

Abstract
Cell cycle progression in eukaryotes is mediated by phosphorylation of protein substrates by the cyclin-dependent kinases (CDKs). We screened a cDNA library by solid-phase phosphorylation and isolated hHR6A as a CDK2 substrate. hHR6A is the human homologue of the product of the Saccharomyces cerevisiae RAD6/UBC2 gene, a member of the family of ubiquitin-conjugating enzymes. hHR6A is phosphorylated in vitro by CDK-1 and -2 on Ser120, a residue conserved in all hHR6A homologues, resulting in a 4-fold increase in its ubiquitin-conjugating activity. In vivo, hHR6A phosphorylation peaks during the G2/M phase of cell cycle transition, with a concomitant increase in histone H2B ubiquitylation. Mutation of Ser120 to threonine or alanine abolished hHR6A activity, while mutation to aspartate to mimic phosphorylated serine increased hHR6A activity 3-fold. Genetic complementation studies in S.cerevisiae demonstrated that hHR6A Ser120 is critical for cellular proliferation. This is the first study to demonstrate regulation of UBC function by phosphorylation on a conserved residue and suggests that CDK-mediated phosphorylation of hHR6A is an important regulatory event in the control of cell cycle progression.
Keywords: cell cycle/phosphorylation/Rad6/ubiquitin
Introduction
In eukaryotes, cell cycle progression is mediated by the sequential activation and inactivation of the cyclin-dependent kinases (CDKs). Active CDKs consist of a protein kinase subunit whose catalytic activity is dependent on the association with a regulatory cyclin subunit (Pines, 1993). In mammalian cells, mitogenic stimulation results in the expression of D-type cyclins which associate with either CDK4 or CDK6 to mediate G1 phase cell cycle progression. Progression from G1 to S phase is controlled by cyclin E–CDK2, with cyclin A–CDK2 mediating S phase progression. Cyclin A–CDC2 is required during the G2 phase of the cell cycle and, finally, transition throughout mitosis is regulated by cyclin B–CDC2 (Grana and Reddy, 1995). Although the mechanisms of cyclin–CDK regulation have largely been unveiled, our understanding of CDK substrates and how phosphorylation alters their function to execute cell cycle transitions remains to be fully defined. In the present study, we have used an in vitro phosphorylation screening method to identify the ubiquitin-conjugating enzyme (UBC), hHR6A, as a CDK substrate.
The ubiquitin pathway plays an important role in the regulation of a broad array of biological processes such as control of cell cycle regulation, signal transduction pathways, apoptosis and DNA repair (Hershko and Ceichanover, 1998). This pathway involves conjugation of the conserved 76 amino acid ubiquitin peptide to substrate proteins via a three-step cascade, which then targets the protein for proteolytic degradation by the 26S proteasome (Ciechanover, 1998; Hershko and Ceichanover, 1998). In this cascade, ubiquitin initially is activated by forming a thioester bond between its C-terminal glycine and a cysteine in the ubiquitin-activating enzyme (E1). After activation, ubiquitin is transferred to a cysteine on an E2 UBC and subsequently covalently attached to the substrate protein either directly, or with the cooperation of a ubiquitin protein ligase (E3). This covalent attachment involves the formation of an isopeptide bond with the N-terminus or an ε-NH2 group of an internal lysine residue.
A central role in the ubiquitin pathway is played by the family of UBC enzymes, which in the yeast Saccharomyces cerevisiae comprises at least 13 members (Ciechanover, 1998). An example of the importance of this family of enzymes is exemplified by the S.cerevisiae Rad6 protein. Genetic studies have shown that yeast rad6 mutants display a pleiotropic phenotype including defects in DNA repair, proteolysis of N-end rule protein substrates, cellular proliferation, cell cycle progression and an inability to sporulate (Watkins et al., 1993; Lawrence, 1994), which are all dependent on its ubiquitin-conjugating activity (Sung et al., 1990). Rad6 can ubiquitylate histones in vitro, and ubiquitylation of histone H2B in S.cerevisiae is Rad6 dependent (Robzyk et al., 2000). In mice and humans, two homologues of RAD6 have been identified, termed HR6A (homologue of RAD6A) and HR6B (homologue of RAD6B) (Koken et al., 1991; Roest et al., 1996). Studies with the human RAD6 homologues show that both can replace the DNA repair functions of S.cerevisiae Rad6 (Koken et al., 1991). In whole animal studies, loss of the mouse HR6B gene caused male infertility due to impaired spermatogenesis (Roest et al., 1996), which was postulated to result from defects in histone ubiquitylation and chromatin modification (Koken et al., 1996). One major function of Rad6 is regulating cell cycle progression, since S.cerevisiae that lack Rad6 display defects in transit through the S/G2 phase of the cell cycle (Ellison et al., 1991).
Our studies show that hHR6A is phosphorylated in vitro by CDK-1 and -2 on serine residue 120, which is conserved in all hHR6A homologues, leading to an increase in the ubiquitin-conjugating activity of this enzyme. Phosphorylation of hHR6A Ser120 is increased during the G2/M phase of the cell cycle when ubiquitylation of its known substrate, histone H2B, is also increased. Site-directed mutagenesis studies identify Ser120 as a critical regulatory site for hHR6A ubiquitin-conjugating activity, and genetic studies demonstrate that this site is important for the in vivo function of this enzyme. These studies provide the first evidence of UBC regulation by phosphorylation on a conserved residue and suggest an interplay between CDKs and hHR6A to mediate cell cycle progression.
Results
hHR6A is phosphorylated by cyclin–CDKs in vitro and during the G2/M phase of the cell cycle in vivo
In order to identify potential CDK substrates, we used purified cyclin A–CDK2 to perform phosphorylation screening (Fukunaga and Hunter, 1997) of a λ phage breast epithelial cell cDNA expression library (Daly et al., 1996). From a screen of 50 000 clones, ∼100 positive clones were identified. Of these, 30 were sequenced, identifying 16 substrates, including the previously described CDK substrate nucleolar protein B23 (Peter et al., 1990; Okuda et al., 2000). We chose to study further a clone encoding hHR6A, also referred to as UBC2 (Koken et al., 1991), since previous work has implicated a cell cycle role for this UBC (Ellison et al., 1991). The consensus phosphorylation sequence for CDKs is a phosphorylated serine or threonine residue immediately flanked by a proline residue C-terminal to the phosphorylated site (Nigg, 1993). Examination of the hHR6A amino acid sequence revealed four potential CDK phosphorylation sites, including threonine residues 3 and 47 and serine residues 97 and 120 (Figure 1). Sequence alignment of the two human RAD6 homologues, hHR6A and hHR6B, with Rad6 homologues from other species showed that Thr47 and Ser120 are entirely conserved, suggesting that these residues may be important for function (Figure 1).

Fig. 1. hHR6A CDK consensus phosphorylation sites and aligned sequences of hHR6A homologues. The CDK consensus phosphorylation sites on hHR6A are boxed. The sequence representing identical amino acids of hHR6A and its homologues, including human RAD6B (hHR6B), Drosophila melanogaster, Neurospora crassa, S.cerevisiae and Schizosaccharomyces pombe Rad6, is in bold and italicized.
To evaluate which cyclin–CDKs phosphorylate hHR6A, recombinant purified hHR6A was phosphorylated with different cyclin–CDKs in vitro. hHR6A was phosphorylated efficiently by cyclin E–CDK2, cyclin A– CDK2, cyclin A–CDC2 and cyclin B–CDC2, but not by cyclin D1–CDK4, when compared with the ability of these kinases to phosphorylate the retinoblastoma protein, a physiological CDK substrate (Grana and Reddy, 1995) (Figure 2A). To determine whether hHR6A was also a phosphoprotein in vivo, we constructed a mammalian expression vector containing hHR6A with an N-terminal FLAG epitope tag. Transient transfection of Chinese hamster ovary (CHO) cells and [32P]phosphate labelling followed by anti-FLAG immunoprecipitation of the expressed hHR6A showed that hHR6A was phosphorylated in vivo (Figure 2B). Phosphorylation of hHR6A was dramatically reduced in the presence of the CDK-1 and -2 inhibitor roscovitine (50 µM) (Meijer et al., 1997) (Figure 2B), indicating that hHR6A phosphorylation in vivo is dependent on cyclin–CDK activity. To determine whether hHR6A is phosphorylated during a particular cell cycle phase, CHO cells transfected with hHR6A were arrested at the G2/M phase of the cell cycle with nocodazole and then induced to re-enter the cell cycle by nocodazole removal. Analysis of DNA content by flow cytometry showed that nocodazole treatment arrested >90% of the cells in G2/M phase (Figure 2C). Following nocodazole removal, the cells initiated synchronous cell cycle progression, peaking in G1 and S phases at 3 and 9 h, respectively, and progressing through G2 after 12 h. Analysis of hHR6A phosphorylation revealed maximal phosphorylation during the G2/M cell cycle phase (Figure 2D). Interestingly, a phosphoprotein co-immunoprecipitating with hHR6A and migrating several kilodaltons slower was also observed. The identity of this phosphoprotein whose phosphorylation peaked during the S/G2 cell cycle phase is currently under investigation.
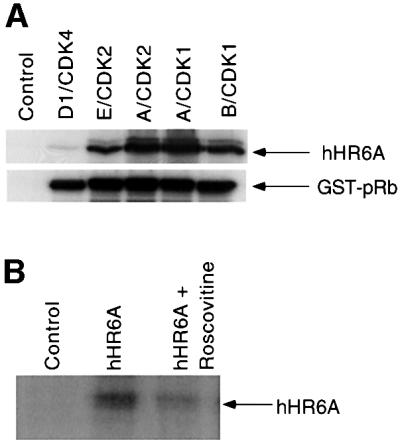

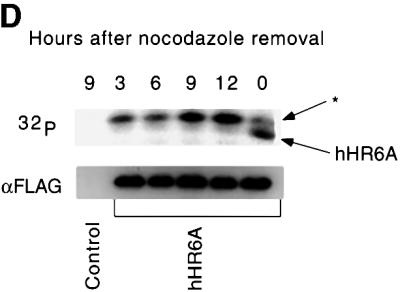
Fig. 2. hHR6A is a CDK substrate in vitro and is phosphorylated during the G2/M cell cycle phase in vivo. (A) Purified His6-hHR6A (top panel) or GST–pRb773–928 (bottom panel) was incubated in the presence of [γ-32P]ATP, in either the absence (Control) or presence of purified cyclin D1–CDK4, cyclin E–CDK2, cyclin A–CDK2, cyclin A–CDK1 or cyclin B–CDK1, resolved by SDS–PAGE and visualized by autoradiography. (B) CHO cells transfected with either pCMV-Tag2 vector (Control) or pCMV-Tag2-hHR6A (hHR6A) were labelled with [32P]phosphate and incubated with either dimethylsulfoxide (left and middle lanes) or 50 µM roscovitine (right lane) during the labelling period. Following labelling, hHR6A was immunoprecipitated, separated by SDS–PAGE and visualized by autoradiography. (C) CHO cells transfected with pCMV-Tag2-hHR6A (hHR6A) were synchronized by blocking in the G2/M cell cycle phase with nocodazole, then initiated to re-enter the cell cycle by nocodazole removal, harvested at various time points for DNA analysis by flow cytometry, and the proportion of cells in G1, S and G2M phases determined. (D) CHO cells transfected with either pCMV-Tag2 vector (Control) or pCMV-Tag2-hHR6A (hHR6A) were synchronized by blocking in the G2/M cell cycle phase with nocodazole (time = 0), initiated to re-enter the cell cycle by nocodazole removal and then lysed at various time points (as indicated). The cells were pulse-labelled with [32P]phosphate for 3 h prior to lysis. Following lysis, hHR6A was immunoprecipitated, separated by SDS–PAGE and visualized by autoradiography (upper panel) or western blotting with an anti-FLAG antibody (lower panel). Phosphorylated hHR6A and the co-immunoprecipitating phosphoprotein (asterisk) are indicated with arrows.
hHR6A is phosphorylated on Ser120 in vivo and on the same site by cyclin–CDKs in vitro
Since hHR6A is phosphorylated during the G2/M phase, we sought to determine whether it is phosphorylated on cyclin–CDK consensus phosphorylation site(s). This was addressed by expressing wild-type and phospho-site mutant hHR6As in [32P]phosphate-labelled CHO cells which were analysed by phosphoamino acid analysis and tryptic phosphopeptide mapping. Phosphoamino acid analysis showed that hHR6A is phosphorylated exclusively on serine in vivo (Figure 3A, upper panel). Since hHR6A contains two potential serine cyclin–CDK phosphorylation sites at residues 97 and 120 (Figure 1), we mutated these amino acids individually to alanine (S97A and S120A) and analysed hHR6A phosphorylation by tryptic phosphopeptide mapping. These studies demonstrated that wild-type hHR6A generated one major phosphopeptide (Figure 3B, upper panel). While the S97A mutant yielded an identical map, this phosphopeptide was abolished in the S120A mutant, indicating that Ser120 is the major phosphorylation site of hHR6A in vivo.

Fig. 3. hHR6A is phosphorylated on Ser120 in vivo and on the same site by cyclin A–CDK2 in vitro. (A) Wild-type hHR6A was transfected into CHO cells. Following [32P]phosphate labelling, hHR6A was immuno precipitated and phosphoamino acid analysis performed (upper panel, In vivo). Recombinant purified wild-type hHR6A was phosphorylated in vitro with cyclin A–CDK2 and subjected to phosphoamino acid analysis (lower panel, In vitro). The positions of phosphoserine (P-Ser), phosphothreonine (P-Thr) and phosphotyrosine (P-Tyr) are indicated with arrows. (B) Wild-type, S97A or S120A hHR6A was transfected into CHO cells. Following [32P]phosphate labelling, hHR6A was immunoprecipitated and subjected to tryptic phosphopeptide mapping (upper panels, In vivo). Recombinant purified wild-type, S120A or T3A hHR6A was phosphorylated with cyclin A–CDK2 in vitro and subjected to tryptic phosphopeptide mapping (lower panels, In vitro). The major phosphopeptides are indicated with arrows. The origin is labelled ‘o’.
Since hHR6A has four potential cyclin–CDK phosphorylation sites (Figure 1), we phosphorylated purified recombinant wild-type and phospho-site mutant hHR6As with cyclin–CDKs in vitro to confirm that Ser120 is phosphorylated. Phosphoamino acid analysis revealed that hHR6A was phosphorylated in vitro on serine and threonine by cyclin A–CDK2 (Figure 3A, lower panel). Phosphorylation of wild-type hHR6A with cyclin A– CDK2 generated four major tryptic phosphopeptides (Figure 3B, lower panel). Phosphorylation of S120A hHR6A resulted in the loss of phosphopeptide 1, indicating that this phosphopeptide results from phosphorylation of Ser120. Since hHR6A is also phosphorylated on threonine in vitro, we mutated Thr3 to alanine (T3A) to determine whether this site is phosphorylated. Phosphorylation of hHR6A T3A resulted in the loss of phosphopeptides 2–4, indicating that Thr3 is also phosphorylated by cyclin A–CDK2 in vitro. The same data were obtained when hHR6A was phosphorylated with cyclin E–CDK2, cyclin A–CDK1 or cyclin B–CDK1 (data not shown). The generation of three phosphopeptides from phosphorylation of Thr3 is probably due to the generation of partial digests by tryptic cleavage at arginine residues 6–8 (Figure 1). These studies therefore reveal that hHR6A Ser120 and Thr3 are phosphorylated by CDK-1 and -2 in vitro.
Cyclin–CDK-mediated phosphorylation of hHR6A Ser120 increases its ubiquitin-conjugating activity in vitro
In order to define whether there are any functional consequences on hHR6A ubiquitin-conjugating activity following CDK phosphorylation, we established a two-step phosphorylation/ubiquitylation assay. Purified hHR6A was first phosphorylated by cyclin A–CDK2 and then assayed for its ubiquitin-conjugating activity towards histone H2A, a known in vitro substrate of Rad6 (Sung et al., 1988). Following phosphorylation of hHR6A and prior to initiation of ubiquitylation, the cyclin A–CDK2 was removed by immunodepletion with an anti-CDK2 antibody (data not shown), to ensure that any change in ubiquitylation was not due to phosphorylation of other components in the ubiquitylation reaction, such as E1 or histone H2A. These studies revealed that the level of histone ubiquitylation was increased 4-fold when hHR6A was phosphorylated by cyclin A–CDK2 (Figure 4A, upper panel), in addition to an increased level of hHR6A auto-ubiquitylation (Figure 4A, lower panel).


Fig. 4. Ser120 is critically important for ubiquitin-conjugating activity, and phosphorylation of this site increases hHR6A activity in vitro. (A) hHR6A was either unphosphorylated (lanes 1, 3, 4 and 6) or phosphorylated with cyclin A–CDK2 (lane 2). Following phosphorylation, cyclin A–CDK2 was immunodepleted and the ubiquitylation reaction performed. Lanes 3–6 are control lanes lacking histone H2A, GST–ubiquitin, hHR6A and E1, respectively (upper panel). hHR6A was either unphosphorylated or phosphorylated with cyclin A–CDK2 (as indicated) and the ubiquitylation reaction performed for either 20, 40 or 60 min (lower panel). Ubiquitylated histone H2A (H2A-Ub) and auto-ubiquitylated hHR6A (hHR6A-Ub) are indicated with arrows. (B) The ubiquitylation activity of wild-type, S120A, S120T, S120D and S120E hHR6As towards histone H2A. Ubiquitylated histone H2A (H2A-Ub) is indicated with an arrow. (C) Wild-type or S120E hHR6A was either unphosphorylated or phosphorylated with cyclin A–CDK2 (A/CDK2). Following phosphorylation, cyclin A–CDK2 was immunodepleted and the ubiquitylation reaction performed (30 min for wild-type hHR6A and 60 min for S120E hHR6A). Ubiquitylated histone H2A is indicated with an arrow.
We next sought to determine whether this increase in ubiquitin-conjugating activity was due to phosphorylation of hHR6A Ser120, since cyclin A–CDK2 phosphorylated both Ser120 and Thr3 in vitro. To determine the importance of Ser120 to hHR6A ubiquitin-conjugating activity, we generated hHR6A mutants where Ser120 was substituted for either threonine (S120T), alanine (S120A), aspartate (S120D) or glutamate (S120E). The rationale for generating these mutations was that S120A, S120D and S120E represent mutants that cannot be phosphorylated at this site and therefore should not be activated by cyclin A– CDK2, while substitution to the closely related threonine represents hHR6A similar to the wild-type form. The S120D mutant was generated potentially to mimic phosphorylated serine, due to the negatively charged carboxyl group on aspartate. The S120E mutant was generated to determine whether the conformation and size in addition to the charge of the side group was important for activity, since the carboxyl-containing side group of glutamate is larger than aspartate. Substitution of Ser120 to alanine or threonine almost completely abolished the ubiquitin-conjugating activity of hHR6A (Figure 4B). The S120E mutant displayed 35% of the activity of wild-type hHR6A. Conversely, the S120D mutant displayed 3-fold higher activity than wild-type hHR6A, similar to the 4-fold increase observed following phosphorylation of hHR6A (Figure 4B). These data indicate that a negative charge in this position is preferred, consistent with the notion that introduction of a negative charge at this site by phosphorylation of serine is responsible for increased hHR6A ubiquitin-conjugating activity. Interestingly, the S120D hHR6A mutant was 9-fold more active than the S120E mutant despite their similar structures, demonstrating that the size and conformation of the side group at position 120 are also important determinants of hHR6A ubiquitin-conjugating activity. To confirm directly that phosphorylation of this site by cyclin A–CDK2 is responsible for the increased ubiquitin-conjugating activity of hHR6A, we measured the ubiquitin-conjugating activity of the S120E mutant that cannot be phosphorylated on this site. These studies revealed that phosphorylation of wild-type and not S120E hHR6A by cyclin A–CDK2 resulted in increased ubiquitin-conjugating activity (Figure 4C), confirming that this is due to phosphorylation of Ser120.
Histone H2B ubiquitylation is increased in vivo during the G2/M phase of the cell cycle
Since our studies demonstrated that phosphorylation of hHR6A Ser120 can increase its ubiquitin-conjugating activity in vitro, we sought to determine whether increased hHR6A phosphorylation during G2/M phase leads to increased substrate ubiquitylation in vivo. We analysed the level of histone H2B ubiquitylation since previous studies have shown that, while Rad6 can ubiquitylate both histones H2A and H2B in vitro (Sung et al., 1988), ubiquitylation of histone H2B is Rad6 dependent in S.cerevisiae (Robzyk et al., 2000). Basic nuclear proteins containing histones were prepared from CHO cells at different stages of the cell cycle, and histone H2B was analysed by immunoblotting. Immunoblotting with an anti-histone H2B antibody revealed the presence of a major immunoreactive band migrating at ∼22 kDa at all cell cycle stages, corresponding to unmodified histone H2B (Figure 5, upper panel). Another band migrating at ∼29 kDa was also detected, corresponding to the predicted size of mono-ubiquitylated histone H2B. Unlike the 22 kDa unmodified histone H2B where levels were invariant at the different cell cycle stages, the level of the 29 kDa band was significantly increased in G2/M phase cells. To confirm that this band represents ubiquitylated histone H2B, we also performed immunoblotting of the same histone preparations with an anti-ubiquitin antibody (Figure 5, lower panel). These results demonstrated that the 29 kDa protein recognized by the histone H2B antibody was also cross-reactive with a ubiquitin antibody and, in agreement with the anti-histone H2B immunoblot, the intensity of this band was significantly increased at the G2/M cell cycle phase. These results demonstrate that ubiquitylation of the known Rad6 substrate, histone H2B, is increased during the G2/M phase, supporting the view that CDK-mediated phosphorylation and activation of hHR6A during this cell cycle phase (Figure 2) leads to increased histone H2B ubiquitylation.

Fig. 5. Increased ubiquitylation of histone H2B during the G2/M cell cycle phase. CHO cells were synchronized by blocking in the G2/M cell cycle phase with nocodazole, then initiated to re-enter the cell cycle by nocodazole removal, harvested at various time points (as indicated) and histones prepared. Histones were separated by SDS–PAGE and then western blotted with an anti-histone H2B (top panel) or anti-ubiquitin antibody (lower panel).
hHR6A Ser120 is critical for cellular proliferation
To determine whether Ser120 is important for hHR6A function in vivo, we adopted a genetic approach using S.cerevisiae, since previous work has shown that hHR6A can functionally complement some functions of the yeast homologue, Rad6 (Koken et al., 1991). We tested wild-type and mutant hHR6As for complementation of the RAD6 cell proliferation phenotype, since S.cerevisiae lacking Rad6 display a markedly reduced rate of cellular proliferation due to defects in cell growth and cell cycle progression (Ellison et al., 1991; Freiberg et al., 2000). Wild-type, S120A, S120T, S120D or S120E mutants of hHR6A were transformed into S.cerevisiae rad6Δ, lacking the yeast RAD6 gene. Immunoblotting of lysates prepared from the transformants revealed that all forms of hHR6A were expressed to equivalent levels at either 30 or 37°C (Figure 6A). The growth of the transformants was then tested at 30 and 37°C, since the proliferative defect of S.cerevisiae lacking Rad6 is significantly more pronounced at 37°C (Ellison et al., 1991; McDonough et al., 1995; Freiberg et al., 2000). These studies showed that at 30°C the wild-type S.cerevisiae and all of the S.cerevisiae rad6Δ transformants grew at a similar rate (Figure 6B). However, at 37°C, the S.cerevisiae rad6Δ strain transformed with empty plasmid displayed significantly reduced growth. The S.cerevisiae rad6Δ transformants expressing S120A or S120T hHR6A also grew poorly and at the same rate as the S.cerevisiae rad6Δ strain transformed with plasmid vector alone. Conversely, the transformants expressing wild-type and S120D hHR6A restored the growth of the S.cerevisiae rad6Δ strain to a similar extent to that of wild-type S.cerevisiae. Interestingly, the transformant expressing S120E hHR6A grew at an intermediate rate. Therefore, the ability of the wild-type and hHR6A mutants to complement the proliferative defect of the S.cerevisiae rad6Δ strain at 37°C paralleled their ubiquitin-conjugating activity (Figure 4B), suggesting that modulating hHR6A enzymatic activity is important for controlling the rate of cellular proliferation.
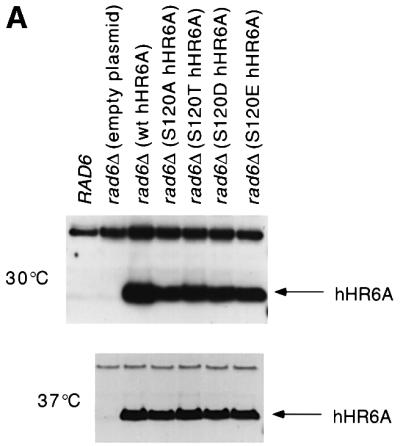

Fig. 6. Complementation of the S.cerevisiae rad6Δ growth defect with wild–type and mutant alleles of hHR6A. The S.cerevisiae rad6Δ strain was transformed with either empty plasmid or plasmid expressing either wild-type, S120A, S120T, S120D or S120E hHR6A. (A) The expression of wild-type and mutant hHR6As in S.cerevisiae rad6Δ. Lysates were prepared from either wild-type S.cerevisiae (RAD6) or the S.cerevisiae rad6Δ transformants grown at either 30 (top panel) or 37°C (lower panel). Equal amounts of protein were separated by SDS–PAGE, transferred to nitrocellulose and immunoblotted using an anti-hHR6A polyclonal antibody. (B) Wild-type S. cerevisiae (RAD6) or S.cerevisiae rad6Δ transformants were grown at either 30 or 37°C. Ten-fold serial dilutions (from left to right) of exponential phase cultures were spotted on plates and incubated at the indicated temperatures.
Discussion
In this study, we used a phosphorylation screening approach to identify hHR6A as a CDK substrate. This method has been utilized previously to identify physiological protein kinase substrates including the MAPK substrate MNK1 (Fukunaga and Hunter, 1997) and the CDK substrate PRC1 (Jiang et al., 1998). Using cyclin A– CDK2, we isolated 16 substrates from a partial screen of a breast epithelial cell cDNA library, including the known CDK substrates nucleophosmin/B23 (Okuda et al., 2000) and topoisomerase II (Cardenas et al., 1992). This study characterizes hHR6A, since previous work has shown that the hHR6A yeast homologue, Rad6, is implicated in regulation of cell cycle progression (Ellison et al., 1991) and functions in DNA repair (Koken et al., 1991). Since CDKs are essential for cell cycle progression and in the initiation and completion of DNA synthesis, this raised the possibility that hHR6A function may be regulated by CDK-mediated phosphorylation during the cell cycle.
Since we isolated hHR6A using an in vitro phosphorylation screen, we sought to determine whether hHR6A is a phosphoprotein in vivo, and whether it is phosphorylated on CDK consensus phosphorylation sites. Phosphoamino acid analysis of hHR6A transfected into CHO cells revealed that the protein was phosphorylated exclusively on serine residues, and mutation of the two potential serine CDK consensus phosphorylation sites revealed that Ser120 was phosphorylated in vivo. The CDK-1 and -2 inhibitor roscovitine (Meijer et al., 1997) significantly decreased phosphorylation of Ser120 in vivo, indicating that phosphorylation was CDK mediated. Analysis of hHR6A phosphorylation in synchronized cells demonstrated that phosphorylation was dramatically increased during the G2/M phase of cell cycle transition. Interestingly, in addition to Ser120, cyclin A–CDK2 also phosphorylated Thr3 in vitro. The reason for this differential phosphorylation in vivo and in vitro is unclear, but may be due to other proteins interacting with hHR6A at or near potential phosphorylation sites to preclude their phosphorylation by CDKs in vivo. Support for this concept comes from studies showing that the N-terminal amino acids 1–9 of Rad6 bind to the Ubr1 E3 protein in vivo (Watkins et al., 1993). A recent study showing that protein kinase A phosphorylates free histone H3 but not when it is bound within nucleosomes, confirms that association of a substrate protein with other proteins can influence its phosphorylation by a protein kinase (Hsu et al., 2000).
Sequence alignment of hHR6A with all its homologues from different species shows that Ser120 and the three amino acids immediately C-terminal to this site, encoding the sequence SPAN, are entirely conserved (Worthylake et al., 1998) (Figure 1), suggesting that this region plays a critical role in hHR6A function. Our in vitro phosphorylation studies show that phosphorylation of Ser120 increases the ubiquitin-conjugating activity of hHR6A 4-fold. Mutation of this site revealed that it is critically important for hHR6A enzymatic activity. Conservative mutation to the closely related threonine or alanine almost completely abolished hHR6A activity. Conversely, substitution to an aspartic acid to introduce a negatively charged carboxyl side group, to mimic phosphoserine, increased hHR6A activity 3-fold, similar to that observed when wild-type hHR6A was phosphorylated. Surprisingly, substitution to the closely related glutamic acid resulted in 65% loss of hHR6A enzymatic activity. Therefore, the presence of a negative charge in the side group is not the sole determinant of increased activity, but the size and conformation of the side group are also important. This is the first study to demonstrate that mutation of a conserved residue can both increase and decrease the activity of a UBC belonging to the Rad6 family and therefore underscores the importance of Ser120 as a hHR6A regulatory site. Previous studies have shown that mutation of conserved residues in S.cerevisiae Rad6 either have no effect or significantly reduce the activity of Rad6, both in vitro (McDonough et al., 1995) and in vivo (Freiberg et al., 2000). For example, mutation of the three arginine residues in the N-terminus of Rad6 to alanine significantly reduced the proliferation of S.cerevisiae at 37°C (McDonough et al., 1995). Interestingly, our in vitro ubiquitylation assays demonstrated that hHR6A auto-ubiquitylates, as has been described previously for yeast UBC3 (Banerjee et al., 1993), suggesting that it may regulate its own degradation. The level of auto-ubiquitylation was significantly lower in the presence of histone H2A, suggesting that hHR6A auto-ubiquitylation in vivo is dependent on the presence of its substrates.
The crystal structure of S.cerevisiae Rad6 provides an insight into the potential importance of Ser120 phosphorylation. These studies show that residues 115–121 form a mobile loop above the catalytic cysteine that can adopt different conformations, thereby affecting the exposure of this residue (Worthylake et al., 1998). Our results suggest that phosphorylation of Ser120 favours a conformation with a more exposed active site cysteine, resulting in higher catalytic activity. Apart from the regulation of hHR6A, our data have raised issues that may be of general importance to the structure and function of other UBCs. All UBCs have a core catalytic domain of ∼150 amino acids with at least 25% sequence identity (Cook et al., 1993; Cook, 1997). Examination of the aligned sequences of 12 different UBC enzymes from several different species, including human, rabbit, wheat and yeast, reveals minimal divergence at the site equivalent to hHR6A Ser120, with either a serine or an aspartic acid in this position (Figure 7). The minimal variance of amino acids in this position suggests that this site is crucial to the function of most UBCs, in agreement with our studies on hHR6A. This is supported further by the deduced three-dimensional structure of UBC4 (Cook et al., 1993) and UBC7 (Cook, 1997) from S.cerevisiae, which demonstrates that the conserved aspartate or serine residue located within the loop structure in these UBCs is immediately proximal to the catalytic cysteine. Further studies will be needed to establish whether the equivalent site in other UBCs is important for their activity and regulation.

Fig. 7. Aligned sequences of hHR6A and other UBCs encompassing the region of hHR6A Ser120 from several species, including human (Homo sapiens), rabbit (Oryctolagus cuniculus), wheat (Triticum aestivum) and yeast (S.cerevisiae). Protein sequences were obtained from the Swiss-Prot database. Ser120 of hHR6A and the proline residue immediately C-terminal to it, and the equivalent aligned residues in the other UBCs are in bold and italicized.
Previous work in S.cerevisiae has shown that ubiquitylation of histone H2B in vivo is dependent on the hHR6A homologue Rad6 (Robzyk et al., 2000). Our studies demonstrate that increased hHR6A phosphorylation during G2/M phase is associated with a concomitant increase in histone H2B ubiquitylation, providing strong support for the view that this is due to increased hHR6A activity following CDK-mediated phosphorylation. These results are consistent with previous studies demonstrating that ubiquitylation of histones H2A and H2B increases as cells progress through to prophase of the cell cycle and then decreases during the metaphase–anaphase transition (Mueller et al., 1985). Furthermore, neither the mRNA nor the protein levels of hHR6A and hHR6B vary throughout the cell cycle in HeLa cells (Koken et al., 1996), indicating that increased histone ubiquitylation is not due to increased levels of these enzymes. Although the function of cell cycle-dependent histone ubiquitylation is unclear, it has been postulated to play a role in the condensation of specific chromosomal regions (Mueller et al., 1985). Studies in a mutant strain of S.cerevisiae containing histone H2B, where the conserved lysine ubiquitylation site was mutated to arginine, exhibited a reduced growth rate (Robzyk et al., 2000). This was due to delays in the late S or G2/M phases of the cell cycle, suggesting that histone ubiquitylation plays important roles during cell cycle progression.
The importance of Ser120 to hHR6A function in vivo was evaluated by genetic complementation studies in a S.cerevisiae rad6Δ strain lacking the hHR6A functional homologue, Rad6. The rationale for this approach was based on studies demonstrating that hHR6A can complement Rad6 functions in S.cerevisiae (Koken et al., 1991). Previous studies have shown that S.cerevisiae lacking Rad6 display a significantly reduced rate of proliferation due to defects in cell cycle and cell growth functions, with defects in the S/G2 phase of the cell cycle (Ellison et al., 1991). We therefore investigated whether hHR6A can complement the role of Rad6 in cellular proliferation and evaluated the importance of Ser120 towards this function. Our studies demonstrated that wild-type and S120D hHR6A restored the growth of the S.cerevisiae rad6Δ to an extent similar to that of wild-type S.cerevisiae. In contrast, the hHR6A S120A and S120T mutants failed to restore normal growth of S.cerevisiae rad6Δ. Interestingly, the S120E mutant restored some growth, but not to the level observed with wild-type and S120D hHR6A. Therefore, the ability of hHR6A to rescue the growth of S.cerevisiae rad6Δ closely paralleled the ubiquitin-conjugating activity of hHR6A, indicating that the rate of cellular proliferation can be regulated by modulating the level of hHR6A ubiquitin-conjugating activity. These studies show that CDK-mediated phosphorylation of hHR6A Ser120 plays an important role in the regulation of the activity of this enzyme during the G2/M phase cell cycle transition and cellular proliferation. Further identification of hHR6A substrates will be important in increasing our understanding of the roles of this enzyme in cell proliferation.
Materials and methods
In vitro phosphorylation screening of a λ phage breast epithelial cell cDNA library for cyclin–CDK substrates
Cyclin–CDKs were prepared as described previously (Sarcevic et al., 1997). Prior to phosphorylation of a human breast epithelial cell cDNA library (Daly et al., 1996), cyclin A–CDK2 was assessed for activity using GST–pRB773–928 (Sarcevic et al., 1997) as substrate to ensure equivalent amounts of kinase activity were added in different experiments. The phosphorylation screening protocol was performed as described previously (Fukunaga and Hunter, 1997), with some modifications. The cDNA library was plated onto Escherichia coli BL21(De3)pLysE cells at 5 × 103 plaques/150 mm plate. The plates were incubated at 37°C and, following the appearance of plaques, overlaid with nitrocellulose (BA85, Schleicher and Schuell) pre-equilibrated in 10 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The plates were incubated overnight at 34°C, and the filters were removed and immersed in blocking solution [20 mM Tris–HCl pH 8.0, 150 mM NaCl, 3% bovine serum albumin (BSA)] for 60 min at room temperature with gentle agitation. The filters were washed 3 × 20 min in Triton wash buffer [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 10 mM EDTA, 1 mM EGTA, 0.5% Triton X-100, 1 mM dithiothreitol (DTT) and 0.2 mM phenylmethylsulfonyl fluoride (PMSF)] and then in CDK reaction buffer (20 mM HEPES pH 7.5, 10 mM MgCl2, 5 mM β-glycerophosphate, 5 mM NaF, 50 µM Na3VO4, 2 mM DTT and 0.1% Triton X-100) for 10 min prior to incubation in the same buffer containing 25 µM ATP for 60 min. The nitrocellulose was washed for 10 min in CDK reaction buffer without ATP. The filters were then subjected to phosphorylation for 60 min at room temperature with gentle agitation by adding 100 U/ml of cyclin A–CDK2 (1 U of activity is capable of transferring 1 pmol phosphate/µg GST–pRb773–928/min at 37°C), 25 µM ATP and 5 µCi/ml [γ-32P]ATP in CDK reaction buffer. Following phosphorylation, the nitrocellulose was washed 6 × 5 min with wash buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 10 mM EDTA, 1 mM EGTA, 20 mM NaF and 0.1% Triton X-100) and then for 5 min in the same buffer lacking Triton X-100. The nitrocellulose was dried and exposed to X-ray film for autoradiography. Positive clones were subjected to secondary and, in some instances, tertiary screening to obtain individual clones. The DNA insert from the recombinant λEXlox phage was excised into the corresponding λEXlox plasmid by infecting E.coli strain BM25.8 cells. The plasmid isolated from these cells was transformed into E.coli DH5α cells, prior to preparation of plasmid DNA for sequencing at the Australian Genome Resource Facility, Brisbane, Australia.
Construction of hHR6A plasmids and generation of recombinant proteins
For bacterial expression, hHR6A was cloned into the pTAT vector to produce a protein containing an N-terminal His6 tag (Nagahara et al., 1998). Wild-type hHR6A was amplified by PCR using the library cDNA clone H5.2 as a template. The PCR amplicon was digested with NcoI and XhoI and cloned into these sites in the pTAT vector. The mutant hHR6A (Thr3→Ala; T3A) was generated by the same protocol except that the forward primer was designed to change Thr3 to alanine. The Ser120→Ala (S120A), Ser120→Thr (S120T), Ser120→Asp (S120D) and Ser120→Glu (S120E) hHR6A mutants were generated by site-directed mutagenesis according to the manufacturer’s instructions (Stratagene) using the wild-type hHR6A-TAT construct as the PCR template. For expression in mammalian cells, hHR6A was cloned into the pCMV-Tag2 vector (Stratagene) to produce hHR6A with an N-terminal FLAG epitope tag. Wild-type hHR6A and the T3A mutant were subcloned from the hHR6A-pTAT constructs. The hHR6A-pTAT plasmid was linearized with NcoI and the 5′ overhang filled using Klenow enzyme (Boehringer Mannheim). The hHR6A cDNA was then liberated with XhoI and ligated into the pCMV-Tag 2A vector cut with EcoRV and XhoI. The Ser97→Ala and Ser120→Ala mutant hHR6As were generated by site-directed mutagenesis (Stratagene) using the wild-type hHR6A-pCMV-Tag2 construct as the template. All constructs were verified by DNA sequencing.
Wild-type and mutant His6-hHR6A fusion proteins were generated from cultures of E.coli BL21(DE3)pLysS transformed with the appropriate plasmids. Cultures were grown to an OD600 nm of 0.6–0.8 and then induced with 1 mM IPTG for 4 h at 37°C. The cells were centrifuged and the pellets resuspended in lysis buffer [phosphate-buffered saline (PBS) supplemented with an extra 100 mM NaCl, 10 µg/ml leupeptin, 1 mM PMSF and 0.2 mM DTT] followed by sonication. The lysate was centrifuged at 20 000 g for 20 min at 4°C. The supernatant was incubated with nickel-NTA–agarose (Qiagen) at 4°C for 90 min and the nickel-NTA–agarose was then washed six times with 20 mM HEPES pH 8.0, 250 mM NaCl, 0.2 mM DTT and 20 mM imidazole. hHR6A was eluted stepwise with the same buffer containing increasing concentrations of imidazole, up to 500 mM. The eluates containing the highest levels of hHR6A were pooled, dialysed against 20 mM HEPES pH 7.5, 500 mM NaCl, 0.2 mM DTT at 4°C, and then stored in aliquots at –80°C.
Phosphorylation of hHR6A in vitro, phosphoamino acid analysis and phosphopeptide mapping
Phosphorylation of hHR6A in vitro was performed in 30 µl consisting of 20 mM Tris–HCl pH 7.5, 0.2 mM DTT, 10 mM MgCl2, 20 µM ATP and 10 µCi of [γ-32P]ATP for 30 min at 37°C in the presence or absence of cyclin–CDK. Reactions were terminated by the addition of 15 µl of 3× stop buffer [187 mM Tris–HCl pH 6.8, 30% (w/v) glycerol, 6% SDS, 15% β-mercaptoethanol]. The samples were then heated at 100°C for 2 min, centrifuged and electrophoresed on a 12% SDS–polyacrylamide gel. Following electrophoresis, proteins were stained with 0.5% Coomassie Blue, the gel dried under vacuum and exposed to X-ray film for autoradiography. Phosphoamino acid analysis and tryptic phosphopeptide mapping were performed as described previously (Campbell et al., 1999).
In vivo [32P]phosphate labelling of hHR6A
CHO cells were seeded at 1.0 × 106 cells/150 mm dish and transfected with 30 µg of plasmid DNA using FuGENE 6 (Boehringer Mannheim) according to the manufacturer’s instructions. For in vivo 32P labelling, CHO cells transiently transfected with pCMV-hHR6A were grown to ∼70% confluence. The cells were pre-incubated for 3 h at 37°C in 8 ml of phosphate-free Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL) containing 10% dialysed fetal bovine serum (FBS), then replaced with 10 ml of the same medium containing 0.6 mCi/ml [32P]phosphate (ICN) and incubated for 6 h at 37°C. The cells were washed twice in PBS and harvested by scraping in 1 ml of RIPA buffer [20 mM Tris pH 7.6, 300 mM NaCl, 2 mM EDTA, 1% (w/v) Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS] supplemented with 1 mM DTT, 10 µg/ml leupeptin, 10 µg/ml aprotonin, 300 µM PMSF and incubated for 5 min on ice. The lysates were syringed through a 23 gauge needle and centrifuged at 17 500 g for 5 min at 4°C. The supernatants were removed and immunoprecipitated with 40 µl of a 1:1 suspension of anti-FLAG M2 beads (Sigma)/CL-6B beads (ratio 1:3) and incubated at 4°C overnight with gentle rotation. The immunoprecipitates were washed five times with ice-cold RIPA buffer supplemented with 1 mM DTT, 300 µM PMSF and once with 20 mM HEPES pH 7.0, 1 mM DTT. The immunoprecipitates were resuspended in 30 µl of stop buffer, boiled at 100°C for 2 min and separated by SDS–PAGE on 12% gels.
In vitro assay for hHR6A histone H2A ubiquitin-conjugating activity
To measure hHR6A ubiquitin-conjugating activity following cyclin– CDK phosphorylation, hHR6A was first phosphorylated by cyclin A– CDK2. Phosphorylation was initiated by adding hHR6A, in a final volume to 30 µl consisting of 20 mM Tris–HCl pH 7.5, 0.2 mM DTT, 10 mM MgCl2, 1 mM ATP, in the absence or presence of cyclin A–CDK2 and incubating for 60 min at 37°C. Following phosphorylation, cyclin A– CDK2 was immunodepleted by sequentially incubating the sample for 4 × 30 min on ice with 10 µl of protein A–Sepharose beads coupled to anti-CDK2 polyclonal antibody (M2; Santa Cruz Biotechnology). The supernatant (10 µl) containing hHR6A was removed and used for the ubiquitylation reaction. The ubiquitylation mixture contained hHR6A, 1 µg of GST–ubiquitin and 10 µg of histone H2A, and was initiated by the addition of 100 nM purified rabbit E1 (Affiniti Research Products, Exeter, UK) to a final volume of 22 µl consisting of 20 mM Tris–HCl pH 7.5, 0.2 mM DTT, 10 mM MgCl2 and 1 mM ATP. The reaction was performed for 30 min at 37°C and then terminated by adding 11 µl of 3× stop buffer and heating for 2 min at 100°C. The samples were separated by SDS–PAGE on 10% gels, transferred to nitrocellulose and immunoblotted with an anti-GST antibody (B-14; Santa Cruz Biotechnology) to determine the level of histone ubiquitylation, since the ubiquitin is GST tagged. Immunoreactivity was visualized by enhanced chemiluminescence (Amersham, Australia). The mutant hHR6As were assayed in the same manner, except they were not subjected to the pre-phosphorylation with cyclin A–CDK2 and immunodepletion steps, unless otherwise stated.
Cell cycle-dependent analysis of hHR6A phosphorylation and histone ubiquitylation
CHO cells were blocked in G2/M phase by incubating for 12 h in medium containing 200 ng/ml nocodazole (Sigma). The medium was removed and the cells lightly trypsinized for 30–45 s to remove G2/M blocked cells. The cells were re-seeded in flasks with fresh medium and then har vested at 0, 3, 6, 9 or 12 h for analysis of hHR6A phosphorylation or histone H2B ubiquitylation. For hHR6A phosphorylation, 0.6 mCi/ml of [32P]phosphate was added to the medium 3 h prior to harvest. The cells were harvested, lysed and hHR6A immunoprecipitated as already described. For isolation of histones, nuclei were prepared as described previously (Sarcevic et al., 1997). Basic nuclear proteins including histones were isolated by resuspending the nuclei in 0.2 M H2SO4 overnight at 4°C. The sample was centrifuged to clarify the acid-soluble proteins, which were then precipitated with 25% trichloroacetic acid. The precipitated histones were redissolved in stop buffer, boiled at 100°C for 2 min and separated by SDS–PAGE on 12% gels. The proteins were transferred to nitrocellulose and immunoblotted with either anti-histone H2B (FL-126: Santa Cruz Biotechnology) or anti-ubiquitin (13-1600: Zymed Laboratories) antibodies. Prior to addition of the anti-ubiquitin antibody, the nitrocellulose was incubated in 6 M guanidine HCl to denature the bound proteins.
Assay for complementation of S.cerevisiae growth by hHR6A
The S.cerevisiae rad6 deletion strain (S.cerevisiae rad6Δ) was KMY618 [Mata, rad6::LEU2, ura3-52, lys2-801, his3-Δ200, trp1-1(am),leu2-3, 2-11]. The congenic wild-type strain was BBY45 [Mata, ura3-52, lys2-801, his3-Δ200, trp1-1(am),leu2-3, 2-11] (Bartel et al., 1990). Yeast strains were propagated and transformed by standard methods (Sambrook et al., 1989). Plasmids expressing wild-type or mutant human hHR6A were constructed by ligating the hHR6A open reading frames into the high-copy plasmid pCS8 (Schauber et al., 1998) but with a TRP1 marker, under the control of the copper-inducible CUP1 promoter. Induction was performed with 10 µM CuSO4 in either solid or liquid media. For growth assays, 10-fold serial dilutions of exponential phase cultures grown at 30°C were spotted on plates and incubated at either 30 or 37°C.
Acknowledgments
Acknowledgements
We thank Dr Kiran Madura, Robert Wood Johnson Medical School-UMDNJ, New Jersey for supplying the S.cerevisiae rad6Δ strain, and Xiao-Wen Wang for technical assistance. This work was supported by the National Health and Medical Research Council of Australia, The Cancer Council of New South Wales and The National Breast Cancer Foundation of Australia.
References
- Banerjee A., Gregori,L., Xu,Y. and Chau,V. (1993) The bacterially expressed yeast cdc34 gene product can undergo autoubiquitination to form a multiubiquitin chain-linked protein. J. Biol. Chem., 268, 5668–5675. [PubMed] [Google Scholar]
- Bartel B., Wunning,I. and Varshavsky,A. (1990) The recognition component of the N-end rule pathway. EMBO J., 9, 3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell D.H., Sutherland,R.L. and Daly,R.J. (1999) Signaling pathways and structural domains required for phosphorylation of EMS1/cortactin. Cancer Res., 59, 5376–5385. [PubMed] [Google Scholar]
- Cardenas M.E., Dang,Q., Glover,C.V.C. and Gasser,S.M. (1992) Casein kinase II phosphorylates the eukaryote-specific C-terminal domain of topoisomerase II in vivo. EMBO J., 11, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. (1998) The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J., 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook W.J. (1997) Crystal structure of a class I ubiquitin conjugating enzyme (Ubc7) from Saccharomyces cerevisiae at 2.9 Å resolution. Biochemistry, 36, 1621–1627. [DOI] [PubMed] [Google Scholar]
- Cook W.J., Jeffrey,L.C., Xu,Y. and Chau,V. (1993) Tertiary structures of class I ubiquitin-conjugating enzymes are highly conserved: crystal structure of yeast Ubc4. Biochemistry, 32, 13809–13817. [DOI] [PubMed] [Google Scholar]
- Daly R.J., Sanderson,G.M., Janes,P.J. and Sutherland,R.L. (1996) Cloning and characterization of GRB14, a novel member of the GRB7 family. J. Biol. Chem., 271, 12502–12510. [DOI] [PubMed] [Google Scholar]
- Ellison K.S., Gwozd,T., Prendergast,J.A., Paterson,M.C. and Ellison,M.J. (1991) A site-directed approach for constructing temperature-sensitive ubiquitin-conjugating enzymes reveals a cell cycle function and growth function for RAD6. J. Biol. Chem., 266, 24116–24120. [PubMed] [Google Scholar]
- Freiberg G., Mesecar,A.D., Huang,H., Hong,J.H. and Liebman,S.W. (2000) Characterization of novel rad6/ubc2 ubiquitin-conjugating enzyme mutants in yeast. Curr. Genet., 37, 221–233. [DOI] [PubMed] [Google Scholar]
- Fukunaga R. and Hunter,T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J., 16, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grana X. and Reddy,E.P. (1995) Cell cycle control in mammalian cells: roles of cyclins, cyclin-dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene, 11, 211–219. [PubMed] [Google Scholar]
- Hershko A. and Ceichanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hsu J. et al. (2000) Mitotic phosphorylation of histone H3 is governed by Ip1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell, 102, 279–291. [DOI] [PubMed] [Google Scholar]
- Jiang W., Jimenez,G., Wells,N.J., Hope,T.J., Wahl,G.M., Hunter,T. and Fukunaga,R. (1998) PRC1: a human mitotic spindle-associated cdk substrate protein required for cytokinesis. Mol. Cell, 2, 877–885. [DOI] [PubMed] [Google Scholar]
- Koken M., Reynolds,P., Jaspers-Dekker,I., Prakash,L., Prakash,S., Bootsma,D. and Hoeijmakers,J.H.J. (1991) Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc. Natl Acad. Sci. USA, 88, 8865–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koken M.H.M., Hoogerbrugge,J.W., Jaspers-Dekker,I., Wit,J., Willemsen,R., Roest,H.P., Grootegoed,J.A. and Hoeijmakers,J. (1996) Expression of the ubiquitin-conjugating DNA repair enzymes HHR6A and B suggests a role in spermatogenesis and chromatin modification. Dev. Biol., 173, 119–132. [DOI] [PubMed] [Google Scholar]
- Lawrence C. (1994) The RAD6 DNA repair pathway in Saccharomyces cerevisiae: what does it do and how does it do it? BioEssays, 16, 253–258. [DOI] [PubMed] [Google Scholar]
- McDonough M., Sangan,P. and Gonda,D.K. (1995) Characterization of novel yeast RAD6 (UBC2) ubiquitin-conjugating enzyme mutants constructed by charge-to-alanine scanning mutagenesis. J. Bacteriol., 177, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Borgne,A., Mulner,O., Chong,J.P.J., Blow,J.J., Inagaki,N., Inagaki,M., Delcros,J.G. and Moulinoux,P. (1997) Biochemical and cellular effects of roscovotine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem., 243, 527–536. [DOI] [PubMed] [Google Scholar]
- Mueller R.D., Yasuda,H., Hatch,C.L., Bonner,W.M. and Bradbury,E.M. (1985) Identification of ubiquitinated histones 2A and 2B in Physarum polycephalum. J. Biol. Chem., 260, 5147–5153. [PubMed] [Google Scholar]
- Nagahara H. et al. (1998) Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nature Med., 4, 1449–1452. [DOI] [PubMed] [Google Scholar]
- Nigg E.A. (1993) Targets of cyclin-dependent protein kinases. Curr. Opin. Cell Biol., 5, 187–193. [DOI] [PubMed] [Google Scholar]
- Okuda M. et al. (2000) Nucleophosmin/B23 is a target of cdk2/cyclin E in centrosome duplication. Cell, 103, 127–140. [DOI] [PubMed] [Google Scholar]
- Peter M., Nakagawa,M., Doree,M., Labbe,J.C. and Nigg,E.A. (1990) Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell, 60, 791–801. [DOI] [PubMed] [Google Scholar]
- Pines J. (1993) Cyclins and cyclin-dependent kinases: take your partners. Trends Biochem. Sci., 18, 195–197. [DOI] [PubMed] [Google Scholar]
- Robzyk K., Recht,J. and Osley,M.A. (2000) Rad6-dependent uniquitination of histone H2B in yeast. Science, 287, 501–504. [DOI] [PubMed] [Google Scholar]
- Roest H.P. et al. (1996) Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell, 86, 799–810. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sarcevic B., Lilischkis,R. and Sutherland,R.L. (1997) Differential phosphorylation of T-47D human breast cancer cell substrates by D1-, D3-, E- and A-type cyclin/CDK complexes. J. Biol. Chem., 272, 33327–33337. [DOI] [PubMed] [Google Scholar]
- Schauber C., Chen,L., Tongaonkar,P., Vega,I., Lambertson,D., Potts,W. and Madura,K. (1998) Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature, 391, 715–718. [DOI] [PubMed] [Google Scholar]
- Sung P., Prakash,S. and Prakash,L. (1988) The RAD6 protein of Saccharomyces cerevisiae polyubiquitinates histones and its acidic domain mediates this activity. Genes Dev., 2, 1476–1485. [DOI] [PubMed] [Google Scholar]
- Sung P., Prakash,S. and Prakash,L. (1990) Mutation of cysteine-88 in the Saccharomyces cerevisiae RAD6 protein abolishes its ubiquitin-conjugating activity and its various biological functions. Proc. Natl Acad. Sci. USA, 87, 2695–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins J.F., Sung,P., Prakash,S. and Prakash,L. (1993) The extremely conserved amino terminus of RAD6 ubiquitin-conjugating enzyme is essential for amino-end rule-dependent protein degradation. Genes Dev., 7, 250–261. [DOI] [PubMed] [Google Scholar]
- Worthylake D.K., Prakash,S., Prakash,L. and Hill,C.P. (1998) Crystal structure of the Saccharomyces cerevisiae ubiquitin-conjugating enzyme Rad6 at 2.6 Å resolution. J. Biol. Chem., 273, 6271–6276. [DOI] [PubMed] [Google Scholar]