed that as much as 30% of newly synthesized proteins in various cell types never attains its correct native structure (Schubert et al., 2000). These incorrectly folded proteins, called defective ribosomal products (DRiPs), were found to be ubiquitylated and degraded by the cytosolic proteasomes. The authors also speculated that some proteins might be more predisposed to become DRiPs than others, on the basis of their size and inherent difficulties in folding.
In agreement with this idea, we recently reported that the human δ opioid receptor (hδOR), a member of the GPCR family, is very inefficiently processed to the mature form, so that as little as 40% of the newly synthesized receptors is ultimately transported to the cell surface in human embryonic kidney (HEK)-293S cells (Petäjä-Repo et al., 2000). As proposed for the DRiPs, the receptors that were unable to leave the ER, possibly due to their inability to adopt their correct conformation, are modified by addition of ubiquitin and targeted to proteasomal degradation (Petäjä-Repo et al., 2001). The ER quality control thus becomes one of the key regulatory steps controlling the level of receptor expression at the cell surface.
Given the dramatic effect that the pharmacological chaperones had on the expression of mutant V2Rs, we wondered whether selective pharmacological treatment could also favor folding of non-mutant receptors that have intrinsically low maturation efficiency. We found that both opioid agonists and antagonist could selectively promote maturation of the hδOR and favor ER export of receptor precursors that would otherwise be degraded. This treatment also facilitated maturation and cell surface targeting of an ER-retained mutant (D95A) hδOR. Our data indicate that incompletely folded hδORs are not irreversibly defective and that they may result from a kinetic limitation of a folding intermediate to reach the correct conformation within the required time rather than from errors in translation or post-translational modifications, as has been proposed for the DRiPs. Taken together, our study thus demonstrates that GPCR biosynthesis can be pharmacologically modulated at the ER level, revealing a new site of regulation that could be targeted for controlling cellular responsiveness in therapeutic settings.
Results
A large fraction of newly synthesized hδORs in HEK-293S cells remains in a pre-Golgi compartment and is not transported to the cell surface (Petäjä-Repo et al., 2000). Thus, we set out to determine whether a lipophilic opioid antagonist could have an effect on receptor maturation and increase the proportion of newly synthesized receptors that are able to leave the ER. Metabolic pulse–chase labeling experiments were carried out in stably transfected HEK-293S cells expressing a C-terminally FLAG-epitope tagged hδOR (HEK-293S-hδOR-FLAG cells). Naltrexone (NTX), a non-selective opioid antagonist, was added or not to the chase medium (Figure 1). As previously documented (Petäjä-Repo et al., 2000), two major receptor species corresponding to the core-glycosylated precursor (Mr 45 000) and the mature (Mr 55 000) form of the receptor were detected, the latter of which contains fully processed N-linked and O-linked oligosaccharides. In addition, two minor receptor species (Mr 42 000 and Mr 39 000) were visible at the end of the pulse, representing partially and completely deglycosylated degradation intermediates, respectively (Petäjä-Repo et al., 2001). As can be seen in Figure 1, the efficiency with which the Mr 45 000 precursor was converted to the Mr 55 000 mature receptor was considerably increased in cells treated with the antagonist. Such a treatment led to a 2.2 ± 0.1-fold (n = 6) increase in the amount of mature receptors at 4 h of chase (compare Figure 1A and B). This effect of the antagonist was found to be concentration-dependent (Figure 1C) and 50% of the maximal response (EC50) was achieved at 22.8 ± 8.2 nM (n = 5); a value very similar to the dissociation constant (Ki) of NTX measured for the receptor in competition binding assays using [3H]bremazocine as the radioligand (16.5 ± 0.8 nM; n = 3).

Fig. 1. HδOR synthesis and maturation in untreated and NTX-treated HEK-293S-hδOR-FLAG cells. Cells were labeled for 60 min with 150 µCi/ml of [35S]methionine/cysteine and then chased in medium supplemented with 5 mM methionine in the absence (A) or presence (B) of 10 µM NTX for the indicated times. Alternatively, the cells were chased for 4 h in the presence of NTX at the indicated final concentration (C). Cellular membranes were isolated and solubilized in DDM and the receptors immunoprecipitated using the immobilized anti-FLAG M2 antibody as described in Materials and methods. The samples were analyzed by SDS–PAGE and fluorography. Molecular weights of the markers used to calibrate the gels are indicated on the right. The different receptor forms are indicated by arrows and symbols (filled circle, Mr 55 000; open circle, Mr 45 000). The graphs in (A) and (B) describe the time course of appearance and disappearance of the Mr 45 000 and Mr 55 000 receptor species. Intensities of these species were obtained by densitometric scanning and the values were normalized to the maximum labeling of the mature receptor at 4 h of chase in untreated cells. In (C), the intensity of the mature hδOR species (Mr 55 000) was normalized to the maximum labeling of that species and the data were fitted to a sigmoidal dose response equation using the GraphPad Prism program version 2.01.
The increase in the amount of mature receptors promoted by NTX was accompanied by a significant reduction in the half-life of the Mr 45 000 precursor (Table I), consistent with the notion that the treatment facilitates processing of the precursor. In contrast, the turnover rate of the mature Mr 55 000 species was not affected by the treatment (Table I), suggesting that the antagonist does not stabilize the mature receptor at the cell surface. The effect of the antagonist on the kinetics and efficiency of receptor maturation also led to a steady-state increase in the amount of functional receptors, as revealed by a 1.4-fold increase in the number of [3H]bremazocine binding sites following a 24 h NTX pre-treatment (10.5 ± 0.5 and 14.2 ± 1.1 pmol/mg in untreated and NTX-treated cells, respectively; n = 6).
Table I. Half-lives of the hδOR species in untreated and NTX-treated HEK-293S-hδOR-FLAG cellsa.
| Receptor species | Half-life |
P value | |
|---|---|---|---|
| untreated | NTX-treated | ||
| Mr 55 000 | 23 ± 3 h | 24 ± 4 h | n.s. |
| Mr 45 000 | 150 ± 7 min | 108 ± 10 min | <0.01 |
n.s., not significant.
aRelative intensities of the labeled receptor species were quantified from fluorographs of pulse–chase experiments, an example of which is presented in Figure 1A and B, by densitometric scanning. The half-lives of the different receptor species were determined using the GraphPad Prism program, version 2.01, and the data represent the mean ± SE of four independent experiments. Statistical significance of the differences was assessed using the two-tailed Student’s t-test.
The above data suggest that NTX may mediate its effects intracellularly by interacting with the precursor form of the receptor in the ER. To test this hypothesis further, we assessed the ability of a membrane impermeable ligand, Leu-enkephaline (LE), to either mimic or block the effects of NTX. Treatment with LE alone did not increase the efficiency of receptor maturation (Figure 2). In fact, at the longest chase time tested (4 h chase), this agonist caused a decrease in the amount of mature receptors, most likely as a result of down-regulation of the receptors that had reached the cell surface (Figure 2C, compare lanes 1 and 3). When used at a 200-fold molar excess over NTX (a concentration that should completely saturate the cell surface sites), the agonist did not block the initial effects (2 h chase) of the lipophilic antagonist (Figure 2B, compare lanes 2 and 4), suggesting that NTX most likely acted intracellularly where its effects could not be blocked by LE. At the longest chase time studied (4 h chase), LE again caused down-regulation (Figure 2C, compare lanes 2 and 4).
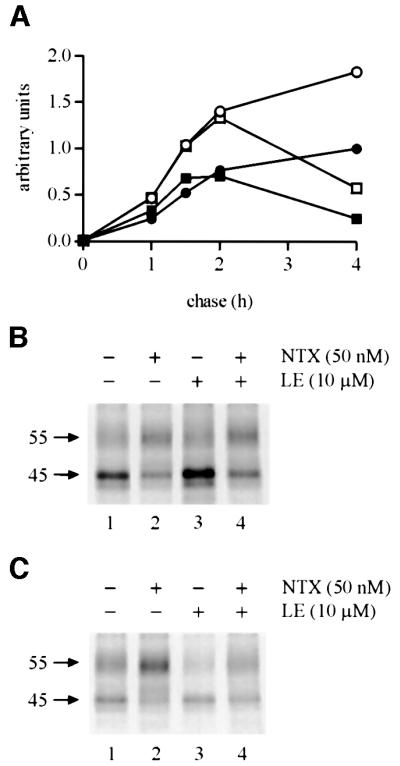
Fig. 2. The inability of a membrane-impermeable opioid ligand to block or mimic the effect of NTX on hδOR maturation. HEK-293S-hδOR-FLAG cells were labeled with [35S]methionine/cysteine and then chased for the indicated times in the absence (filled circles) or presence of 50 nM NTX (open circles), 10 µM LE (filled squares) or 50 nM NTX + 10 µM LE (open squares). Cellular membranes were solubilized and the receptors immunoprecipitated and analyzed as described in Figure 1. The graph in (A) describes the time course of appearance and disappearance of the mature Mr 55 000 receptor. Intensity of this species was obtained by densitometric scanning and the values were normalized to the maximum labeling at 4 h of chase in untreated cells. The fluorographs in (B) and (C) correspond to the samples at 2 and 4 h of chase, respectively.
As a second approach to assess the site of action of NTX, its effects on hδOR maturation were tested in cells treated or not with brefeldin A (BFA), a compound that blocks transport of newly synthesized proteins to the plasma membrane by causing collapse of the Golgi (Lippincott-Schwartz et al., 1989). Upon treatment with BFA, the Mr 45 000 receptor precursor was processed to an endo-β-N-acetylglucosaminidase H (Endo H)-resistant form that migrates as a Mr 46 000 protein (Figure 3A). As previously documented, this species contains fully processed O-linked but incompletely processed N-linked oligosaccharides (Petäjä-Repo et al., 2000). Cell surface biotinylation assay confirmed that the Mr 46 000 species, unlike the Mr 55 000 receptor species in untreated cells, was retained intracellularly and did not reach the cell surface (Figure 3B and C, compare lanes 1 and 3). As can be seen by comparing lanes 3 and 4 in Figure 3B, NTX promoted a dramatic increase in the amount of the Mr 46 000 receptor species in BFA-treated cells, confirming the fact that it functions as a pharmacological chaperone by promoting maturation of the receptor precursor in the ER. Interestingly, the NTX treatment that leads to an increase in the amount of the Mr 55 000 mature receptor species in the absence of BFA is accompanied by the appearance of a Mr 48 000 receptor form, which also reaches the cell surface (Figure 3C; see also Figures 1B, C and 6A). This form was found to be Endo H resistant (data not shown) and thus most likely represents a differentially glycosylated mature form of the receptor that is distinct from the ER-retained Mr 45 000 precursor.

Fig. 3. HδOR synthesis and maturation in BFA-treated HEK-293S-hδOR-FLAG cells. Cells were pulse-labeled with [35S]methionine/ cysteine and chased for the indicated times (A) or for 6 h (B, C). BFA (5 µg/ml) was added to the medium 60 min prior to labeling and NTX (10 µM) at the beginning of the chase. In (A), receptors were immunoprecipitated as described in Figure 1 and incubated for 16 h at 37°C in the absence or presence of 25 mU/ml of Endo H as indicated. In (B) and (C), cell surface proteins were biotinylated by incubating the intact metabolically labeled cells with sulfo-NHS-biotin (0.5 mg/ml) and the cellular membranes were isolated and solubilized in DDM. Receptors from one-quarter of the extract were immunoprecipitated using the immobilized anti-FLAG M2 antibody to isolate the total pool of labeled receptors (B). The rest of the extract was used to isolate the cell surface receptors by sequential precipitations using the immobilized streptavidin and anti-FLAG M2 antibody (C). The purified receptors were analyzed by SDS–PAGE and fluorography.

Fig. 6. Rescue of the cMyc-tagged wild-type hδOR and the D95A mutant by pharmacological chaperones. (A) Western blot analysis of the steady-state levels of the wild-type hδOR and the D95A mutant in stably transfected HEK-293S cells treated or not with 10 µM NTX for 24 h. Immunoblotting was carried out using the polyclonal anti-cMyc antibody following immunoprecipitation of the receptor with the immobilized monoclonal anti-cMyc antibody. (B) Maturation of the wild-type hδOR and the D95A mutant in BFA-treated cells. Stably transfected HEK-293S cells were pulse-labeled with [35S]methionine/cysteine and chased for 6 h in the absence (lane 1 and 3) or presence (lanes 2 and 4) of 10 µM NTX. BFA (5 µg/ml) was added to the medium 60 min prior to labeling. Receptors were isolated by immunoprecipitation using the immobilized monoclonal anti-cMyc antibody and analyzed as in Figure 1. (C) FACS analysis of the cell surface receptors. The HEK-293S cells stably expressing either the cMyc-tagged wild-type hδOR (upper panel) or the D95A mutant (lower panel) were treated for 24 h in the absence or presence of 10 µM of the following opioid ligands: naltriben (green), naltrindole (red), NTX (orange), naloxone (blue) or SNC-80 (yellow). The cell surface receptors were labeled with the monoclonal anti-cMyc antibody and quantitated by FACS as described in Materials and methods. The purple curve represents untreated cells; the background signal obtained in the absence of the primary antibody is shown in black. The values presented in the insets indicate the fold increase in the mean cell surface fluorescence promoted by each ligand and represent the mean ± SE of four independent experiments.
To determine whether the hδOR precursors that have been retained in the ER for an extended period of time could still be rescued by the pharmacological chaperone, NTX was added at 1, 2 or 3 h after the beginning of the chase period and the extent of receptor maturation assessed after 4 h of chase. A detectable increase in the amount of mature receptors could be observed even when the antagonist was added 3 h into the chase (Figure 4, compare lanes 1 and 2). In fact, there was almost no difference in the amount of mature receptors whether NTX was added immediately at the beginning of the chase or 2 h later (Figure 4, compare lanes 5 and 3, respectively). This suggests that the degradation rate of the incompletely folded precursors is relatively slow and that those receptors that remain in the ER maintain their capacity to be processed correctly.
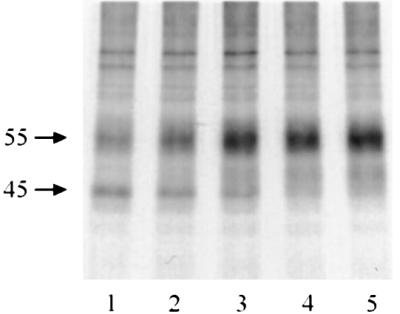
Fig. 4. Rescue of ER-retained hδOR precursors by NTX treatment. HEK-293S-hδOR-FLAG cells were pulse-labeled with [35S]methionine/cysteine and chased for 4 h in the absence (lane 1) or presence of 10 µM NTX (lanes 2–5). NTX was added to the chase medium at the beginning of the chase (lane 5), or at 1 h (lane 4), 2 h (lane 3) or 3 h (lane 2) after the beginning of the chase. Receptors were then isolated by immunoprecipitation and analyzed as described in Figure 1.
To assess whether the effect of NTX on hδOR maturation is unique to this opioid ligand, a series of antagonists were tested for their ability to promote receptor maturation in pulse–chase labeling experiments (Figure 5A). In each case, the amount of mature receptors was examined 6 h after the beginning of the chase period. Although to different extents, all of the non-peptidic opioid antagonists (alkaloids) promoted maturation of the receptor (Figure 5A, upper panel, lanes 2–5). When the peptidic antagonists were considered, only TICP(ψ) led to a consistent effect (Figure 5A, upper panel, lane 7). This peptide derivative contains a highly lipophilic cyclohexylalanine residue, and thus, unlike the other peptides, might be able to permeate cellular membranes. This notion is supported by the observation that TICP(ψ), like the alkaloids, also had an effect when receptor transport to the cell surface was inhibited by BFA (Figure 5A, lower panel, lane 7). The pharmacological selectivity was confirmed by the lack of effect shown by the β-adrenergic and V2R antagonists, propranolol and SR121463A, respectively (Figure 5A, lanes 9 and 10). The lack of effect of SR121463A is particularly important, since this compound was previously shown to rescue maturation and cell surface expression of ER-retained V2R mutants (Morello et al., 2000a). Reciprocally, NTX was without effect on the maturation of the V2R mutants (data not shown). Taken together, these data indicate that the pharmacological chaperone action of the compounds is linked to their ability to bind to the receptor.

Fig. 5. Both membrane-permeable opioid antagonists and agonists can function as pharmacological chaperones. HEK-293S-hδOR-FLAG cells were pulse-labeled with [35S]methionine/cysteine and chased for 6 h in the presence of 10 µM of the indicated opioid antagonists (A) or agonists (B). BFA (5 µg/ml) or the vehicle (–BFA) was added to the medium 60 min prior to labeling. Receptors were then isolated by immunoprecipitation and analyzed as described in Figure 1. The abbreviations used are: BUBU, Tyr-D-Ser[-O-C(CH3)3]-Gly-Phe-Leu-Thr-O-C(CH3)3; DPDPE, cyclic[D-Pen2,D-Pen5]enkephalin; ICI-174864, N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH; SNC-80, (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamidine; SR121463A, 1-[4-(N-tert-butylcarbamoyl)-2- methoxybenzenesulfonyl]-5-ethoxy-3-spiro-[4-(2-morpholinoethoxy)cyclohexane]indol-2-one, fumerate; TAN-67, 2-methyl-4aα-(3-hydroxyphenyl)-1,2,3,4,4a,5,12,12aα-octahydroquinolino-[2,3,3-g]isoquinoline); TICP(ψ), Tyr-Ticψ[CH2NH]Cha-Phe-OH; TIPP, Tyr-Tic-Phe-Phe-OH.
When opioid agonists were tested for their potential pharmacological chaperone action, only buprenorphine was found to have a significant positive effect when tested in control cells (Figure 5B, upper panel, lane 2). In contrast, all the non-peptidic agonists had a very dramatic effect in cells treated with BFA (Figure 5B, lower panel, lanes 2–7). This clearly indicates that an agonist-promoted down-regulatory process at the cell surface counterbalanced the effect exerted by these drugs on receptor maturation. In the BFA-treated cells, this negative regulatory process cannot hamper the pharmacological chaperone effect since the receptors are not transported to the cell surface. In the absence of BFA treatment, down-regulation of the mature Mr 55 000 receptor was most evident following treatment with TAN-67 and SNC-80 (Figure 5B, upper panel, lanes 3 and 4, respectively) that are full agonists for the hδOR (as assessed in the [35S]GTPγS binding assay; data not shown). Bupre norphine, which is a relatively weak partial agonist in the [35S]GTPγS binding assay, is expected to promote less down-regulation. This may explain why its pharmacological chaperone effects could be detected even in the absence of BFA.
As expected, none of the membrane impermeable peptidic agonists had a pharmacological chaperone effect, whether or not cells were treated with BFA (Figure 5B, lanes 8–10). However, their ability to promote down-regulation was readily apparent in control cells (Figure 5B, upper panel, lanes 8–10). Interestingly, when all the non-peptidic opioid ligands were considered together, a significant correlation (P = 0.002) was found between their pharmacological chaperone efficacy and their affinity (log Ki) [determined in competition binding assays against [125I]deltorphin II (data not shown)], indicating that the binding energy may play an important role in the pharmacological chaperone action of the compounds.
Several GPCR mutations have previously been shown to lead to retention of the mutant protein inside the cell where it is retained by the ER quality control (Kaushal and Khorana, 1994; Themmen and Huhtaniemi, 2000; Morello and Bichet, 2001). Given that we recently showed that V2R antagonists could rescue cell surface expression of ER-retained mutants (Morello et al., 2000a), the effects of the opioid pharmacological chaperones were assessed also on a mutant hδOR. The highly conserved aspartate in the transmembrane (TM) domain II of the hδOR was mutated to alanine and the resultant mutant, containing an N-terminal cMyc epitope (cMyc-D95A-hδOR), was stably transfected in HEK-293S cells. As can be seen in Figure 6A, the steady-state ratio of the mature Mr 55 000 receptor to the Mr 45 000 precursor was much smaller for the cMyc-D95A-hδOR than for the corresponding wild-type, as assessed by western blot (compare lanes 3 and 1, respectively), indicating that the mutation led to a significant ER retention of the protein. A 24 h treatment with NTX promoted a significant increase in the amount of the mature mutant Mr 55 000 receptor (Figure 6A, compare lanes 3 and 4). In fact, the extent of increase was even more dramatic for the mutant than for the wild-type cMyc-hδOR (3.9- and 1.9-fold elevation, respectively). A similar NTX-promoted increase in the cMyc-D95A-hδOR maturation was also observed in metabolic labeling experiments. As was the case for the wild-type hδOR-FLAG, the action of the antagonist was not inhibited by BFA treatment (Figure 6B, compare lanes 3 and 4), demonstrating that it acted intracellularly and promoted maturation of the mutant receptor precursor in the ER. This increase in maturation was accompanied by an elevation in cell surface expression, as assessed by fluorescence-assisted cell sorting (FACS) (Figure 6C). When a series of antagonists was tested, the relative efficiency of promoting cell surface expression of the cMyc-D95A-hδOR was indistinguishable from that of the wild-type receptor (Figure 6C, compare insets in lower and upper panel), consistent with the normal antagonist binding properties previously reported for this mutant (Bot et al., 1998). In contrast, although the agonist SNC-80 promoted a significant 55% loss in cell surface expression of the wild-type receptor (most likely as a result of down-regulation), its pharmacological chaperone action could be detected for the cMyc-D95A-hδOR, as it promoted a 1.3-fold increase in cell surface expression of the protein. This is consistent with the finding that the D95A mutation significantly alters the receptor signaling efficacy (Bot et al., 1998) and thus leads to reduced agonist-promoted down-regulation. For NTX, the effect of a 24 h treatment on the number of [3H]bremazocine binding sites was also assessed. In agreement with the biochemical and FACS data, the treatment promoted a 3-fold increase in binding sites in HEK-293S-cMyc-D95A-hδOR cells (1.33 ± 0.43 and 4.03 ± 1.5 pmol/mg in untreated and NTX-treated cells, respectively; n = 4).
Discussion
An increasing number of studies suggest that acquisition of the native conformation and export from the ER represent limiting factors for the expression of membrane-bound proteins (Kopito, 1999; Bermak et al., 2001). For the hδOR, we recently reported that only a fraction of the synthesized receptors is ultimately transported to the cell surface (Petäjä-Repo et al., 2000), the rest being targeted for degradation by the proteasomes (Petäjä-Repo et al., 2001). In the present study, we demonstrate that membrane-permeable opioid ligands are able to facilitate transport of newly synthesized receptors out of the ER and thus decrease the portion of precursors that would otherwise be targeted for degradation. We propose that these ligands act as pharmacological chaperones for the hδOR and, like molecular chaperones, interact with the nascent protein in the ER to minimize off-pathway interactions and increase the yield of the native correctly folded protein. Furthermore, we show that the receptor precursors retained in the ER represent fully competent folding intermediates and not permanently misfolded off-pathway products, and they remain in the ER because of limitations in the folding kinetics. Thus, direct regulation of receptor folding and trafficking represents a novel way by which receptor ligands could control cellular responsiveness.
Our hypothesis that opioid ligands act as pharmacological chaperones by binding to newly synthesized hδOR in the ER is supported by several lines of evidence. (i) The non-selective opioid antagonist, NTX, significantly increased the turnover rate of the Mr 45 000 receptor precursor, leading to a 2-fold increase in the processing of this species to the mature Mr 55 000 receptor. (ii) The NTX-mediated enhancement in receptor maturation was dose-dependent and the EC50 was similar to the estimated Ki for this ligand, indicating requirement for receptor occupancy. (iii) The antagonist was added to the chase medium following removal of the label, thus demonstrating that it mediates its effects post-translationally, ruling out the possibility of changes in protein synthesis. (iv) The NTX-promoted increase in the amount of the mature receptor occurred without any change in half-life of this receptor species, excluding the possibility that this effect could result from stabilization of the protein once it has reached the cell surface. (v) NTX was able to enhance receptor maturation even when protein transport to the cell surface was blocked by BFA and a membrane-impermeable peptidic opioid agonist, LE, at saturating concentration, was not able block its effects, confirming the intracellular site of action of the antagonist. (vi) In addition to NTX, a series of membrane-permeable opioid antagonists and agonists favored maturation and cell surface expression of the hδOR, suggesting that any ligand that is able to occupy the binding pocket of the newly synthesized receptor could act as a pharmacological chaperone. (vii) The chaperone action was found to be pharmacologically selective, since unrelated GPCR ligands, including SR121463A, a compound that was previously shown to promote rescue of ER-retained V2R mutants (Morello et al., 2000a), were unable to mimic the effects mediated by the opioid ligands. (viii) Reciprocally, NTX was unable to promote cell surface expression of the ER-retained V2R mutants, confirming the requirement for receptor binding.
The effects observed with the selective ligands in the present study are reminiscent of the action of other small molecules that were shown to increase export of newly synthesized proteins from the ER. These chemical chaperones include glycerol, trimethylamine-N-oxide and dimethyl sulfoxide (Brown et al., 1996; Sato et al., 1996; Tamarappoo and Verkman, 1998; Burrows et al., 2000). Agents that are known to bind specifically to ER-retained proteins and promote their export out of this cellular compartment have also been identified (Loo and Clarke, 1997; Fan et al., 1999; Wang and White, 1999; Zhou et al., 1999; Halaban et al., 2001). For example, Loo and Clarke (1997) reported that drug substrates and modulators of the P-glycoprotein allowed mutant forms of this ATP-dependent drug pump to attain its ER export-competent form. Likewise, the tyrosinase substrates, DOPA and tyrosine, were found to enhance maturation and trafficking of this enzyme (Halaban et al., 2001).
As has been suggested for both specific and non-specific chemical chaperones, the most likely mechanism by which opioid ligands could mediate their action is stabilization of the newly synthesized hδOR in the native or intermediate state of its folding pathway. Binding of a ligand may alter the thermodynamic equilibrium in favor of the stable correctly folded protein. This would then decrease the possibility of premature degradation, ultimately leading to an increase in the steady-state level of functional receptors at the cell surface. This hypothesis is consistent with the observed correlation between the magnitude of the enhancement in receptor maturation in response to a series of opioid ligands and their binding affinity (Ki).
The precise molecular mechanisms by which opioid ligands could enhance conformational stabilization of the newly synthesized hδOR are as yet unknown. Never theless, it can be hypothesized that binding of these compounds to the receptor may promote stable packing of its TM α-helices. According to the recently reported crystal structure of rhodopsin, the α-helical bundle of GPCRs is stabilized by several interhelical hydrogen bonds (Palczewski et al., 2000), an observation that is supported by abundant functional and structural data (Gether, 2000). Three-dimensional computer modeling and mutagenesis data have proposed that opioid ligands could establish direct hydrogen bonding with amino acid residues within the hydrophobic core formed by the TMs (Pogozheva et al., 1998; Quock et al., 1999). Thus, it is reasonable to speculate that binding of a ligand to the newly synthesized receptor might stabilize the labile protein by inducing additional conformational constraints within the α-helical bundle. Since all the tested membrane-permeable opioid ligands were found to enhance hδOR maturation, both the inactive receptor conformation, stabilized by antagonists or inverse agonists, and the active one(s), stabilized by agonists, appear to be recognized as export competent forms by the ER quality control.
Long-term antagonist and, in some cases, agonist treatments have previously been shown to increase the number of mutant or wild-type GPCRs, including opioid receptors, in vivo or in vitro (Zadina et al., 1995; Gether et al., 1997; Lee et al., 1997; Samama et al., 1997; Alewijnse et al., 2000; Wilson and Limbird, 2000; Wilson et al., 2001). Despite the abundant reported examples of ligand-promoted receptor up-regulation, the mechanism underlying this phenomenon has remained elusive and several possible explanations have been proposed. These include activation of cryptic receptors, decrease in receptor degradation, increase in receptor stability and in hibition of endogenous agonist-induced down-regulation. Although these different mechanisms may all contribute, our present results suggest that the pharmacological chape rone action of the drugs, involving enhanced processing of receptor precursors, is an important component in receptor up-regulation following chronic agonist or antagonist administration. It remains to be determined whether other GPCR antagonists and agonists, in addition to those that bind to δORs and V2Rs (Morello et al., 2000a), could act as pharmacological chaperones for their cognate receptors. One study supporting this possibility showed that addition of 11-cis-retinal into culture medium increased the amount of mature mutant rhodopsin molecules that were expressed at the plasma membrane of stably transfected HEK-293 cells (Li et al., 1998). Both this and our own studies were conducted in model cellular systems that do not truly recapitulate all the characteristics of cells endogenously expressing the receptors. Unfortunately, the tools currently available do not allow direct assessment of whether the pharmacological chaperone mode of action described herein also applies to neuronal cells endogenously expressing the hδOR.
Potential clinical implications of chemical and pharmacological chaperones have already been evoked in the case of diseases that result from the inappropriate ER retention of mutant proteins. For example, the fact that the ΔF508 mutant form of CFTR, which is normally unable to exit the ER, can be functionally rescued by treating cells with chemical chaperones (Brown et al., 1996; Sato et al., 1996) has obvious implications for cystic fibrosis patients. Similarly, patients that suffer from nephrogenic diabetes insipidus could benefit from treatment with pharmacological chaperones, since ER-retained mutant V2Rs that cause this disease were targeted to the cell surface as functional receptors upon exposure to V2R antagonists (Morello et al., 2000a). The present study indicates that the clinical implications of pharmacological chaperones might not be limited to conditions resulting from mutated genes. Indeed, modulating cell responsiveness to specific drugs by regulating the expression level of their targets could prove to be a useful therapeutic strategy. For proteins such as opioid receptors, pharmacological modulation of their responsiveness already has well known consequences in the management of pain. For instance, desensitization and down-regulation of these receptors, leading to tolerance and dependence, limit the chronic use of opioid analgesics in the clinical setting (Reisine and Pasternak, 1993). The pharmacological chaperone activity of opioid ligands may thus prove to be a useful parameter in the design of better therapeutic analgesics and may help to understand and predict the functional consequences that follow chronic treatment with these agents in vivo. For example, partial agonists could increase the sensitivity of the system not only by promoting less desensitization and down-regulation but also by increasing the number of receptors via their pharmacological chaperone action. For antagonists, one could predict that the beneficial effect that would be gained by increasing receptor density would be dampened by their antagonistic activity, which could even increase nociception. However, several studies have reported that low doses of NTX can augment opioid-induced analgesia and inhibit the development of tolerance (Shen and Crain, 1997; Powell et al., 2002). Whether these paradoxical effects could be in part mediated by the pharmacological chaperone action of NTX remains to be investigated.
In conclusion, we have demonstrated that opioid ligands can increase the efficiency with which wild-type and mutant hδORs are transported out of the ER. We propose that these ligands act as pharmacological chaperones and by stabilizing the correct native conformation of the receptor precursor enable the newly synthesized proteins to leave the ER more efficiently. In addition to providing important insight into the post-translational mechanisms that regulate folding and ER export of GPCRs, the present study describes a novel mechanism by which agonists and antagonists might mediate changes in receptor density and cell responsiveness to drugs.
Materials and methods
Materials
EXPRE35S35S protein labeling mix (1175 Ci/mmol) and [9-3H(N)]-bremazocine (26.6 Ci/mmol) were purchased from PerkinElmer LifeSciences and [125I]deltorphin II (2200 Ci/mmol) was synthesized as described previously (Fraser et al., 1999). Endo H was from Roche Molecular Biochemicals and cell culture reagents either from Life Technologies or Wisent. The anti-FLAG M2 antibody and the FLAG peptide were from Sigma. Anti-cMyc antibodies were purchased from Santa Cruz and the phycoerythrin-conjugated goat anti-mouse IgG from Immunotech. Protein G–Sepharose was from Amersham Pharmacia Biotech and BFA from Calbiochem. EZ-linkedTM sulfo-N-hydroxysuccinimide (NHS)–biotin and immunopure® immobilized streptavidin were obtained from Pierce. The opioid agonists and antagonists were from Sigma, Tocris or Neosystem, or were generously provided by Dr C.Serradeil-LeGal (Sanofi Research, Toulouse Cedex, France; SR121463A) or Dr P.Schiller [Clinical Research Institute of Montreal, Montreal, Canada; TICP(ψ), TIPP]. All the other reagents were of analytical grade and obtained from various commercial suppliers.
DNA constructs and site-directed mutagenesis
The hδOR cDNA was subcloned into the pcDNA3 expression vector (Invitrogen) as described previously (Valiquette et al., 1996). The receptor was tagged either at the C-terminal end with the FLAG epitope (DYKDDDDK) or at the N-terminal end with the cMyc epitope (MEQKLISEEDLNA) using the Clontech site-directed mutagenesis kit, which is based on the method of Deng and Nickoloff (1992). The same technique was also used to create the D95A mutant of the N-terminally tagged receptor. The constructs were verified and confirmed by restriction enzyme mapping and DNA sequencing.
Cell culture and transfections
HEK-293S cells were grown and maintained as described previously (Petäjä-Repo et al., 2000). Cells were transfected by a modified calcium-phosphate precipitation method (Chen and Okayama, 1987) and stable clones (hδOR-FLAG: 10.5 pmol; cMyc-hδOR: 2.6 pmol and cMyc-D95A-hδOR: 1.3 pmol of receptor/mg of membrane protein) were selected using 400 µg/ml of geneticin.
Radioligand binding assays
Saturation and competition binding assays were carried out as described previously (Fraser et al., 1999; Petäjä-Repo et al., 2000).
Metabolic labeling with [35S]methionine/cysteine
Metabolic pulse–chase labeling was performed as described previously (Petäjä-Repo et al., 2000). When labeling was performed in the presence of BFA, the reagent was added 60 min before the pulse labeling. The opioid ligands were added at the beginning (Figures 1–3, 5 and 6) or at the indicated time (Figure 4) during the chase at final concentrations specified in the figures and the text.
Immunoprecipitation of the FLAG- and cMyc-tagged hδORs
Crude membranes were prepared as described previously (Petäjä-Repo et al., 2000). Briefly, cells were sonicated in buffer A (25 mM Tris–HCl pH 7.4, 2 mM EDTA, 5 µg/ml leupeptin, 5 µg/ml soybean trypsin inhibitor, 10 µg/ml benzamidine, 2 µg/ml aprotinin, 0.5 mM phenylmethylsulfonyl fluoride and 2 mM 1,10-phenantroline) and centrifuged in a Beckman L8-70M ultracentrifuge at 27 000 g for 20 min. For cells expressing the cMyc-tagged receptor, the buffer also contained 20 mM N-ethylmaleimide. Membranes were solubilized in buffer A containing 0.5% n-dodecyl-β-d-maltoside (DDM) (w/v) and 140 mM NaCl for 60 min. After centrifugation in a Beckman L8-70M ultracentrifuge at 100 000 g for 60 min, the FLAG-tagged receptor was immunoprecipitated from the supernatant fraction using immobilized anti-FLAG M2 antibody as described previously (Petäjä-Repo et al., 2000), while the cMyc-tagged receptors were purified by a two-step immunoprecipitation (Petäjä-Repo et al., 2001) using immobilized anti-cMyc-antibody (9E10).
Biotinylation and isolation of cell surface receptors
Cell surface proteins were biotinylated and isolated using immobilized streptavidin as described previously (Petäjä-Repo et al., 2000); receptors were purified by immunoprecipitation as described above.
Deglycosylation of the hδOR
The receptors were deglycosylated following elution from the immobilized anti-FLAG M2 or the anti-cMyc antibodies as described previously (Petäjä-Repo et al., 2000) using Endo H at a final concentration of 25 mU/ml.
SDS–PAGE and western blotting
For SDS–PAGE (10% separating gels), samples were denatured by heating at 95°C for 2 min in the absence (cMyc-epitope tagged hδOR) or presence (FLAG-epitope tagged hδOR) of 50 mM dithiothreitol. For detection of radioactivity, the gels were treated with En3hance® (PerkinElmer LifeSciences) according to the manufacturer’s instructions, dried and exposed at –80°C for 1–15 days, using the Biomax MR film and intensifying screens (Kodak). For western blotting, the proteins resolved in SDS–PAGE were transferred electrophoretically to Immobilon P membrane (Millipore) and the bound proteins were probed using the polyclonal anti-cMyc antibody as described previously (Petäjä-Repo et al., 2000). The relative intensities of the bands on the autoradiograms were analyzed by densitometric scanning with Agfa Arcus II lazer scanner and the data quantified using NIH image software version 1.61, substracting the local background from each lane.
FACS analysis
The HEK-293S cells stably transfected with the cMyc-hδOR or the cMyc-D95A-hδOR cDNAs were subcultured in six-well culture plates, grown to ∼70% confluency and treated or not with opioid ligands (10 µM) for 24 h, as specified in Figure 6. The cells were then prepared for FACS analysis as described previously (Morello et al., 2000a).
Acknowledgments
Acknowledgements
We are grateful to Dr Manon Valiquette and Huy Vu for generating and providing us the hδOR constructs for the cMyc-tagged wild type and D95A mutant. We are also indebted to Dr Kemal Payza and Stephane St-Onge for fruitful discussions and for providing the data on the pharmacological characterization of the opioid ligands (Ki values), and to Dr Sultan Ahmad for the critical reading of the manuscript. We thank Dr Peter Schiller for the generous gift of TIPP and TICP(ψ), and Dr C.Serradeil-LeGal for SR121463A. This work was supported by a grant from the Canadian Institute for Health Research (to M.B.). M.B. is the holder of the Hans Selye Chair in Molecular and Cell Biology and holds a Canada Research Chair in Molecular Pharmacology. U.E.P.-R was supported by grants from the Ella and Georg Ehrnrooth Foundation, the Helsinki University Pharmacy and the Oulu University Scholarship Foundation.
References
- Alewijnse A.E., Timmerman,H., Jacobs,E.H., Smit,M.J., Roovers,E., Cotecchia,S. and Leurs,R. (2000) The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H2 receptor. Mol. Pharmacol., 57, 890–898. [PubMed] [Google Scholar]
- Aridor M. and Balch,W.E. (1999) Integration of endoplasmic reticulum signaling in health and disease. Nature Med., 5, 745–751. [DOI] [PubMed] [Google Scholar]
- Bermak J.C., Li,M., Bullock,C. and Zhou,Q.-Y. (2001) Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nature Cell Biol., 3, 492–498. [DOI] [PubMed] [Google Scholar]
- Bot G., Blake,A.D., Li,S. and Reisine,T. (1998) Mutagenesis of the mouse delta opioid receptor converts (–)-buprenorphine from a partial agonist to an antagonist. J. Pharmacol. Exp. Ther., 284, 283–290. [PubMed] [Google Scholar]
- Brown C.R., Hong-Brown,L.Q., Biwersi,J., Verkman,A.S. and Welch,W.J. (1996) Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones, 1, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J.A.J., Willis,L.K. and Perlmutter,D.H. (2000) Chemical chaperones mediate increased secretion of mutant α1-antitrypsin (α1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc. Natl Acad. Sci. USA, 97, 1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. and Okayama,H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol., 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W.P. and Nickoloff,J.A. (1992) Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem., 200, 81–88. [DOI] [PubMed] [Google Scholar]
- Ellgaard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Fan J.-Q., Ishii,S., Asano,N. and Suzuki,Y. (1999) Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nature Med., 5, 112–115. [DOI] [PubMed] [Google Scholar]
- Fraser G.L., Labarre,M., Godbout,C., Butterworth,J., Clarke,P.B.S., Payza,K. and Schmidt,R. (1999) Characterization of [125I]AR-M100613, a high-affinity radioligand for δ opioid receptors. Peptides, 20, 1327–1335. [DOI] [PubMed] [Google Scholar]
- Gether U. (2000) Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev., 21, 90–113. [DOI] [PubMed] [Google Scholar]
- Gether U., Ballesteros,J.A., Seifert,R., Sanders-Bush,E., Weinstein,H. and Kobilka,B.K. (1997) Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J. Biol. Chem., 272, 2587–2590. [DOI] [PubMed] [Google Scholar]
- Halaban R., Cheng,E., Svedine,S., Aron,R. and Hebert,D.N. (2001) Proper folding and endoplasmic reticulum to Golgi transport of tyrosinase are induced by substrates, DOPA and tyrosinase. J. Biol. Chem., 276, 11933–11938. [DOI] [PubMed] [Google Scholar]
- Kaushal S. and Khorana,H.G. (1994) Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry, 33, 6121–6128. [DOI] [PubMed] [Google Scholar]
- Kim P.S. and Arvan,P. (1998) Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev., 19, 173–202. [DOI] [PubMed] [Google Scholar]
- Kopito R.R. (1999) Biosynthesis and degradation of CFTR. Physiol. Rev., 79, S167–S173. [DOI] [PubMed] [Google Scholar]
- Lee T.W., Cotecchia,S. and Milligan,G. (1997) Up-regulation of the levels of expression and function of a constitutively active mutant of the hamster α1B-adrenoceptor by ligands that act as inverse agonists. Biochem. J., 325, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Sandberg,M.A., Pawlyk,B.S., Rosner,B., Hayes,K.C., Dryja,T.P. and Berson,E.L. (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17→methionine and proline-347→serine in transgenic mice and in cell cultures. Proc. Natl Acad. Sci. USA, 95, 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Yuan,L.C., Bonifacino,J.S. and Klausner,R.D. (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell, 56, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T.W. and Clarke,D.M. (1997) Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem., 272, 709–712. [DOI] [PubMed] [Google Scholar]
- Morello J.-P. and Bichet,D.G. (2001) Nephrogenic diabetes insipidus. Annu. Rev. Physiol., 63, 607–630. [DOI] [PubMed] [Google Scholar]
- Morello J.-P. et al. (2000a) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest., 105, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello J.-P., Petäjä-Repo,U.E., Bichet,D.G. and Bouvier,M. (2000b) Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol. Sci., 21, 466–469. [DOI] [PubMed] [Google Scholar]
- Palczewski K. et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Petäjä-Repo U.E., Hogue,M., Laperrière,A., Walker,P. and Bouvier,M. (2000) Export from the endoplasmic resticulum represents the limiting step in the maturation and cell surface expression of the human δ opioid receptor. J. Biol. Chem., 275, 13727–13736. [DOI] [PubMed] [Google Scholar]
- Petäjä-Repo U.E., Hogue,M., Laperrière,A., Bhalla,S., Walker,P. and Bouvier,M. (2001) Newly synthesized human δ opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated and degraded by the proteasome. J. Biol. Chem., 276, 4416–4423. [DOI] [PubMed] [Google Scholar]
- Pogozheva I.D., Lomize,A.L. and Mosberg,H.I. (1998) Opioid receptor three-dimensional structures from distance geometry calculations with hydrogen bonding constrains. Biophys. J., 75, 612–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell K.J., Abul-Husn,N.S., Jhamandas,A., Olmstead,M.C., Beninger,R.J. and Jhamandas,K. (2002) Paradoxical effects of the opioid antagonist naltrexone on morphine analgesia, tolerance and reward in rats. J. Pharmacol. Exp. Ther., 300, 588–596. [DOI] [PubMed] [Google Scholar]
- Quock R.M. et al. (1999) The δ-opioid receptor: molecular pharmacology, signal transduction and the determination of drug efficacy. Pharmacol. Rev., 51, 503–532. [PubMed] [Google Scholar]
- Reisine T. and Pasternak,G. (1993) Opioid analgesics and antagonists. In Hardman,J.G., Limbird,L.E., Molinoff,P.B., Ruddon,R.W. and Gilman,A.G. (eds), The Pharmacological Basis of Therapeutics. McGraw-Hill, New York, NY, pp. 521–555.
- Samama P., Bond,R.A., Rockman,H.A., Milano,C.A. and Lefkowitz,R.J. (1997) Ligand-induced overexpression of a constitutively active β2-adrenergic receptor: pharmacological creation of a phenotype in transgenic mice. Proc. Natl Acad. Sci. USA, 94, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Ward,C.L., Krouse,M.E., Wine,J.J. and Kopito,R.R. (1996) Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J. Biol. Chem., 271, 635–638. [DOI] [PubMed] [Google Scholar]
- Schubert U., Antón,L.C., Gibbs,J., Norbury,C.C., Yewdell,J.W. and Bennink,J.R. (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature, 404, 770–774. [DOI] [PubMed] [Google Scholar]
- Shen K.F. and Crain,S.M. (1997) Ultra-low doses of naltrexone or etorphine increase morphine’s antinociceptive potency and attenuate tolerance/dependence in mice. Brain Res., 757, 176–190. [DOI] [PubMed] [Google Scholar]
- Tamarappoo B.K. and Verkman,A.S. (1998) Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Invest., 101, 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themmen A.P.N. and Huhtaniemi,I.T. (2000) Mutations of gonado tropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr. Rev., 21, 551–583. [DOI] [PubMed] [Google Scholar]
- Valiquette M., Vu,H.K., Yue,S.Y., Wahlestedt,C. and Walker,P. (1996) Involvement of Trp-284, Val-296 and Val-297 of the human δ-opioid receptor in binding of δ-selective ligands. J. Biol. Chem., 271, 18789–18796. [DOI] [PubMed] [Google Scholar]
- Wang J. and White,A.L. (1999) 6-aminohexanoic acid as a chemical chaperone for apolipoprotein(a). J. Biol. Chem., 274, 12883–12889. [DOI] [PubMed] [Google Scholar]
- Wilson M.H. and Limbird,L.E. (2000) Mechanisms regulating the cell surface residence time of the α2A-adrenergic receptor. Biochemistry, 39, 693–700. [DOI] [PubMed] [Google Scholar]
- Wilson M.H., Highfield,H.A. and Limbird,L.E. (2001) The role of a conserved inter-transmembrane domain interface in regulating α2a-adrenergic receptor conformational stability and cell-surface turnover. Mol. Pharmacol., 59, 929–938. [DOI] [PubMed] [Google Scholar]
- Zadina J.E., Kastin,A.J., Harrison,L.M., Ge,L.-J. and Chang,S.L. (1995) Opiate receptor changes after chronic exposure to agonists and antagonists. Ann. N.Y. Acad. Sci., 757, 353–361. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Gong,Q. and January,C.T. (1999) Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem., 274, 31123–31126. [DOI] [PubMed] [Google Scholar]