

Abstract
Most mitochondrial mRNAs are edited in Trypano soma brucei by a series of steps that are catalyzed by a multienzyme complex that is in its initial stages of characterization. RNA interference (RNAi)-mediated repression of the expression of TbMP81, a zinc finger protein component of the complex, inhibited growth of bloodstream and insect forms, and blocked in vivo RNA editing. This repression preferentially inhibited insertion editing compared with deletion editing in vitro. It resulted in reduced specific endoribonucleolytic cleavage and a greater reduction of U addition and associated RNA ligation activities than U removal and associated RNA ligation activities. The repressed cells retained 20S editing complexes with several demonstrable proteins and adenylatable TbMP52 RNA ligase, but adenlyatable TbMP48 was not detected. Elimination of TbMP48 by RNAi repression did not inhibit cell growth or in vivo editing in either bloodstream or procyclic forms. These results indicate that TbMP81 is required for RNA editing and suggest that the editing complex is functionally partitioned.
Keywords: editosome/endoribonuclease/mitochondria/RNA editing
Introduction
Mitochondrial mRNAs in Kinetoplastida are edited by a multiprotein complex (Pollard et al., 1992; Corell et al., 1996) that catalyzes a series of enzymatic steps, which result in insertion or deletion of uridylates as specified by guide RNAs (gRNAs) (for reviews see Kable et al., 1997; Stuart et al., 1997; Estevez and Simpson, 1999). The most extensive RNA editing has been found in Trypanosoma brucei, where, in some cases, more than 50% of the mRNA sequence was remodeled by editing (Feagin, 1990). RNA editing is crucial for the survival in both bloodstream form (BF) (Schnaufer et al., 2001) and procyclic form (PF) (Stuart, 1971) life cycle stages. However, the pattern of the edited mRNAs that are present in each stage reflects the differences in their energy metabolism (Feagin et al., 1986).
Several characteristics of the editing mechanism have been revealed using an in vitro RNA editing system (Kable et al., 1996; Seiwert et al., 1996; Igo et al., 2000). Editing proceeds generally from the 3′ to 5′ end of mRNA (Maslov and Simpson, 1992) at editing sites that are selected by gRNA. Each gRNA has three functional domains (Hinz and Goringer, 1999). The 5′ end forms a 4–11 bp anchor duplex with the mRNA immediately 3′ to the block of mRNA that will be edited. The central domain is ∼35 nucleotides (nt) long, which is a sufficient sequence to specify the editing of ∼10 editing sites. The 3′ end has an oligo(U) tail that is added post-transcriptionally (Blum and Simpson, 1990). The role of the oligo(U) tail is unknown but it has been proposed to stabilize the interaction between the gRNA and mRNA. Only a few gRNAs have anchor domains that can form duplexes with unedited RNA. The rest can form an anchor duplex with sequences that are generated by editing. RNA editing is accomplished by a sequence of coordinated catalytic steps. The mRNA is first cleaved by an endoribonuclease at an editing site that is specified by the gRNA. Then, depending on the guiding sequence, uridines are either added by a 3′ terminal uridylyl transferase (TUTase) or removed by a 3′ terminal uridylyl exonuclease (exoUase). In the last step, mRNA is rejoined by an RNA ligase (for reviews see Kable et al., 1997; Stuart et al., 1997; Estevez and Simpson, 1999).
The enzymatic activities co-sediment in glycerol gradients at ∼20S. This indicates that the proteins participating in RNA editing form a complex, which has been referred to as the editosome (Pollard et al., 1992; Corell et al., 1996). The complex has been partially purified by a variety of biochemical methods resulting in fractions that contain seven (Rusche et al., 1997), 13 (Madison-Antenucci et al., 1998) or ∼20 major proteins (Panigrahi et al., 2001a). The composition of the functional editing complex is not yet known although several candidate components have been identified (Koller et al., 1997; Madison-Antenucci et al., 1998; Vanhamme et al., 1998; Hayman and Read, 1999). Some components have been shown to be part of the editing complex (Panigrahi et al., 2001a,b), two of which, TbREL1 (TbMP52) and TbREL2 (TbMP48), have RNA ligase activities (McManus et al., 2001; Rusche et al., 2001; Schnaufer et al., 2001). TbMP63, a zinc finger protein, interacts with TbMP52 (Panigrahi et al, 2001b). Monoclonal antibodies (mAbs) specific to TbMP63 efficiently immunoprecipitate RNA editing activity and proteins from whole-cell lysates as well as more purified material (Panigrahi et al, 2001b). However, none except for the TbREL1 (TbMP52) RNA ligase (Schnaufer et al., 2001) and the mHel61p RNA helicase (Missel et al., 1997) have been shown to play a role in editing.
Here we report on genetic and functional studies that show the TbMP81 protein to have an essential role in RNA editing in both BF and PF T.brucei. TbMP81 is present in the editing complex, since it co-fractionates in glycerol gradients with in vitro deletion and insertion editing, and is among the proteins immunoprecipited with anti-TbREL1 monoclonal antibody. It contains a zinc finger motif, but no related proteins were found in database searches and hence its function is uncertain (Panigrahi et al., 2001b). RNA interference (RNAi)-mediated inactivation of TbMP81 in vivo results in the loss of endoribonuclease, TUTase and RNA ligase activities associated with insertion editing. However, some exoUase and RNA ligase activities associated with deletion editing are retained.
Results
Loss of TbMP81 inhibits growth and in vivo RNA editing
Double-stranded RNA (dsRNA) directed against the TbMP81 coding region was expressed from a tetracycline (tet)-controled construct that was stably integrated into the non-transcribed spacer of the rDNA locus of the parasite genome (Wirtz and Clayton, 1995). Two expression constructs, pHD677, which has a VSG 3′UTR and is generally used in BFs, and pHD678, which has an actin 3′ UTR and is used in PFs (Biebinger et al., 1997), were equally efficient in dsRNA expression. However, only cells transfected using pHD1297, a derivative of pHD677, were used in this study. Two BF clones and 15 PF clones were obtained. Northern blot analysis was performed on RNA from both BF clones and five of the PF clones that were grown in the presence or absence of 100 ng/ml tet for 16 h. All clones showed strong tet-induced reduction of intact TbMP81 mRNA and a broad smear that probably represents degraded RNA. The levels of mRNA from cells grown without tet were comparable with those in a parental strain (see Materials and methods) (Figure 1A). Western blot analysis of both stages revealed that the TbMP81 protein was greatly reduced but still detectable after 1 day of induction, but was not detectable after 2 or 3 days (Figure 1B).

Fig. 1. Inactivation of TbMP81 expression. (A) Northern blot analysis of TbMP81 RNA from PF and BF in which RNAi was not induced (–) or was induced by growth with tet (+). RNA from the 449 parental strain cells (P) was used as a positive control, ribosomal RNA (rRNA) was used as a loading control. (B) Western analysis showing the inactivation of TbMP81 expression of upon induction of RNAi (+) in PF and BF. Cells of the parental strain and non-induced cells were used as positive controls. The cytoplasmic protein TbCSM1 (Guerra-Giraldez et al., 2002) was used as a loading control.
Inactivation of TbMP81 expression inhibited cell proliferation. As shown in Figure 2, growth over 12 days of both BF clone 1 and PF clone 6 in the continuous presence of 100 ng/ml tet was less than that of parental strain and in the absence of tet. Cells of the parental strain and cells without tet grew at the same rate. After ∼3 days, the growth of the BFs stopped (Figure 2A) and that of the PFs slowed down (Figure 2B). After about an additional 6 days, growth of both the BF and PF returned closer to the cells of the parental strain and –tet level. Withdrawal of tet after day 6 had no detectable effect on the growth rate (data not shown).

Fig. 2. Growth inhibition of (A) BF and (B) PF upon induction of TbMP81 RNAi. Cells were counted and diluted (BF to 2.5 × 105, PF to 106 cells/ml) daily. tet was replenished in cultures in which RNAi was induced (solid squares) and omitted in the control (solid circles) and parental strain (crosses) cultures.
Repression of TbMP81 expression blocked RNA editing in vivo in both BF and PF (Figure 3). RNA that was isolated from cells grown after 3 days with tet had a substantial reduction in fully edited A6, ND7 and RPS12 RNA and a reduction in the size profile of partially edited RNA compared with parental cells and cells grown without tet (Figure 3A and B; data not shown). There was no substantial increase in the unedited RNA, although this was the case upon inactivation of the REL1 RNA ligase gene (Schnaufer et al., 2001), which may reflect the functional difference between these two proteins. The abundance of ND4 RNA, which does not get edited, was unaffected in the presence of tet (Figure 3C). Poisoned primer analysis of Cyb RNA, which is edited by U insertion only, showed that while about half of the RNA was edited in the parental cells and before addition of tet, somewhat less than a third was edited 5 days after the addition of tet, i.e. there was a 40% reduction of edited RNA (Figure 3D). Thus, editing was substantially inhibited, but not totally eliminated, upon repression of TbMP81 expression.

Fig. 3. Loss of edited RNA in vivo upon induction of TbMP81 RNAi. RT–PCR products of (A) A6, (B) ND7 and (C) ND4 RNAs from parental strain cells (P) and cells in which TbMP81 RNAi was not induced (–) or was induced by growth in tet for 3 days. PE, partially edited. (D) The fraction of Cyb mRNA that is edited, as determined by primer extension analysis. P1 and P2 indicate clones of the 449 parental line. See Materials and methods for details.
Preferential loss of insertion editing and endonuclease activities
Repression of TbMP81 expression resulted in preferential loss of U addition (TUTase) and the associated RNA ligase activities as determined by in vitro pre-cleaved insertion assays of immunoprecipitates with anti-TbMP63 mAb from total cell lysates (Figure 4). After 3 or 4 days with tet, the activities that added one or two Us and produced edited RNA or ligated input 5′ and 3′ substrate fragments were diminished by ∼80–95% in PFs and BFs (Figure 4A). However, pre-cleaved deletion assays showed that the activities that specifically remove four Us and produce edited RNA or ligation of input 5′ and 3′ fragments were less affected, being diminished by 5–55% (Figure 4B). Negative control immunoprecipitates from whole-cell lysates using anti-pyruvate dehydrogenase monoclonal antibody do not exhibit ligase, TUTase or exonuclease RNA editing activities (data not shown). Thus, loss of TbMP81 resulted in substantial loss of TUTase and ligase activity with an insertion substrate but retention of much of the exonuclease and RNA ligase activities with a deletion substrate. In vitro editing assays that require endonuclease cleavage are not sufficiently sensitive to detect editing in gradient fractions or immunoprecipitates of total cell lysates (data not shown). No endoribonuclease, TUTase, exonuclease or RNA ligase activities were detected upon direct analysis of in vitro translated TbMP81 (data not shown).
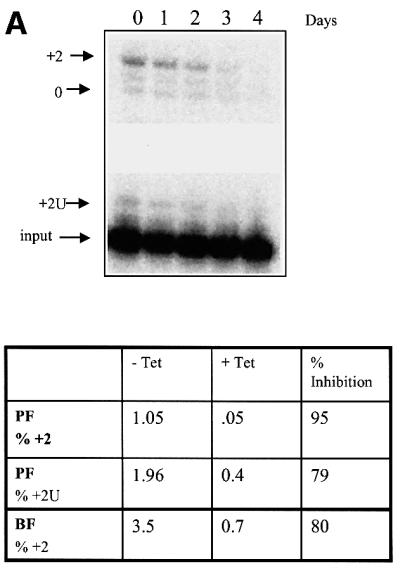
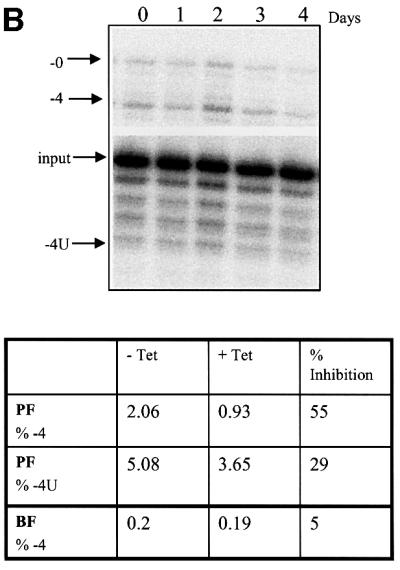
Fig. 4. The effect of repression of TbMP81 expression on in vitro pre-cleaved U insertion and deletion editing. Editing complexes were immunoprecipitated with MAb P1H3-D7 (specific for TbMP63) from total cell lysates that were grown in the absence of tet (no RNAi) or in the presence of tet (RNAi). (A) Pre-cleaved insertion editing by the immunoprecipitates. Products from PF immunoprecipitates with two added Us (+2U), edited by the insertion of two Us (+2), or the ligated input (0) RNAs are indicated in the top panel. (B) Pre-cleaved deletion editing by the immunoprecipitates. Products with 4 Us removed (–4U), edited by deletion of 4 Us (–4), and the ligated input (–0U) RNAs are indicated in the top panel. The percentage of +2 or –4 edited and +2U addition or –4 removal products as a function of total input RNA in PF after 4 days and in BF after 3 days, as well as the percentage inhibition upon RNAi induction (+tet), are indicated in the lower panels. Similar experiments were performed with BF immunoprecipitates. Pre-cleaved insertion and deletion editing experiments were repeated four and three times, using PF and BF immunoprecipitates, respectively.
Repression of TbMP81 expression in PFs resulted in editing complexes with substantially diminished gRNA-directed endoribonucleolytic cleavage of cytochrome b (CYb) pre-mRNA at editing site 1, an activity that co-purifies with the editing complex (Harris and Hajduk, 1992; Pollard et al., 1992; Piller et al., 1995; Salavati et al., 2002) (Figure 5). TbMP81 protein was not detected in editing complexes that were immunoprecipitated from whole PF cell lysates using anti-TbMP63 mAb 36–42 h in tet (Figure 5A). Similarly, this cleavage activity was progressively reduced up to 96 h (arrow, Figure 5B). This specific cleavage activity is evident in the background of other nuclease activities that are present in these immunoprecipitates from whole-cell lysates as is the product resulting from ligation of the cleaved RNA with gRNA (asterisk). Glycerol-gradient sedimentation analysis of total cell lysates after 3 days in tet showed that this specific cleavage activity sedimented in fractions 7–15 (Figure 5C). This is where the editing complex normally sediments (Figure 6) and is away from the non-specific cleavage activity fractions.

Fig. 5. Loss of endonuclease activity from PF upon repression of TbMP81 expression. (A) Western blot analysis showing the loss of TbMP81 (arrow) after the number of indicated hours in tet. (B) Endonuclease analysis of editing complexes that were immuno precipitated from total cell lysates with mAb P1H3–D7, which is specific for TbMP63. Note the diminishment of cleavage product (arrow) and this product ligated with the gRNA (asterisk) in RNAi. (C) Endonuclease analysis of glycerol-gradient fractions from total cell lysates of induced RNAi (+tet) or non-induced (–tet) cells of the parental strain and those containing the RNAi construct. C1, 5′ Cyb RNA; C2, 5′ Cyb RNA + gRNA; C3, 5′ Cyb RNA + purified editing complex (superose 6 fraction); C4, Cyb RNA + gRNA + purified editing complex.

Fig. 6. Retention of editing complexes after loss of TbMP81. Western blot analyses of glycerol gradient fractions from BF and PF of the parental strain (P) and cells expressing dsRNA with or without treatment with tet for 3 days. The arrow indicate the location of TbMP81 protein which is lost upon RNAi induction with tet (asterisk).
Complex retention despite TbMP81 repression
While repression of TbMP81 expression for 3 days resulted in the loss of the TbMP81 protein, the editing complex was retained (Figure 6). Total cell lysates that were fractionated on glycerol gradients were analyzed for the presence of the TbMP81, TbMP63, TbMP52 and TbMP42 components of the editing complex using a cocktail of mAbs specific for these proteins. Western blot analyses revealed all four proteins, primarily in fraction 7–15 in PF parental cells and non-repressed strains, but TbMP81was not detected when expression of the gene was inactivated by RNAi. A similar result was obtained with BFs although the complex was consistently higher in the gradient than with the PFs. Tet appeared to induce expression of a protein below 66 kDa that reacts non-specifically with the antibody. Thus, while TbMP81 protein expression was inhibited, this did not block expression of other protein components of the editing complex nor the presence of editing complexes. Hence, TbMP81 does not appear to be required for interactions among editing complex proteins and formation of the complex.
Adenylatable REL2 (TbMP48) RNA ligase was lost from editing complexes that were immunoprecipitated with the MAb against TbMP63 (MAb P1H3-D7) when expression of TbMP81 was repressed (Figure 7). While the amount of adenylatable TbREL1 (TbMP52) remained unchanged over the course of 3 days in the absence of tet, adenylatable TbMP48 diminished by ∼10-fold when the cells were grown with tet. However, TbMP48 mRNA levels did not change after 16 h following inactivation of TbMP81 (data not shown). Growth of cells of the parental strain with tet for the same period had no effect on the abundance of the adenylatable RNA ligases (data not shown). Thus, either TbMP48 protein was reduced in abundance in the complex or its ability to be adenylated was diminished.

Fig. 7. Loss of adenylatable REL2 (TbMP48) upon RNAi repression of TbMP81 expression. (A) PF cells of the parental strain and (B) those in which TbMP81 expression was repressed by growth in tet for 1, 2 and 3 days. (C and D) The graphs indicate the proportion of radiolabel normalized against total radioactivity in the parental and RNAi, respectively.
REL2 is not essential for editing in BFs or PFs
Repression of TbMP48 expression by regulated expression of TbMP48 RNAi resulted in loss of TbMP48 mRNA and adenylatable TbMP48 protein (Figure 8). Both BFs and PFs were transfected with pHD1300 that had been linearized using ClaI (see Methods and materials for details). Northern blot analysis of three BF and five PF clones showed that all PF and two BF clones showed strong tet-dependent reduction of the TbMP48 mRNA as illustrated in Figure 8A. No TbMP48 mRNA was detectable in one BF clone even when cells were grown in the absence of tet, suggesting leakage in the control of transcription of the dsRNA. Analysis of one BF and one PF clone revealed that both had greatly reduced adenylatable TbMP48 compared with TbMP52 (Figure 8B). Despite the reduction of TbMP48 mRNA and adenylatable protein, both BFs and PFs of these cells grew at the same rate in the presence or absence of tet (Figure 8C) and the ability to detect in vivo edited RNA by RT–PCR was indistinguishable between cells grown in the presence or absence of tet (Figure 8D). Similarly, no differences were detected in endonuclease activity, in vitro precleaved U addition editing or in vitro pre-cleaved deletion editing between cells grown in the presence or absence of tet (data not shown).

Fig. 8. Retention of editing and normal growth upon loss of adenylatable TbMP48 and its RNA. (A) Northern analysis showing loss of TbMP48 RNA upon induction of TbMP48 RNAi in BF (top) and PF (bottom). Ribosomal RNA (rRNA) and tubulin were used as probes for loading controls. Cells of the parental strain (P) were used as positive controls. (B) Adenylation assay showing loss of adenylatable TbMP48. (C) Growth of BF and PF with and without RNAi repression of TbMP48 expression. (D) RT–PCR analysis showing the presence of in vivo edited RNA despite RNAi repression of adenylatable TbMP48 production. RNA abbreviations are as in the legend for Figure 3, except S12 refers to RPS12.
Discussion
This report shows that TbMP81 is a functional protein component of the RNA editing complex of T.brucei. Regulated production of TbMP81 RNAi inactivated expression of the RNA and protein products of the gene. The loss of TbMP81 protein was accompanied by a reduction of the growth rate in both BF and PF life cycle stages, loss of editing, but not transcription of mitochondrial RNAs in vivo, and loss of some, but not all, editing-associated in vitro catalytic activities. Loss of TbMP81 protein resulted in preferential loss of insertion editing compared with deletion editing, and of adenylatable TbMP48 RNA ligase in both BFs and PFs. However, while RNAi inactivation of TbMP48 expression resulted in loss of mRNA and adenylatable TbMP48, it did not affect cell growth or editing. Cells with diminished TbMP81 protein retained a complex with sedimentation characteristics that were not distinguishable from that of the parental strain.
The inhibition of growth of both BFs and PFs upon inhibition of TbMP81 production (Figure 2) indicates that editing is critical in both life cycle stages. This is consistent with the finding that editing is normally required in BFs since regulated inactivation of the editing RNA ligase REL1 (TbMP52) gene expression resulted in cell death (Schnaufer et al., 2001). The inhibition of growth of PFs by TbMP81 RNAi inactivation of editing is not surprising since PFs edit mRNAs for components of the oxidative phosphorylation system (Stuart et al., 1997) that they depend on for energy (Hill, 1976). The differential effect on the growth of the PFs versus BFs may reflect growth rate and/or physiological difference between these life cycle stages. It remains to be determined whether the recovery of growth in both life cycle stages is a consequence solely of mutations that eliminate RNAi inhibition or whether there is genetic or physiological compensation for the loss of editing. Such compensation may have occurred in mutants that lack editing as a result of kDNA deletions and can grow as BFs but not PFs (Hajduk, 1978; Stuart, 1980; Zweygarth et al., 1990).
The greater loss of insertion editing-associated activities compared with deletion editing-associated activities upon TbMP81 repression suggests that TbMP81 is essential for insertion but not deletion editing. Endonucleolytic cleavage of a Cyb/gRNA insertion editing substrate (Figure 5), and U addition and ligation in pre-cleaved insertion editing assays (Figure 4A) were inhibited upon repression of TbMP81 expression, but U deletion and RNA ligation activities associated with pre-cleaved U deletion activity were more retained (Figure 4B). One possibility is that TbMP81 is normally restricted to the editing complex, or a subunit or domain thereof, which catalyzes insertion editing. This would imply that some editing complexes, or subunits or domains thereof, catalyze insertion editing while others catalyze deletion editing. The tetrameric particles observed in fractions of purified editing complexes (Stuart et al., 2002) are consistent with this possibility. The loss of deletion, but not insertion, editing upon overexpression of a functionless mutant REL1 (TbMP52) (Huang et al., 2001) also suggests segregation of insertion and deletion editing. These data together imply that REL1 ligase function is associated with deletion editing and REL2 (TbMP48) is associated with insertion editing. The loss of adenylatable REL2 (Figure 7) and insertion editing (Figure 4) upon the loss of TbMP81 is consistent with this possibility. However, inactivation of REL2 ligase expression has no effect on cell growth or RNA editing (Figure 8) suggesting an additional complexity. This result implies that REL1 can compensate in vivo for the loss of REL2 but REL2 cannot compensate for the loss of REL1. Perhaps the simplest explanation for this result is that REL1 can be inserted into functional complexes in the absence of REL2 while REL2 cannot be inserted into functional complexes in the absence of REL1. Alternatively, REL1 and REL2 may have functional differences, such as differing in their stringency of substrate recognition.
The specific function of TbMP81 is uncertain. The inability to detect endoribonuclease, TUTase, exonuclease or RNA ligase activity with recombinant TbMP81 suggests that it does not have a direct catalytic function. However, the level of activity of in vitro produced protein may be below the sensitivity of the assays or a catalytic activity of TbMP81 may require association with another protein. The loss of endonuclease activity upon TbMP81 repression and the lack of detectable endonuclease activity in immunoprecipitates using anti-TbMP81 mAb (Panigrahi et al., 2001b) suggests that TbMP81 may be an endoribonuclease. Alternatively, TbMP81 may not have endonuclease activity itself but be required for such activity (e.g. have a structural role), not only for endonuclease activity but also for U addition and ligation. In addition, we cannot yet exclude the possibilities that the absence of endonuclease activity in the anti-TbMP81 immunoprecipitates is due to steric interference or a low recovery with that antibody, although all other activities were readily detectable. TbMP81 may have a more structural role, perhaps in binding or positioning RNA and/or a protein within the insertion editing complex (or subunit or domain thereof). Such a role is consistent with both its zinc finger and zinc finger-like domains and the loss of adenylatable REL2 upon loss of TbMP81. Nevertheless, such a structural role is not essential for the assembly, general composition and integrity of the editing complex since a 20S complex that lacks adenylatable REL2 is retained in cells that have lost TbMP81 protein. The abundance of TbMP63, TbMP52 and TbMP42 in these complexes appears similar to that in complexes of the parental strain, based on western analyses of total and immunoprecipitated complexes (Figures 5 and 6). The loss of adenylatable REL2 may be due to the loss of protein (no anti-TbMP48 antibody was available) or its ability to be adenylated.
Overall, TbMP81 has been shown to be a critical component of the RNA editing complex, although no specific catalytic or other direct function has been identified. It is not essential for complex integrity, but is essential for the presence of adenylatable REL2 ligase, RNA editing in vivo and cell survival. The differential effect on insertion versus deletion editing suggests a partitioning of insertion versus deletion editing within the complex.
Materials and methods
Plasmid constructs, transfections and induction of RNAi
Two vectors for RNAi were constructed by modifying pSP72 stuffer plasmid (a gift from E.Ullu, Yale University, New Haven, CT). The TbMP81 vector was constructed as follows. A 715 nt fragment of TbMP81 was amplified by PCR from genomic DNA using 5′-ACGAGACGCCTCACCCGCCG as the forward primer and 5′-CATCAACCACCGCCGCTTTT as the reverse primer. The product was then cloned into TOPO pCR II vector (Invitrogen). An EcoRI fragment from this clone was subcloned into EcoRI-digested pSP72 stuffer plasmid. The resultant product was digested with HindIII and SphI and cloned into the first construct so that the two inserts were in the opposite orientation to each other and separated by the stuffer fragment. This product was digested with ClaI, the ends were filled using Klenow fragment, and then it was purified and digested with HindIII. The resulting fragment was ligated into pHD677 digested with HindIII and HpaI to produce the final construct (pHD1297).
To create the TbMP48 RNAi vector, a 746 nt fragment of TbMP48 was amplified from genomic DNA using 5′-ATATTGCGTCGCCTCGGTG as the forward primer and 5′-ACAAGCCCCTCAGCCCAGTT as the reverse primer. The PCR product was cloned into TOPO pCR II vector (Invitrogen) and subcloned into pHD677, using the same strategy and restriction enzymes as before, yielding pHD1300.
The pHD449 TetR PF and BF T.brucei cell lines of strain 427 (MiTat 1) were used for all transfections, and are referred to as the parental strains throughout the article. Both PF and BF trypanosomes were transfected with 10–20 µg plasmid DNA as described previously (Biebinger et al., 1996). The construct was linearized with ClaI. The BF cells were cloned immediately after electroporation by splitting cells into 24 wells of a microtiterplate. PF cells were cloned one day after transfection by limiting dilution. BF transfectants were selected in 15 µg/ml hygromycin and 0.2–0.5 µg/ml phleomycin, and PF cells in 50 µg/ml hygromycin and 0.5 µg/ml phleomycin. The BF cells were grown in HMI-9 medium (Hirumi and Hirumi, 1989) at 37°C and were generally passaged every second day to maintain the log phase growth. The PF cells were grown either in Mem–Pros or in SDM-79 medium at 27°C (Brun and Schonenberger, 1979). Cultures were split into two flasks, only one of which contained tet, and the number of cells was determined daily by manual counting to obtain growth curves. The BF parasites were diluted to 0.25 × 105 cells/ml and PFs to 1.0 × 106 cells/ml in 5 or 10 ml of appropriate medium. In cases where tet was removed, the cells were spun down at 2500 g for 10 min, washed three times with appropriate medium without tet, resuspended in small amount of medium, counted and diluted to the desired density.
RNA analysis
Total RNA was isolated from both stages of parasites using Trizol (Gibco-BRL) or TriFast (peqGOLD TriFast, peqLab Biotechnology, Erlangen, Germany) reagents. At the time of RNA isolation, the BF cell densities did not exceed 106 cells/ml and PF cell densities did not exceed 5 × 106 cells/ml (Hausler and Clayton, 1996). To assess RNAi-mediated elimination of RNA, northern blot analysis was performed using cells that were grown for 16 h with 100 ng/ml of tet. Preparation of the digoxigenin-labeled RNA probe, transfer of RNAs to membrane (Nytran N, Schleicher and Schuell, Dassel, Germany), hybridization and detection were performed according to manufacturer’s protocol (Boehringer, Mannheim, Germany). RNA for the RT–PCR was isolated from cells induced with 100 ng/ml tet for 3 days. Contaminating DNA was removed by treatment with RQ1 RNase-free DNase (Promega) prior to RNA extraction. The RT–PCRs were carried out essentially as in Schnaufer et al. (2001). The upstream/downstream primer combinations used for RT–PCR were 5′-AAAAATAAGTATTTTGATATTATTAAAG/5′-TATTATTAACTTATTTGATC for A6, 5′-TGTGTGACTACCAGAGAT/5′-ATCCTATACCCGTGTGTA for ND4 and 5′-ATGACTACATGATAAGTA/5′-CGGAAGACATTGTTCTACAC for ND7. The A6 RT– PCR products were resolved on 1% TAE/agarose gels while the ND7 and ND4 products were resolved on 3% gels. The products were cloned into TOPO pCR II vector (Invitrogen) and several clones were sequenced (ZMBH, Heidelberg, Germany).
Primer extensions were performed with 50 µg of whole-cell steady-state RNA and 0.1 pmol of 5′-32P-labeled oligodeoxynucleotide (Cyb–RT AACCTGACATTAAAAGAC) (Estevez et al., 1999). Samples were mixed and precipitated with 0.3 M sodium acetate and ethanol. The pellet was resuspended in 0.01 M Tris–HCl (pH 8.3)–0.15 KCl and incubated for 90 min at 42°C. Primer extension was performed in 50 mM Tris–HCl (pH 8.3)–75 mM KCl–30 mM MgCl2–1 mM dithiothreitol (DTT), 250 µM each dATP, dCTP and dTTP, 500 µM ddGTP, 0.3 U rRNasin (Promega) and 100 U of Superscript II (Gibco-BRL) at 42°C for 60 min. RNA was removed by alkaline hydrolysis and precipitated with 5 µg of linear acrylamide (Ambion), 0.37 M ammonium acetate and ethanol, and the products were separated on a 6% (19:1 w:w acrylamide:bis) denaturing polyacrylamide gel containing 7.5 M urea and 1× TBE with 0.025% each of Bromophenol Blue and xylene cyanol as tracking dyes. Reaction products were visualized on a BAS1000 phosphorimager (Fujix, Fuji, Tokyo, Japan). Quantification was performed using macBAS V2 software and the edited RNA was expressed as a percentage of total input.
The sequencing ladder was made by sequencing pBS II KS+ (Stratagene) with 5′-32P-labeled T7 primer using Cyclist Taq DNA sequencing kit (Stratagene).
Analysis of the editing complex
For glycerol gradient analyses, 6 × 108 cells were lysed in 500 µl of the IP300 buffer (10 mM Tris pH 7.6, 10 mM MgCl2, 300 mM KCl, 0.1% Triton X-100, 1% bovine serum albumin) containing protease inhibitors (10 µg/ml Leupeptin, 5 µg/ml Pepstatin, 1 mM Pefabloc). The lysates were cleared by centrifugation, loaded onto linear 10–30% glycerol gradients. The gradients were centrifuged at 38 000 r.p.m. for 5 h at 4°C (SW40 rotor; Beckman), 500 µl fractions were collected from the top, flash-frozen in liquid nitrogen and stored at –80°C. Ten microlitres of every second fraction were resolved by 10% SDS–PAGE, transferred to PVDF membranes and examined by western blot analyses, using mAbs specific for TbMP81, TbMP63, TbMP52 and TbMP42 (Panigrahi et al., 2001b).
Editing complexes were immunoprecipitated from glycerol-gradient fractions (Panigrahi et al., 2001b) or from 6 × 108 PF and BF cells that were harvested by centrifugation, washed once with PBS and lysed in IP300 buffer containing protease inhibitors. MAbs against TbMP63 (mAb P1H3-D7) were conjugated to anti-mouse IgG coated immunomagnetic beads (Dynabeads M-450; Dynal) and incubated with the whole cell lysate as described (Panigrahi et al., 2001b). The samples bound to the beads were directly assayed for pre-cleaved deletion and insertion editing (Seiwert et al., 1996; Igo et al., 2000, 2002), adenylation (Sabatini and Hajduk, 1995) and endonuclease activity (Salavati et al., 2002).
Assays of enzymatic activities
Self-adenylatable RNA ligases were detected as described (Sabatini and Hajduk, 1995). Briefly, the beads from the immunprecipitations were incubated for 15 min at 28°C with 2.5 µCi [α-32P]ATP in 25 mM HEPES pH 7.9, 10 mM Mg(OAc)2, 50 mM KCl, 0.5 mM DTT and 10% dimethylsulfoxide. The proteins were resolved by 10% SDS–PAGE and the radiolabeled proteins were detected by phosphorImaging. Deletion and insertion editing were assayed in vitro using 5′-labeled 5′CL18, 3′CL13pp substrates with gPCA6-2A RNAs for pre-cleaved insertion assays and 5′CL3U, 3′CLpp and gPCA6-0A, as well as 5′-labeled U5 5′CL and U5 3′CL with gA6[14]PC-del gRNA for pre-cleaved deletion assays (Igo et al., 2000). The reaction products were resolved on polyacrylamide–urea gels and visualized on Storm PhosphorImager screens (Molecular Dynamics). The intensity of bands was quantified by ImageQuant software. Endonuclease activity was assayed using pre-edited CYb 5′end-labeled mRNA with 1 pmol CYb gRNA (30 nt) that directs insertion of 2, 1 and 3 U residues at ES1, ES2 and ES3, respectively, and 5 µl of glycerol-gradient immunoprecipitate as described previously (Salavati et al., 2002).
Acknowledgments
Acknowledgements
We thank Robert P.Igo,Jr and Achim Schnaufer for critical reading of the manuscript. This work was supported by the NIH grant AI14102 and the Human Frontiers of Science Program Organization grant RG0316/1997M.
References
- Biebinger S., Rettenmaier,S., Flaspohler,J., Hartmann,C., Pena-Diaz,J., Wirtz,L.E., Hotz,H.R., Barry,J.D. and Clayton,C. (1996) The PARP promoter of Trypanosoma brucei is developmentally regulated in a chromosomal context. Nucleic Acids Res., 24, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebinger S., Wirtz,L.E., Lorenz,P. and Clayton,C. (1997) Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol., 85, 99–112. [DOI] [PubMed] [Google Scholar]
- Blum B. and Simpson,L. (1990) Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo(U) tail involved in recognition of the preedited region. Cell, 62, 391–397. [DOI] [PubMed] [Google Scholar]
- Brun R. and Schonenberger,M. (1979) Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop., 36, 289–292. [PubMed] [Google Scholar]
- Corell R.A. et al. (1996) Complexes from Trypanosoma brucei that exhibit deletion editing and other editing-associated properties. Mol. Cell. Biol., 16, 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez A.M. and Simpson,L. (1999) Uridine insertion/deletion RNA editing in trypanosome mitochondria—a review. Gene, 240, 247–260. [DOI] [PubMed] [Google Scholar]
- Estevez A.M., Kierszenbaum,F., Wirtz,E., Bringaud,F., Grunstein,J. and Simpson,L. (1999) Knockout of the glutamate dehydrogenase gene in bloodstream Trypanosoma brucei in culture has no effect on editing of mitochondrial mRNAs. Mol. Biochem. Parasitol., 100, 5–17. [DOI] [PubMed] [Google Scholar]
- Feagin J.E. (1990) RNA editing in kinetoplastid mitochondria. J. Biol. Chem., 265, 19373–19376. [PubMed] [Google Scholar]
- Feagin J.E., Jasmer,D.P. and Stuart,K. (1986) Differential mitochondrial gene expression between slender and stumpy bloodforms of Trypanosoma brucei. Mol. Biochem. Parasitol., 20, 207–214. [DOI] [PubMed] [Google Scholar]
- Guerra-Giraldez C., Quijada,L. and Clayton,C. (2002) Compartmentation of enzymes in a microbody, the glycosome, is essential in Trypanosoma brucei. In press. [DOI] [PubMed]
- Hajduk S.L. (1978) Influence of DNA complexing compounds on the kinetoplast of trypanosomatids. In Hahn,F.G. (ed.) Progress in Molecular and Subcellular Biology. Springer, Heidelberg, Germany, Vol 6, pp. 158–200.
- Harris M.E. and Hajduk,S.L. (1992) Kinetoplastid RNA editing: in vitro formation of cytochrome b gRNA–mRNA chimeras from synthetic substrate RNAs. Cell, 68, 1091–1099. [DOI] [PubMed] [Google Scholar]
- Hausler T. and Clayton,C. (1996) Post-transcriptional control of hsp70 mRNA in Trypanosoma brucei. Mol. Biochem. Parasitol., 76, 57–71. [DOI] [PubMed] [Google Scholar]
- Hayman M.L. and Read,L.K. (1999) Trypanosoma brucei RBP16 is a mitochondrial Y-box family protein with guide RNA binding activity. J. Biol. Chem., 274, 12067–12074. [DOI] [PubMed] [Google Scholar]
- Hill G.C. (1976) Electron transport systems in kinetoplastida. Biochim. Biophys. Acta, 456, 149–193. [DOI] [PubMed] [Google Scholar]
- Hinz S. and Goringer,H.U. (1999) The guide RNA database (3.0). Nucleic Acids Res., 27, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H. and Hirumi,K. (1989) Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol., 75, 985–989. [PubMed] [Google Scholar]
- Huang C.E., Cruz-Reyes,J., Zhelonkina,A.G., O’Hearn,S., Wirtz,E. and Sollner-Webb,B. (2001) Roles for ligases in the RNA editing complex of Trypanosoma brucei: band IV is needed for U-deletion and RNA repair. EMBO J., 20, 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo R.P. Jr, Palazzo,S.S., Burgess,M.L., Panigrahi,A.K. and Stuart,K. (2000) Uridylate addition and RNA ligation contribute to the specificity of kinetoplastid insertion RNA editing. Mol. Cell. Biol., 20, 8447–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo R.P. Jr, Weston,D., Ernst,N., Panigrahi,A.K., Salavati,R. and Stuart,K. (2002) Role of uridylate-specific exoribonuclease activity in kinetoplastid RNA editing. Eukaryotic Cell, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable M.L., Seiwert,S.D., Heidmann,S. and Stuart,K. (1996) RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science, 273, 1189–1195. [DOI] [PubMed] [Google Scholar]
- Kable M.L., Heidmann,S. and Stuart,K.D. (1997) RNA editing: getting U into RNA. Trends Biochem. Sci., 22, 162–166. [DOI] [PubMed] [Google Scholar]
- Koller J., Muller,U.F., Schmid,B., Missel,A., Kruft,V., Stuart,K. and Goringer,H.U. (1997) Trypanosoma brucei gBP21. An arginine-rich mitochondrial protein that binds to guide RNA with high affinity. J. Biol. Chem., 272, 3749–3757. [DOI] [PubMed] [Google Scholar]
- Madison-Antenucci S., Sabatini,R.S., Pollard,V.W. and Hajduk,S.L. (1998) Kinetoplastid RNA-editing-associated protein 1 (REAP-1): a novel editing complex protein with repetitive domains. EMBO J., 17, 6368–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov D.A. and Simpson,L. (1992) The polarity of editing within a multiple gRNA-mediated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell, 70, 459–467. [DOI] [PubMed] [Google Scholar]
- McManus M.T., Shimamura,M., Grams,J. and Hajduk,S.L. (2001) Identification of candidate mitochondrial RNA editing ligases from Trypanosoma brucei. RNA, 7, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missel A., Souza,A.E., Norskau,G. and Goringer,H.U. (1997) Disruption of a gene encoding a novel mitochondrial DEAD-box protein in Trypanosoma brucei affects edited mRNAs. Mol. Cell. Biol., 17, 4895–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi A.K. et al. (2001a) Association of two novel proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA editing complex. Mol. Cell. Biol., 21, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi A.K. et al. (2001b) Four related proteins of the Trypanosoma brucei RNA editing complex. Mol. Cell. Biol., 21, 6833–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller K.J., Decker,C.J., Rusche,L.N., Harris,M.E., Hajduk,S.L. and Sollner-Webb,B. (1995) Editing domains of Trypanosoma brucei mitochondrial RNAs identified by secondary structure. Mol. Cell. Biol., 15, 2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard V.W., Harris,M.E. and Hajduk,S.L. (1992) Native mRNA editing complexes from Trypanosoma brucei mitochondria. EMBO J., 11, 4429–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche L.N., Cruz-Reyes,J., Piller,K.J. and Sollner-Webb,B. (1997) Purification of a functional enzymatic editing complex from Trypanosoma brucei mitochondria. EMBO J., 16, 4069–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche L.N., Huang,C.E., Piller,K.J., Hemann,M., Wirtz,E. and Sollner-Webb,B. (2001) The two RNA ligases of the Trypanosoma brucei RNA editing complex: cloning the essential band IV gene and identifying the band V gene. Mol. Cell. Biol., 21, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini R. and Hajduk,S.L. (1995) RNA ligase and its involvement in guide RNA/mRNA chimera formation. Evidence for a cleavage-ligation mechanism of Trypanosoma brucei mRNA editing. J. Biol. Chem., 270, 7233–7240. [DOI] [PubMed] [Google Scholar]
- Salavati R., Panigrahi,A.K., Morach,B.A., Palazzo,S.S., Igo,R.P.,Jr and Stuart,K. (2002) Endoribonuclease activities of Trypanosoma brucei mitochondria. Mol. Biochem. Parasitol., 120, 23–31. [DOI] [PubMed] [Google Scholar]
- Schnaufer A., Panigrahi,A.K., Panicucci,B., Igo,R.P.,Jr, Salavati,R. and Stuart,K. (2001) An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science, 291, 2159–2162. [DOI] [PubMed] [Google Scholar]
- Seiwert S.D., Heidmann,S. and Stuart,K. (1996) Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell, 84, 831–841. [DOI] [PubMed] [Google Scholar]
- Stuart K.D. (1971) Evidence for the retention of kinetoplast DNA in an acriflavine-induced dyskinetoplastic strain of Trypanosoma brucei which replicates the altered central element of the kinetoplast. J. Cell Biol., 49, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K. (1980) Cultivation of dyskinetoplastic Trypanosoma brucei. J. Parasitol., 66, 1060–1061. [PubMed] [Google Scholar]
- Stuart K., Allen,T.E., Heidmann,S. and Seiwert,S.D. (1997) RNA editing in kinetoplastid protozoa. Microbiol. Mol. Biol. Rev., 61, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K., Panigrahi,A.K., Schnaufer,A., Drozdz,M., Clayton,C. and Salavati,R. (2002) Composition of the editing complex of Trypanosoma brucei. Philos. Trans. R. Soc. Lond. B Biol. Sci., 357, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhamme L. et al. (1998) Trypanosoma brucei TBRGG1, a mitochondrial oligo(U)-binding protein that co-localizes with an in vitro RNA editing activity. J. Biol. Chem., 273, 21825–21833. [DOI] [PubMed] [Google Scholar]
- Wirtz E. and Clayton,C. (1995) Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science, 268, 1179–1183. [DOI] [PubMed] [Google Scholar]
- Zweygarth E., Kaminsky,R. and Webster,P. (1990) Trypanosoma brucei evansi: dyskinetoplasia and loss of infectivity after long-term in vitro cultivation. Acta Trop., 48, 95–99. [DOI] [PubMed] [Google Scholar]