

Abstract
Thioredoxin-2 (Trx-2) is a mitochondria-specific member of the thioredoxin superfamily. Mitochondria have a crucial role in the signal transduction for apoptosis. To investigate the biological significance of Trx-2, we cloned chicken TRX-2 cDNA and generated clones of the conditional Trx-2-deficient cells using chicken B-cell line, DT40. Here we show that TRX-2 is an essential gene and that Trx-2-deficient cells undergo apoptosis upon repression of the TRX-2 transgene, showing an accumulation of intracellular reactive oxygen species (ROS). Cytochrome c is released from mitochondria, while caspase-9 and caspase-3, but not caspase-8, are activated upon inhibition of the TRX-2 transgene. In addition, Trx-2 and cytochrome c are co-immunoprecipitated in an in vitro assay. These results suggest that mitochondrial Trx-2 is essential for cell viability, playing a crucial role in the scavenging ROS in mitochondria and regulating the mitochondrial apoptosis signaling pathway.
Keywords: apoptosis/cytochrome c/mitochondria/redox/thioredoxin-2
Introduction
Thioredoxin (Trx) is a small, multi-functional protein that has a redox-active disulfide/dithiol group within the conserved active site sequence Cys–Gly–Pro–Cys. Reduced Trx catalyzes the reduction of disulfide bonds in multiple substrate proteins, and oxidized Trx is reversibly reduced by the action of Trx reductase and NADPH (Holmgren, 1985). As Trx-dependent peroxidase was identified as peroxiredoxin, the Trx/peroxiredoxin system is considered to act as an endogenous antioxidant system in addition to the glutathione (GSH)/GSH peroxidase (GPx) system (Chae et al., 1994a,b). Trx is widely present in prokaryotes and eukaryotes, and appears to be ubiquitous in essentially all living cells (Holmgren, 1985). During our study of adult T-cell leukemia (ATL) transformed by retrovirus human T-cell leukemia virus type-I (HTLV-I), we purified an active component as ADF (ATL-derived factor) (Teshigawara et al., 1985). After cloning, we then identified ADF as human thioredoxin (Tagaya et al., 1989), and reported that human Trx plays important roles in the redox regulation of signal transduction and in cytoprotection against oxidative stress (Yodoi and Uchiyama, 1992; Nakamura et al., 1997). In a previous study, we have shown that homozygotes carrying a targeted disruption of the mouse TRX gene die shortly after implantation, suggesting that TRX is essential for early differentiation and morphogenesis of the mouse embryo (Matsui et al., 1996).
Mammalian thioredoxin 2 (TRX-2) gene was cloned and its product was shown to localize in the mitochondria (Spyrou et al., 1997). All previously described thioredoxins in mammals have two to three cysteine residues in addition to the two located in the active site. Oxidation of these structural cysteine residues leads to a loss of its enzymatic activity (Holmgren and Bjornstedt, 1995). In contrast, Trx-2 is more resistant to oxidative stress since it lacks the corresponding structural cysteine residues, as is the case with prokaryotic Trx (Spyrou et al., 1997).
The mitochondrion is the major physiological source of reactive oxygen species (ROS) generated during respiration. Therefore, the production of ROS in mitochondria is strictly regulated by mitochondrial antioxidant systems. Recent studies have shown that mitochondrial Trx reductase, Trx-2, and mitochondrial thioredoxin peroxidase (peroxiredoxin III), which was originally identified as MER5 (Yamamoto et al., 1989) or SP-22 (Watabe et al., 1997; Araki et al., 1999), compose a mitochondrial antioxidant system.
To study the function of Trx-2 in vivo, we cloned chicken TRX-2 cDNA and generated conditional Trx-2-deficient cells expressing a tetracycline (tet)-repressible TRX-2 transgene (TRX-2–/–/Trx-2 cells), using the hyper-recombinogenic chicken B-cell line DT 40 (Buerstedde and Takeda, 1991). Upon the inhibition of the TRX-2 transgene by doxycycline (dox), which is a tetracycline derivative, TRX-2–/–/Trx-2 cells showed an increased accumulation of intracellular ROS, while the total and mitochondrial GSH levels decreased. Trx-2-deficient cells fall into apoptosis and Trx-2-deficient cells treated with buthionine–sulfoximine (BSO) augmented cell death, possibly by increasing the number of necrotic cells. Interestingly, Trx-2 and cytochrome c are co-immunoprecipitated in in vitro assay. These results suggest that Trx-2 regulates the generation of ROS through the Trx-2/Prx system in mitochondria and plays a crucial role in the mitochondrial apoptosis-signaling pathway.
Results
Isolation of chicken TRX-2 cDNA
A cDNA fragment encoding a chicken TRX-2 homolog was isolated by RT–PCR from chicken testes RNA. An open reading frame (ORF) in the cDNA encoded a protein consisting of 151 amino acids, which contained a mitochondrial translocation signal peptide at the N-terminus, as shown in Figure 1. Although the length of this targeting signal peptide in chicken Trx-2 is shorter than that in the mammalian one, the consensus motif for the cleavage by mitochondria signal peptide protease (Hendrick et al., 1989) was conserved in chicken Trx-2. Matured chicken Trx-2 is highly homologous (91.6% identical) to the human counterpart. Southern blot analysis of BamHI-digested chicken genomic DNA showed a single band hybridizing to a chicken TRX-2 cDNA probe containing the entire ORF (data not shown), indicating that there is a single TRX-2 gene locus in the chicken genome.

Fig. 1. Alignment of the predicted amino acid sequences of chicken, human, mouse and rat Trx-2. The vertical arrow shows the probable mitochondrial translocation signal peptide cleavage site, similar to the consensus motif for the cleavage site (open box), based on the two- protease model (Hendrick et al., 1989). The active sites are shown in black boxes.
Deletion of TRX-2 results in cell death
To reveal the biological function of Trx-2, we utilized the strategy of gene disruption in the chicken B lymphocyte cell line, DT40. TRX-2 genomic fragments were isolated by long-range PCR based on the chicken TRX-2 cDNA sequences and were partially sequenced to determine the position of exons. To make TRX-2 disruption constructs, TRX-2–BsrR (the gene for blasticidin-S resistance) and TRX-2–HisR (the gene for histidinol resistance) were inserted between 2 and 5 kb from the TRX-2 locus (Figure 2A). Targeted integration of this genomic DNA was expected to disrupt the active site of Trx-2 and targeting events were defined by the presence of a 5 kb band after Southern blot analysis of BamHI-digested genomic DNA hybridized to an external probe (Figure 2A and B). DT40 cells were transfected with the TRX-2–BsrR construct and 17 blasticidin-resistant clones were isolated and screened for homologous recombination by Southern blotting (Table I). Hybridization with the probe showed that BamHI-digested DNA from five of the 17 clones gave rise to a new 5-kb band, the size expected following homologous recombination (Figure 2B). These results indicate that targeted disruption of one allele of the TRX-2 gene occurred at a frequency of ∼30%. One of the five heterozygous clones that were transfected using the TRX-2–BsrR construct was transfected with the TRX-2–HisR construct and selected in medium containing both blasticidin-S and histidinol. DNAs from 83 resistant clones were analyzed by Southern blotting. The results showed that none of them underwent homologous recombination.

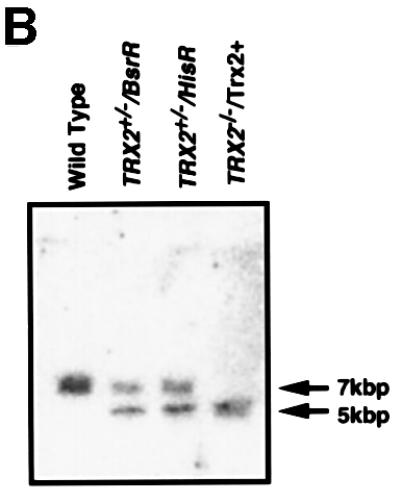
Fig. 2. (A) Schematic representation of part of the TRX-2 locus, the gene disruption constructs, TRX-2–HisR, TRX-2–BsrR, and the configuration of the targeted loci. Open boxes indicate the exon positions. The relevant BamHI restriction enzyme sites are labeled ‘B’. (B) Generation of a TRX-2–/– clone. Southern blot analysis of wild-type (TRX-2+/+) and two heterozygous mutants derived from targeting with TRX-2–BsrR construct (TRX-2+/–/BsrR) or with TRX-2–HisR construct (TRX-2+/–/HisR), and homozygous mutant (TRX-2–/–) clones. BamHI- digested genomic DNA was hybridized with the probe DNA (black band).
Table I. Frequencies of homologous recombination in TRX-2–HisR- and TRX-2–BsrR-transfected wild-type and heterozygous DT40 cells.
| Transfected vector | TRX-2–HisR | TRX-2–BsrR |
|---|---|---|
| Wild-type DT40 cells | 12/35 (34%) | 5/17 (29%) |
| Heterozygous DT40 cells | 0/83a (0%) | 0/63b (0%) |
aHeterozygous DT40 cells with one TRX-2 allele disrupted with TRX-2–BsrR were transfected.
bHeterozygous DT40 cells with one TRX-2 allele disrupted with TRX-2–HisR were transfected.
DT40 cells were transfected with the indicated plasmids and drug-resistant clones were analyzed for homologous integration by Southern blotting.
To confirm that the TRX-2–HisR construct was capable of homologous recombination, we transferred wild-type DT40 cells with this construct. Twelve heterozygous clones with one TRX-2 allele disrupted with the TRX-2–HisR construct were isolated from 35 histidinol-resistant clones. Moreover, heterozygous DT40 cells with one allele disrupted by TRX-2–HisR construct were transfected with TRX-2–BsrR construct. None of 63 resistant clones underwent homologous recombination. We were unable to isolate any homozygous TRX-2–/– mutant clones. This result suggested that TRX-2 is an essential gene in DT40 cells.
Proliferation properties of Trx-2-deficient cells
To investigate the cause of cell death, we tried to generate a conditional Trx-2-deficient cell line in which the Trx-2 transgene is controlled by a tetracycline (tet)-repressible promoter. The amount of Trx-2 protein expressed from the transgene was examined by western blot analysis, using anti-Trx-2 polyclonal antibody. This exogenous Trx-2 protein was expressed 6-fold more than endogenous Trx-2 (data not shown). The exogenous Trx-2 was reduced to one-eighth at day 1 after treatment with doxycycline (dox; a tetracycline derivative). It was hardly detected at day 3, subsequently becoming undetectable at day 5 (Figure 3A). The half-life of Trx-2 was ∼8 h. The localization of Trx-2 expressed from the transgene was also confirmed by immunofluoresence with anti-Trx-2 polyclonal antibody. Confocal microscope images showed that the Trx-2 protein expressed from the transgene was localized in the mitochondria as in the wild-type cells (Figure 3B). The proliferative properties of TRX-2–/– cells after the addition of dox were monitored using growth curves. The growth curve of TRX-2–/– cells cultured without dox was indistinguishable from that of wild-type cells (Figure 3C). In contrast, after the addition of dox, cell proliferation was delayed at day 4, and subsequently ceased after day 5, in the TRX-2–/– cells. Dox had no effect on the growth or viability of wild-type cells (data not shown). These results suggested that the exogenously expressed transgene was capable of supporting the viability of TRX-2–/–/Trx-2 cells.
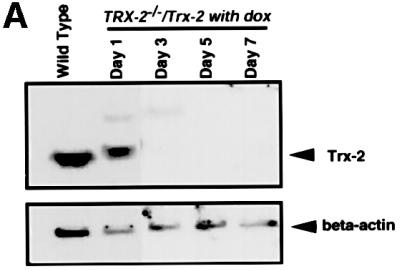


Fig. 3. (A) Western blotting analysis of chicken Trx-2 protein expressed in wild-type DT40 (wild type) and TRX-2–/– cells carrying the chicken TRX-2 transgene (TRX-2–/–/Trx-2) at days 1, 3, 5 and 7 after the addition of dox. Whole-cell lysates of wild-type cells (40 µg), and TRX-2–/–/Trx-2 day-1, -3, -5 and -7 cells (40 µg) were separated by SDS–PAGE (15% polyacrylamide) and transferred to a filter. Trx-2 was visualized by incubation with mouse anti-Trx-2 polyclonal antibody and subsequent incubation with labeled secondary antibody. (B) Immunofluorescence double staining using the MitoTracker Green® and anti-Trx-2 antibody to detect TRX-2 protein expressed from the transgene or wild type. Wild-type cells were stained with mouse anti-Trx-2 polyclonal antibody together with a labeled secondary antibody (a) and the MitoTracker Green® (b). The merged image is shown in (c). The same experiment was carried out in TRX-2–/–/Trx-2 cells on day 0 (d–f) and day 5 (g–i) after addition of dox with mouse anti-Trx-2 polyclonal (d and g) and the MitoTracker Green® (e and h); the merged images are shown in (f) and (i), respectively. (C) TRX-2–/–/Trx-2 cells cultured in the absence (open squares) or presence (closed squares) of dox. The numbers of cells not stained with Trypan Blue were counted. Wild-type DT40 cells are represented by closed circles. The data shown are the average results from three separate experiments.
Increased intracellular ROS in Trx-2-deficient cells
The mitochondria are a major source of ROS generation and the Trx-2/Prx-III system is a mitochondria-specific antioxidant system. The malfunction of the antioxidant system in mitochondria may lead to an increase of intracellular ROS. We determined intracellular accumulation of ROS by flow cytometric analysis with 2′,7′-dichlorofluorescein diacetate (DCFH-DA), and found the level of intracellular ROS was increased (2- to 3-fold) in TRX-2–/–/Trx-2 cells at day 5 after dox treatment compared with the untreated cells (Figure 4A). Since the GSH/GPx system plays a major role as an antioxidant in mitochondria as well as in the cytoplasm, we investigated the compensatory effect of the GSH/GPx system in TRX-2–/–/Trx-2 cells at day 5 after dox treatment. Total and mitochondrial GSH levels were separately measured in TRX-2–/–/Trx-2 cells after dox treatment and were found to decrease by one-half and one-seventh, respectively, compared with control cells (Figure 4B). Moreover, cell injury was induced when the GSH system was inhibited by the addition of BSO to the medium (Figure 4C). The ratio of propidium iodide (PI)-positive dead cells in TRX-2–/–/Trx-2 cells at day 3, after dox treatment, increased from 16.0 to 39.0% due to the addition of BSO. In contrast, the one in untreated TRX-2–/–/Trx-2 cells slightly increased (from 9.3 to 11.5%). These results suggest that the deficiency of Trx-2 leads to an increase of intracellular ROS, but that the majority of ROS accumulation is suppressed by the compensatory effect of the GSH/GPx system.
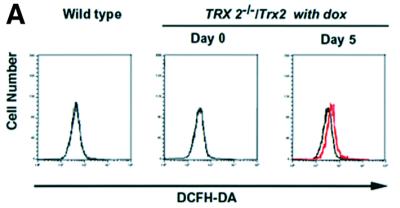


Fig. 4. (A) Flow cytometric analysis of intracellular redox state estimated by the level of intracellular ROS. The cells were cultured in medium with 5 µM DCF-DA for 15 min at 39°C. After washing the cells twice with PBS, the fluorescence intensity was measured by flow cytometry. The generation of ROS in the TRX-2–/–/Trx-2 cells at day 5 after dox treatment (Day 5, red line) was slightly increased compared with that in the untreated cells (Day 0, black line). (B) Glutathione level analysis. Shown are levels of total and mitochondrial glutathione in TRX-2–/–/Trx-2 cells, which were cultured for 5 days with dox (Day 5). Total and mitochondrial GSH levels at day 5 were found to be decreased by one-half and one-seventh, respectively, compared with control cells (Day 0). (C) Flow cytometric analysis of cell death after BSO treatment. TRX-2–/–/Trx-2 cells were cultured with 0.5 mM BSO for 12 h after the addition of BSO, for 3 days after dox treatment (Day 3) or not (Day 0), and the expression of phospholipid phosphatidylserine was analyzed with Annexin V–FITC. The necrotic cells were stained by PI.
Deficiency of Trx-2 promotes apoptosis
It is known that ROS act as mediators of apoptosis, and therefore we investigated whether the suppression of Trx-2 induces apoptosis. To examine the apoptotic properties, we analyzed TRX-2–/–/Trx-2 cells cultured with dox on a flow cytometer after double staining with PI and Annexin-V–FITC (fluorescein isothiocyanate) (Figure 5A). The proportion of the TRX-2–/–/Trx-2 cells undergoing apoptosis (Annexin-V–FITC-positive and PI-negative) was 10.0% at day 0, 14.2% at day 3, 24.6% at day 5 and 45.3% at day 7, after treatment with dox. Flow cytometric analysis was performed using PhiPhiLux G1D2 (OncoImmunin, Inc., Gaithersburg, MD) for detection of caspase-3 protease activity (Figure 5B). Caspase-3 activity was increased in TRX-2–/–/Trx-2 cells at day 5, after the addition of dox, compared with untreated cells. These results strongly suggest that a deficiency of Trx-2 promotes apoptosis, implying that the deficiency of Trx-2 transduces the positive signal for apoptosis. Therefore, we investigated caspase activity for the signal transduction of apoptosis in Trx-2-deficient cells. Checking the activity of caspase-8 and caspase-9 is a useful tool to examine whether apoptosis occurs via the mitochondrial pathway or not. In the mitochondrial apoptosis pathway, cytochrome c, released from the mitochondria into the cytoplasm, forms the ‘apoptosome’, which is composed of cytochrome c, Apaf-1 and procaspase-9. This then results in the activation of caspase-9, which in turn induces the activation of death-effector caspases such as caspase-3 to orchestrate the biochemical execution of cells. We investigated the activity of caspase-8 and -9 in TRX-2–/–/Trx-2 cells during culture with dox. Caspase-9 activity significantly increased at day 3 and day 5 after the addition of dox (Figure 6A). In contrast, no significant increase in caspase-8 activity was observed (Figure 6B). Moreover, Trx-2 deficiency resulted in the increase of cytochrome c in the cytoplasmic fraction (Figure 6C), suggesting that cytochrome c was released from mitochondria into the cytoplasm in TRX-2–/–/Trx-2 cells at day 3 after dox treatment without stimuli. We then measured mitochondria membrane potential of TRX-2–/–/Trx-2 cells at day 0, day 3 or day 5 after dox treatment using 3,3′-dihexyloxacarbocyanine iodide (DiCO6). These cells were incubated with the caspase inhibitor zVAD-fmk in order to exclude the influence of caspase on mitochondria membrane potential. The mitochondria membrane potential of Trx-2-deficient cells was maintained (Figure 6D). Since the activation of caspase-9 and the release of cytochrome c from the mitochondria indicates mitochondrial involvement in apoptotic signaling, these results indicate that dox-treated TRX-2–/–/Trx-2 cells undergo apoptosis via the mitochondrial pathway. In addition, the disruption of mitochondria membrane potential does not seem to be involved in the mechanism of the mitochondrial apoptosis pathway observed in Trx-2-deficient cells.


Fig. 5. (A) Induction of apoptosis in TRX-2–/–/Trx-2 cells by dox treatment suppressing the expression of Trx-2. Flow cytometric analysis of dox-treated TRX-2–/–/Trx-2 cells stained with PI and Annexin V–FITC. Cells stained positive with annexin V–FITC and negative with PI are apoptotic. Cells did not stain with both annexin V–FITC and PI are alive. Cells stained with PI were necrotic. LR, percentages of apoptotic cells. LL, percentages of alive cells. UL and UR, percentages of necrotic cells. (B) Flow cytometric data of caspase-3 activity at each cell stage. Cells were cultured with 2 µg/ml dox for 3, 5 and 7 days, as indicated. The caspase-3 activity of cells at each stage was evaluated with PhiPhiLux G1D2 by flow cytometry. Caspase-3 activity of TRX-2–/– cells was increased at each stage compared with that at day 0.


Fig. 6. Activities of caspase-8 (A) and caspase-9 (B) in TRX-2–/–/Trx-2 cells. Caspase-8 and -9 activities were determined using the fluorescent substrates IETD–AFC and LEVD–AMC at days 0, 3 and 5 after dox treatment. Etoposide was used as a positive control. (C) Cytochrome c release in the cytoplasm was observed by western blotting using anti-cytochrome c antibody in the cytoplasmic fraction of TRX-2–/–/Trx-2 cells at days 0, 3, 5 and 7 after dox treatment. (D) Analysis of membrane potential by fluorescence-activated cell sorting (FACS) using DiCO6 at days 0, 3 and 5 after dox treatment. CCCP was used as a positive control.
To investigate the relationship between an increase in intercellular ROS and the mitochondrial apoptotic pathway of Trx-2-deficient cells, we examined whether reducing agents such as 2-mercaptoethanol suppress the death of Trx-2-deficient cells. Treatment with 500 nM or 250 nM 2-mercaptoethanol partly ameliorated the decrease in number of TRX-2–/–/Trx-2 cells compared with untreated cells (Figure 7). These results suggest that the increase of intracellular ROS by Trx-2 ablation promotes cell death. However, this increased number of ROS is not the only cause of cell death observed in Trx-2-deficient cells, as 2-mercaptoethanol could not fully rescue cell death.

Fig. 7. Effect of antioxidant agent 2-mercaptoethanol (2ME) on the viability of TRX-2–/–/Trx-2 cells. TRX-2–/–/Trx-2 cells were cultured in the absence (squares) or presence (circles) of 2-mercaptoethanol. Cells were also cultured in the absence (open symbols) or presence (closed symbols and crossed circle) of dox. The numbers of cells not stained with Trypan Blue were counted. Wild-type DT40 cells are represented by closed circles. The data shown are the average results from three separate experiments.
We then tested the effect of Trx-2 deficiency on apoptosis in a physiological condition. DT40 cells resulted in apoptosis when cultured in serum-free medium (Lee et al., 1999). Wild-type cells or TRX-2–/–/Trx-2 cells at day 0 or day 5 after treatment with dox were cultured in serum-free medium for 4 h, and the cell survival ratio was measured using the Trypan Blue dye exclusion test. The viability of the wild-type cells was decreased to 56%. TRX-2–/–/Trx-2 cells without dox treatment, which express higher levels of Trx-2 than wild-type cells, showed greater viability than wild-type cells (76%) in the serum-free condition. TRX-2–/–/Trx-2 cells treated with dox for 5 days were more susceptible to serum withdrawal (42%) (Figure 8). These results suggest that Trx-2-deficient cells are more sensitive to physiological stimuli that induce apoptosis, although we cannot completely rule out the possibility that serum starvation may cause ROS production in mitochondria.

Fig. 8. Effect of serum deprivation on the viability of TRX-2–/–/Trx-2 cells. Wild-type DT40 cells and TRX-2–/–/Trx-2 cells were cultured in the absence of serum in culture medium for 4 h. The ratio of living cells was calculated by dividing the number of living cells after 4 h by the initial number of cells. The numbers of cells not stained with Trypan Blue were counted. The data shown are the average results from four separate experiments.
To analyze further the mechanism of cell death, we analyzed binding molecules of Trx-2 by the immunoprecipitation with anti-Trx-2 antibody using mitochondrial fraction from Jurkat cells. Cytochrome c was co-immunoprecipitated with Trx-2 (Figure 9A). To reconfirm the binding of Trx-2 with cytochrome c, we then tested whether Trx-2 binds to cytochrome c directly or not in an in vitro study. Cytochrome c was co-immunoprecipitated with recombinant Trx-2 protein by anti-Trx-2 antibody, but not by control rabbit serum (Figure 9B). These results suggest that Trx-2 interacts directly with cytochrome c.


Fig. 9. Co-immunoprecipitation of Trx-2 with cytochrome c. (A) Trx-2 and cytochrome c were co-immunoprecipitated in the mitochondrial fraction of Jurkat cells. The mitochondrial fraction was prepared with 0.1% digitonin buffer, and immunoprecipitated using anti-Trx-2 antibody or normal rabbit serum. Cytochrome c–Trx-2 complex was purified using each immunoaffinity column. Lanes 1 and 4, anti-Trx-2 antibody; lanes 2 and 5, normal rabbit serum. Eluted samples were concentrated and analyzed by western blotting using an anti-cytochrome c antibody. Cytochrome c (20 ng/lane) was shown as a positive control in lane 6. (B) Direct interaction between cytochrome c and Trx-2 in vitro. Cytochrome c was mixed with cold recombinant mouse Trx-2 and immunoprecipitated using anti-Trx-2 antibody (lane 1) or normal rabbit serum (lane 2). Translation product of cytochrome c in vitro is shown in lane 3. Cytochrome c and recombinant mouse Trx-2 were prepared by use of an in vitro translation method. Antibody (1.5 µg of each) was used for each sample. Precipitated samples were subjected to western blotting analysis using the anti-cytochrome c antibody.
Discussion
In this study, we have generated TRX-2 gene-disrupted cells carrying a chicken TRX-2 transgene under the control of a dox-repressible promoter. We observed that the growth of TRX-2–/–/Trx-2 cells was significantly retarded after repression of the TRX-2 transgene by dox treatment. ROS generation was slightly elevated and both intracellular and intramitochondrial GSH levels decreased in TRX-2–/–/Trx-2 cells at 5 days after dox treatment. Moreover, BSO treatment induced cell death in TRX-2–/–/Trx-2 cells at 3 days after dox treatment. These results indicate that ROS scavenging activity decreases when Trx-2 becomes deficient in TRX-2–/–/Trx-2 cells.
It is well known that in viable cells mitochondria are the major source of ROS generation during respiration and that this ROS generation is strictly regulated by mitochondrial antioxidant enzymes including Mn-SOD, the mitochondrial GSH/GPx system, and the mitochondrial-specific Trx-2/Prx III system (Miranda-Vizuete et al., 2000; Pedrajas et al., 2000). The present results showing that intracellular and intramitochondrial GSH decreased in Trx-2-deficient cells suggest that GSH was consumed to compensate for the loss of Trx-2/Prx III-dependent antioxidant function.
Next we analyzed the cell death mechanism of Trx-2-deficient cells. When TRX-2–/–/Trx-2 cells were treated with dox, apoptotic signals such as the release of cytochrome c from the mitochondria and the activation of caspases were observed at day 3, and the delay in cell proliferation was detectable by day 4. Trx-2 protein was completely undetectable and most of the Trx-2-deficient cells had apoptosed by day 5. These results suggest that a deficiency of Trx-2 promotes signal transduction for apoptosis. Moreover, we have observed that the content of cytochrome c in the cytoplasm and the activity of caspase-9, but not caspase-8, significantly increased in TRX-2–/–/Trx-2 cells after dox treatment. These results suggest that Trx-2 is involved in the mitochondria-mediated apoptotic pathway.
Caspases are quite sensitive to oxidation (Hampton and Orrenius, 1997; Fadeel et al., 1998). We and others have shown previously that mild oxidative stress induces apoptosis without inactivation of caspases, whereas severe oxidative stress induces necrosis by inactivation of caspases (Hampton and Orrenius, 1997; Ueda et al., 1998). The activation of caspase-3 in Trx-2-deficient cells was lower than in cells treated with etoposide. The apparent inefficiency of apoptosis in Trx-2-deficient cells, compared with etoposide-induced apoptosis, may be explained by the slow-onset suppression of TRX-2 transgene expression in this tet-off system. Alternatively, since some Trx-2-deficient cells become PI positive (necrotic), the accumulated ROS may be too much to inactivate caspase-3 in some Trx-2-deficient cells. In addition, etoposide-induced apoptosis is thought to be initiated by DNA damage, which may explain the fact that it appears to be more efficient than mitochondria-initiated apoptosis in Trx-2-deficient cells.
The mitochondria play a pivotal role in the signal transduction of apoptosis. The permeability transition (PT) pore is regulated by Bcl-2 family proteins and Bcl-xL interacts with the voltage-dependent anion channel (VDAC) (Shimizu et al., 1999). In addition, the PT pore is modulated by a redox-sensitive thiol and the redox state of pyridine nucleotide (Petronilli et al., 1994; Costantini et al., 1996). ROS generation in mitochondria modulates the opening of the PT pore and several antioxidants, including Trx and mitochondrial Trx reductase, have been reported to regulate the ROS-dependent release of cytochrome c (Rigobello et al., 1998; Tan et al., 1998; Ueda et al., 1998).
The present results showing that mitochondrial membrane potential is maintained in Trx-2-deficient cells suggest that ROS generation in mitochondria is not suf ficient to open the PT pore. Bossy-Wetzel and colleagues showed that the translocation of cytochrome c from mitochondria to cytoplasm does not require mitochondrial transmembrane depolarization (Bossy-Wetzel et al., 1998). We have shown that Trx-2 and cytochrome c were co-immunoprecipitated in the mitochondrial fraction of Jurkat cells. These results suggest that Trx-2 deficiency in mitochondria induces the translocation of cytochrome c from mitochondria into cytoplasm without mitochondrial transmembrane depolarization.
The role of this interaction between Trx-2 and cytochrome c is currently unclear. It is possible that Trx-2 regulates mitochondria-mediated apoptosis not only by ROS scavenging, but also by regulating the release of cytochrome c from mitochondria. The precise role of Trx-2 in the regulation of cytochrome c release from mitochondria is now under investigation.
ROS generation is also substantially involved in neurodegenerative disorders such as Alzheimer’s disease and Parkinsonism (Benzi and Moretti, 1995; Jenner and Olanow, 1996). Trx-2 is highly expressed in neurons (Rybnikova et al., 2000), whereas Trx expression is high in glia cells but low in neurons (Hori et al., 1994). Therefore, it is possible to speculate that Trx-2 may be critically involved in the pathogenesis of some neurodegenerative disorders. Further studies in developing conditional TRX-2 knock-out mice or TRX-2 transgenic mice will clarify the role of Trx-2 in the regulation of mitochondria-mediated ROS in oxidative stress-associated disorders, including neurodegenerative diseases.
In conclusion, the present study shows that a deficiency of Trx-2 results in the decrease of ROS scavenging activity and leads to apoptosis via the mitochondria pathway in chicken DT40 cells. These results indicate that TRX-2 is an essential gene and that Trx-2 plays a crucial role, not only in the regulation of ROS generation in the mitochondria as a component of the mitochondrial antioxidant system, but also in the regulation of the mitochondrial apoptotic signaling pathway.
Materials and methods
Cloning of chicken TRX-2 cDNA
A set of degenerate oligonucleotides was designed for PCR amplification of the chicken TRX-2 homolog, based on well conserved regions of the human, mouse and rat Trx-2 protein amino acid sequence. The primer sequences were TGG TG(T/C) GGI CCI TG(T/C) and IAC IAC (A/G)TC ICC (A/G)TT, where ‘I’ represents inosine, and alternative nucleotides are given in parentheses. A partial fragment of chicken TRX-2 cDNA was amplified with these degenerate primers. Subsequently, the cDNA containing the entire ORF of chicken TRX-2 was isolated by 5′- and 3′-RACE with chicken testis RNA (Clontech).
Plasmid constructs
The 7-kb genomic chicken TRX-2 locus was isolated from DT40 genomic DNA by long-range PCR with the chicken TRX-2 cDNA primers 5′-AGGCCGTTGGCCTCTCAGGGCTTACCCGGC-3′ and 5′-GGCGATAAAGTTCTCTTTATTACAGACCTA-3′. The positions of the exons and introns of this genomic locus were determined by sequencing. To produce chicken TRX-2 disruption constructs, the selection marker genes for histidinol resistance (HisR) or blasticidin resistance (BsrR) were inserted between the left and right arms, which were derived by PCR amplification of this TRX-2 disruption construct. To produce the chicken Trx-2 expression construct, the chicken TRX-2 ORF was inserted into pUHG 10-3 (pTet-TRX2; a gift from H.Bujard, Heidelberg). tTA-Hyg vector was a gift from K.Shimizu (Okayama University).
Cell culture and DNA transfections
DT40 cells were cultured in RPMI 1640 medium supplemented with penicillin, streptomycin, 10% fetal calf serum (FCS) and 1% chicken serum (Biosciences) at 39°C. DNA transfection and selection was performed as described previously (Buerstedde et al., 1991). Briefly, 10–30 µg of each linearized plasmid was electroporated into 107 cells with a Gene Pulser apparatus (Bio-Rad) at 550 V and 25 µF. Drug-resistant colonies were selected with medium containing 50 µg/ml blasticidin-S (Calbiochem, La Jolla, CA) or 2.5 mg/ml histidinol (Wako). The tetracycline analog dox (1 µg/ml; Sigma) was used to suppress expression of the TRX-2 transgene.
Jurkat, a human T-cell line, was maintained in RPMI 1640 with penicillin, streptomycin and 10% FCS at 37°C.
Isolation of conditional TRX-2–/– clones
The conditional Trx-2-deficient cells were generated as follows. The TRX-2–BsrR construct was transfected into wild-type DT40 cells, and clones heterozygous for the TRX-2 locus (TRX-2+/–) were isolated. One of the TRX-2+/– clones was co-transfected with tTa-Hyg, which is a construct expressing tet-repressible transactivator (tTA), and pTet-TRX-2, which is a Trx-2 expressing construct with a tTA binding site, to obtain TRX-2+/– clones with tet-repressible Trx-2 expressing cells (TRX-2+/–/Trx-2). The TRX-2–HisR construct was then introduced into several TRX-2+/–/Trx-2 clones to isolate conditional TRX-2–/– clones (TRX-2–/–/Trx-2).
Preparation of recombinant proteins
Bacterially expressed His6-tagged recombinant protein was prepared under denaturing conditions according to the manufacturer’s instructions (Qiagen). Escherichia coli strain XL1 Blue MRF, transformed with a mature form of mouse Trx-2 (amino acids 60–166, ΔTrx-2) expression vector derived from pQE-31, was treated with 1 mM isopropyl-β-d-thiogalactoside for 4 h. The His6-tagged ΔTrx-2 protein was purified using a Ni2+-nitrilotriacetic acid–agarose column. The size and purity of the eluted protein was determined by SDS polyacrylamide gel electrophoresis. The anti-Trx-2 antibody was prepared by immunization of bacterially expressed His6-tagged ΔTrx-2 protein, as described previously (Spyrou et al., 1997). The immune serum was purified further by affinity columns coupled to the His6-tagged ΔTrx-2 protein with CNBr-activated Sepharose 4B (Amersham).
Western blotting
Cells were grown to 8 × 105 cells/ml in 10 ml culture medium, collected, washed twice with cold phosphate-buffered saline (PBS), and lysed with a solubilizing solution (150 mM NaCl, 1.0% Nonidet P-40, 1.0% sodium deoxycolate, 0.1% SDS, 20 mM Tris–HCl pH 7.5, 5 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, 1 µg/ml leupeptin and 5 µg/ml aprotinin) on ice for 30 min. The extracts were cleared by centrifugation and subjected to SDS–PAGE. SDS–PAGE and western blotting were performed as described previously (Ueda et al., 1998).
Flow cytometric estimation of intracellular ROS
Intracellular redox status was estimated by the level of intracellular ROS, which was monitored by flow cytometric analysis with DCFH-DA, as described previously (Ueda et al., 1998). Cells were incubated in the medium with 5 µM DCF-DA for 15 min at 39°C, washed with PBS and resuspended in 1 ml of PBS. The intensity of fluorescence was analyzed using a flow cytometer (FACSCalibur; Becton Dickinson).
Determination of mitochondrial and cellular total glutathione
To prepare the mitochondrial fraction, cells were collected, washed twice with ice-cold PBS, and lysed in 0.25 M sucrose, 10 mM potassium HEPES pH 7.4 and 0.2 mM EDTA pH 7.4 using the French-press method. The lysate was centrifuged at 600 g for 10 min at 4°C to remove the nuclei. The supernatant was centrifuged further at 8000 g for 10 min at 4°C (Beckman TL-100 Ultracentrifuge). The pellet was resuspended in 1% 5-sulfosalicylic acid and shaken overnight at 4°C. Following centrifugation, the supernatant was pooled as mitochondrial fraction. To measure total cellular glutathione, cells were washed twice with ice-cold PBS and lysed in 5-sulfosalicylic acid solution (final concentration 1%), and shaken overnight at 4°C. After centrifugation, the supernatant was pooled as whole cell lysate. Each supernatant was assayed for mitochondrial or total cellular glutathione (reduced GSH and glutathione disulfide), whereas the pellets were subjected to measuring protein concentration. Total glutathione was determined by the enzymatic method as described previously (Tietze, 1969). Protein concentration was measured by Lowry’s method (Bio-Rad protein assay).
Detection of apoptosis and activities of caspases
To determine the proportion of apoptotic cells, cells were analyzed with Annexin V–FITC (Pharmingen) and PI double staining. The cells were washed with PBS, resuspended in 100 µl binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), incubated for 15 min with 5 µl Annexin V–FITC and 50 µg/ml PI in the dark, and subjected to flow cytometric analysis. The activity of caspase-3 was determined by flow cytometric analysis using PhiPhiLux G1D2. The activities of caspase-8 and caspase-9 were measured using IETD–AFC (7-amino-4-trifluoromethyl coumarin) or LEHD–AMC (7-amino-4-methyl coumarin) as substrate (caspase-8 and -9 fluorescent assay kit; Clontech), and detected as the emission shift of AFC or AMC.
Measurement of mitochondria membrane potential by flow cytometry
A mitochondria membrane potential was measured using DiCO6. Briefly, 1 × 105 cells were incubated with caspase inhibitor z-VAD-fmk (Calbiochem). Cells were then cultured with DiCO6 (40 nM) for 15 min in culture medium, in 5% CO2 at 39°C, and then analyzed by cytometry. Data acquisition and analysis was performed using the Cell Quest software. As a positive control for mitochondria membrane potential loss, DT40 cells were incubated with uncoupling agent, carbonyl cyanide m-chlorophenylhydrazone (CCCP).
Immunoprecipitation and protein binding assay
Jurkat cells were grown in RPMI 1640 supplemented with 10% FBS. Mitochondria extracts were lysed with 0.1% digitonin buffer (0.1% digitonin, 10 mM triet hanolamine, 150 mM NaCl, 10 mM iodoacetamide, 1 mM EDTA, 10 µg/ml aprotinine pH 7.5) at 4°C for 30 min. The lysates were cleared by centrifugation at 10 000 g for 15 min. The super natants were removed as samples. Samples were incubated overnight at 4°C with 20 µl protein A–Sepharose (Amersham Pharmacia) and 5 µl normal rabbit serum in order to block any non-specific binding protein.
After pre-clearing using protein A–Sepharose, samples were incubated with anti-Trx-2 antibody or normal rabbit serum at 4°C for 2 h and with protein A–Sepharose for an additional 1 h. Samples were then centrifuged and washed five times with 0.1% digitonin buffer. The precipitated proteins were subjected to western blot analysis and detected by anti-cytochrome c antibody (Pharmingen).
Protein binding assay was performed as follows: cytochrome c and recombinant His6-Trx-2 were incubated overnight in 0.1% digitonin buffer at 4°C. After pre-clearing at 4°C for 2 h, we performed an immunoprecipitation similar to that mentioned above.
Acknowledgments
Acknowledgements
We thank Y.Sato and O.Koga for their excellent technical assistance, N.Kondo and M.Tanito for discussion, and Y.Kanekiyo for secretarial help. This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by a grant-in-aid of Research for the Future from the Japan Society for the Promotion of Science.
References
- Araki M., Nanri,H., Ejima,K., Murasato,Y., Fujiwara,T., Nakashima,Y. and Ikeda,M. (1999) Antioxidant function of the mitochondrial protein SP-22 in the cardiovascular system. J. Biol. Chem., 274, 2271–2278. [DOI] [PubMed] [Google Scholar]
- Benzi G. and Moretti,A. (1995) Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol. Aging, 16, 661–674. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer,D.D. and Green,D.R. (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J., 17, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerstedde J.M. and Takeda,S. (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell, 67, 179–188. [DOI] [PubMed] [Google Scholar]
- Chae H.Z., Chung,S.J. and Rhee,S.G. (1994a) Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem., 269, 27670–27678. [PubMed] [Google Scholar]
- Chae H.Z., Robison,K., Poole,L.B., Church,G., Storz,G. and Rhee,S.G. (1994b) Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl Acad. Sci. USA, 91, 7017–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini P., Chernyak,B.V., Petronilli,V. and Bernardi,P. (1996) Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem., 271, 6746–6751. [DOI] [PubMed] [Google Scholar]
- Fadeel B., Ahlin,A., Henter,J.I., Orrenius,S. and Hampton,M.B. (1998) Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood, 92, 4808–4818. [PubMed] [Google Scholar]
- Hampton M.B. and Orrenius,S. (1997) Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett., 414, 552–556. [DOI] [PubMed] [Google Scholar]
- Hendrick J.P., Hodges,P.E. and Rosenberg,L.E. (1989) Survey of amino-terminal proteolytic cleavage sites in mitochondrial precursor proteins: leader peptides cleaved by two matrix proteases share a three-amino acid motif. Proc. Natl Acad. Sci. USA, 86, 4056–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem., 54, 237–271. [DOI] [PubMed] [Google Scholar]
- Holmgren A. and Bjornstedt,M. (1995) Thioredoxin and thioredoxin reductase. Methods Enzymol., 252, 199–208. [DOI] [PubMed] [Google Scholar]
- Hori K., Katayama,M., Sato,N., Ishii,K., Waga,S. and Yodoi,J. (1994) Neuroprotection by glial cells through adult T cell leukemia-derived factor/human thioredoxin (ADF/TRX). Brain Res., 652, 304–310. [DOI] [PubMed] [Google Scholar]
- Jenner P. and Olanow,C.W. (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology, 47, S161–S170. [DOI] [PubMed] [Google Scholar]
- Lee R.M., Gillet,G., Burnside,J., Thomas,S.J. and Neiman,P. (1999) Role of Nr13 in regulation of programmed cell death in the bursa of Fabricius. Genes Dev., 13, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M., Oshima,M., Oshima,H., Takaku,K., Maruyama,T., Yodoi,J. and Taketo,M.M. (1996) Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol., 178, 179–185. [DOI] [PubMed] [Google Scholar]
- Miranda-Vizuete A., Damdimopoulos,A.E. and Spyrou,G. (2000) The mitochondrial thioredoxin system. Antioxid. Redox Signal., 2, 801–810. [DOI] [PubMed] [Google Scholar]
- Nakamura H., Nakamura,K. and Yodoi,J. (1997) Redox regulation of cellular activation. Annu. Rev. Immunol., 15, 351–369. [DOI] [PubMed] [Google Scholar]
- Pedrajas J.R., Miranda-Vizuete,A., Javanmardy,N., Gustafsson,J.A. and Spyrou,G. (2000) Mitochondria of Saccharomyces cerevisiae contain one-conserved cysteine type peroxiredoxin with thioredoxin per oxidase activity. J. Biol. Chem., 275, 16296–16301. [DOI] [PubMed] [Google Scholar]
- Petronilli V., Costantini,P., Scorrano,L., Colonna,R., Passamonti,S. and Bernardi,P. (1994) The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation–reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J. Biol. Chem., 269, 16638–16642. [PubMed] [Google Scholar]
- Rigobello M.P., Callegaro,M.T., Barzon,E., Benetti,M. and Bindoli,A. (1998) Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radic. Biol. Med., 24, 370–376. [DOI] [PubMed] [Google Scholar]
- Rybnikova E., Damdimopoulos,A.E., Gustafsson,J.A., Spyrou,G. and Pelto-Huikko,M. (2000) Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur. J. Neurosci., 12, 1669–1678. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita,M. and Tsujimoto,Y. (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature, 399, 483–487. [DOI] [PubMed] [Google Scholar]
- Spyrou G., Enmark,E., Miranda-Vizuete,A. and Gustafsson,J. (1997) Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem., 272, 2936–2941. [DOI] [PubMed] [Google Scholar]
- Tagaya Y. et al. (1989) ATL-derived factor (ADF), an IL-2 receptor/Tac inducer homologous to thioredoxin; possible involvement of dithiol-reduction in the IL-2 receptor induction. EMBO J., 8, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S., Sagara,Y., Liu,Y., Maher,P. and Schubert,D. (1998) The regulation of reactive oxygen species production during program med cell death. J. Cell Biol., 141, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshigawara K., Maeda,M., Nishino,K., Nikaido,T., Uchiyama,T., Tsudo,M., Wano,Y. and Yodoi,J. (1985) Adult T leukemia cells produce a lymphokine that augments interleukin 2 receptor expression. J. Mol. Cell. Immunol., 2, 17–26. [PubMed] [Google Scholar]
- Tietze F. (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem., 27, 502–522. [DOI] [PubMed] [Google Scholar]
- Ueda S., Nakamura,H., Masutani,H., Sasada,T., Yonehara,S., Takabayashi,A., Yamaoka,Y. and Yodoi,J. (1998) Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J. Immunol., 161, 6689–6695. [PubMed] [Google Scholar]
- Watabe S., Hiroi,T., Yamamoto,Y., Fujioka,Y., Hasegawa,H., Yago,N. and Takahashi,S.Y. (1997) SP-22 is a thioredoxin-dependent peroxide reductase in mitochondria. Eur. J. Biochem., 249, 52–60. [DOI] [PubMed] [Google Scholar]
- Yamamoto T., Matsui,Y., Natori,S. and Obinata,M. (1989) Cloning of a housekeeping-type gene (MER5) preferentially expressed in murine erythroleukemia cells. Gene, 80, 337–343. [DOI] [PubMed] [Google Scholar]
- Yodoi J. and Uchiyama,T. (1992) Diseases associated with HTLV-I: virus, IL-2 receptor dysregulation and redox regulation. Immunol. Today, 13, 405–411. [DOI] [PubMed] [Google Scholar]