

Abstract
FKHR is phosphorylated by protein kinase B (PKB) at Thr24, Ser256 and Ser319 in response to growth factors, stimulating the nuclear exit and inactivation of this transcription factor. Here we identify two further residues, Ser322 and Ser325, that become phosphorylated in insulin-like growth factor-1 (IGF-1)-stimulated cells and which are mediated by the phosphatidylinositol 3-kinase-dependent PKB-catalysed phosphorylation of Ser319. Phosphorylation of Ser319 forms a consensus sequence for phosphorylation by CK1, allowing it to phosphorylate Ser322, which in turn primes the CK1-catalysed phosphorylation of Ser325. IGF-1 stimulates the phosphorylation of Thr24, Ser256, Ser319, Ser322 and Ser325 in embryonic stem (ES) cells, but not in PDK1–/– ES cells, providing genetic evidence that PDK1 (the upstream activator of PKB) is required for the phosphorylation of FKHR in mammalian cells. In contrast, the phosphorylation of Ser329 is unaffected by IGF-1 and the phosphorylation of this site is not decreased in PDK1–/– ES cells. The cluster of phosphorylation sites at Ser319, Ser322, Ser325 and Ser329 appears to accelerate nuclear export by controlling the interaction of FKHR with the Ran-containing protein complex that mediates this process.
Keywords: CK1/forkhead/nuclear export/PKB/SGK
Introduction
It is now well established that protein kinase B (PKB, also called Akt) plays a critical role in mediating many of the effects of insulin and related growth factors, downstream from phosphatidylinositol (PI) 3-kinase and 3-phosphoinositide-dependent protein kinase-1 (PDK1) (reviewed in Alessi and Cohen, 1998; Datta et al., 1999; Vanhaesebroeck and Alessi, 2000). PKB mediates these effects by phosphorylating key regulatory proteins at serine and threonine residues that lie in Arg–Xaa–Arg– Xaa–Xaa–Ser/Thr motifs (Alessi et al., 1996) and established substrates include glycogen synthase kinase-3 (Cross et al., 1995; van Weeren et al., 1998), and several closely related members of the forkhead family of transcription factors (FKHR, FKHRL1 and AFX) (Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Nakae et al., 1999; Rena et al., 1999; Takaishi et al., 1999; Tang et al., 1999).
FKHR, FKHRL1 and AFX are phosphorylated by PKB at three residues in vitro and in co-transfection experiments, equivalent to Thr24, Ser256 and Ser319 of FKHR (Brunet et al., 1999; Kops et al., 1999; Nakae et al., 1999; Rena et al., 1999; Takaishi et al., 1999; Tang et al., 1999), and many studies have been aimed at understanding the regulatory roles of these phosphorylation events (Biggs et al., 1999; Brunet et al., 1999; del Peso et al., 1999; Guo et al., 1999; Takaishi et al., 1999; Nakae et al., 2000; Brownawell et al., 2001; Rena et al., 2001). It now appears that the phosphorylation of Ser256 inhibits transactivation, probably by inhibiting nuclear import through suppression of a nuclear localization signal (Guo et al., 1999; Brownawell et al., 2001; Rena et al., 2001). In contrast, the phosphorylation of Thr24 induces interaction with 14-3-3 proteins (Brunet et al., 1999; Rena et al., 2001), which has been proposed to sequester FKHR isoforms in the cytosol, contributing to the growth factor-induced nuclear exit of these transcription factors.
There is less information about the role of Ser319 phosphorylation in comparison with Thr24 and Ser256. However, the mutation to alanine of either Thr24 or Ser256 has been reported to be without effect on agonist-induced nuclear exclusion of FKHR (Rena et al., 2001), and to have only a mild effect on FKHRL1 (Brunet et al., 2001). In contrast, the mutation of Ser319 to alanine decreases the rate of nuclear exclusion (Nakae et al., 2000; Brunet et al., 2001), resulting in accumulation of FKHR in the nucleus of unstimulated cells (Brunet et al., 2001) and visibly slower rates of extrusion from the nucleus of live cells (see Supplementary data in Rena et al., 2001) In addition, the serum- and glucocorticoid-induced protein kinase SGK, whose catalytic domain is most similar to that of PKB isoforms, phosphorylates FKHRL1 at Ser316 preferentially (the residue equivalent to Ser319 in FKHR) and stimulates nuclear exit in co-transfection experiments (Brunet et al., 2001).
There is evidence for the presence of additional phosphorylation sites on FKHR isoforms. For example, it has been reported that AFX becomes phosphorylated at other sites when the small G-protein Ral is activated (Kops et al., 1999), now identified as Thr447 and Thr451 (de Ruiter et al., 2001). Recently, we identified Ser329 as a new phosphorylation site in FKHR (Woods et al., 2001b). This residue, which is phosphorylated specifically in vitro by the dual specificity tyrosine phosphorylated regulated kinase 1A (DYRK1A), is highly phosphorylated in unstimulated cells and its phosphorylation in transfected 293 cells is not altered by stimulation with insulin-like growth factor-1 (IGF-1) or other agonists so far tested. Nevertheless, its mutation to alanine increases the proportion of FKHR in the nucleus of unstimulated (but not IGF-1-stimulated) cells, and also enhances transactivation.
Ser329 and the sequence surrounding it are highly conserved in FKHR, FKHRL1 and AFX (Figure 1). One of the conserved residues in this sequence is Ser325, raising the possibility that this residue might also be phosphorylated in vivo, perhaps by GSK3. GSK3 phosphorylates serine or threonine residues that lie in Ser/Thr–Xaa– Xaa–Xaa–pSer/pThr motifs (where pSer and pThr are phosphoserine and phosphothreonine). Such a situation is found in protein synthesis initiation factor eIF2B, where the DYRK-catalysed phosphorylation of the ε-subunit at Ser544 is a prerequisite for phosphorylation of Ser540 by GSK3, which inactivates eIF2B (Woods et al., 2001a). These considerations led us to examine whether Ser325 was phosphorylated in cells. Here we demonstrate that this is indeed the case, but that phosphorylation is not catalysed by GSK3. Our results indicate that the PKB-catalysed phosphorylation of Ser319 creates the pSer–Xaa–Xaa–Ser recognition motif for CK1, allowing it to phosphorylate Ser322. The phosphorylated Ser322 in turn ‘primes’ the phosphorylation of Ser325. The highly acidic patch formed by the phosphorylation of Ser319, Ser322, Ser325 and Ser329 appears to promote nuclear export by regulating the interaction of FKHR with the Ran-containing protein complex that mediates this process.

Fig. 1. Amino acid sequences of mammalian FKHR homologues surrounding the cluster of phosphorylation sites between Ser319 and Ser329. Identities are shown in bold and conservative replacements are underlined. The four phosphorylation sites and the protein kinases thought to phosphorylate them are indicated. Conserved residues include the RxRxxS motif (residues 314–319), which forms the recognition site for PKB/SGK (Alessi et al., 1996), R327 and P330 which are critical for the phosphorylation of Ser329 by DYRK1A (Woods et al., 2001b), and the phosphorylation sites themselves.
Results and discussion
Ser325 is a novel phosphorylation site on FKHR
In order to investigate whether the phosphorylation of Ser329 ‘primes’ FKHR for phosphorylation by GSK3 at Ser325 (see Introduction), we raised a phospho-specific antibody capable of recognizing FKHR only if it was phosphorylated at this residue (Figure 2A). We then phosphorylated bacterially expressed FKHR at Ser329 with DYRK1A in order to ‘prime’ phosphorylation by GSK3. As shown in Figure 2A and B, this did indeed allow GSK3 to phosphorylate Ser325 much more extensively in vitro. The specificity of the antibody was verified by showing that it did not recognize dephosphorylated FKHR or FKHR phosphorylated by PKB (data not shown), but only recognized FKHR phosphorylated at Ser325 by GSK3. Moreover, recognition by the antibody was abolished if it was pre-incubated with the phosphopeptide immunogen, but not by the dephosphopeptide or by a peptide phosphorylated at Ser319 (Figure 2A). GSK3 also induced the phosphorylation of the endogenous FKHR at Ser325 when transfected into 293 cells (data not shown). However, although these experiments suggested that GSK3 might phosphorylate the endogenous FKHR at Ser325 in cells, the experiments presented below demonstrate that this is not the case and that Ser325 is phosphorylated in cells by another mechanism.

Fig. 2. GSK3 phosphorylates FKHR at Ser325 in vitro and in transfected 293 cells. (A) Generation of a phospho-specific antibody that recognizes FKHR phosphorylated at Ser325. Bacterially expressed GST–FKHR was left unphosphorylated (U) or maximally phosphorylated with DYRK (DYRK), 1 U/ml GSK3 (GSK3) or DYRK followed by 1 U/ml GSK3 (DYRK/GSK3), and aliquots spotted onto a nitrocellulose membrane. They were then immunoblotted with the phospho-specific Ser325 antibody (p325) and an antibody that recognizes phosphorylated and dephosphorylated FKHR equally well (FKHR) and in the absence (NONE) or presence of the peptides shown on the right. The sequences of these peptides are given in Materials and methods. The prefix ‘p’ denotes the phosphorylated form of the peptide. (B) Phosphorylation by DYRK1A at Ser329 primes FKHR for phosphorylation by GSK3 at Ser325 in vitro. FKHR (2 µM) was either left unphosphorylated (–) or phosphorylated (+) for 30 min with GST– DYRK1A using unlabelled ATP (Woods et al., 2001b). Buffer (–) or GSK3 (+) were then added to give a final GSK3 concentration of 1 U/ml and phosphorylation continued for a further 30 min after the addition of [γ-32P]ATP. Aliquots of each reaction were subjected to electrophoresis on 4–12% SDS–polyacrylamide gradient gels, and either autoradiographed to show 32P radioactivity (32P) incorporated into FKHR, or transferred to nitrocellulose membranes and immunoblotted with phospho-specific antibodies that recognize FKHR when it is phosphorylated at Ser325 (anti-pSer325) or Ser329 (anti-pSer329), or with an antibody that recognizes the unphosphorylated and phosphorylated forms of FKHR equally well (anti-FKHR). The results shown in this figure are typical of experiments performed in duplicate at least three times.
In initial experiments, we transfected 293 cells with FKHR and studied the effect of IGF-1 on the phosphorylation of Thr24, Ser319 and Ser325. These experiments revealed that Ser325 became phosphorylated in the transfected cells and that the phosphorylation of this site, like the phosphorylation of Thr24 and Ser319, was stimulated by IGF-1 (Figure 3A).

Fig. 3. IGF-1 induces the phosphorylation of Ser325 in 293 cells; evidence that this residue is not phosphorylated by GSK3. (A) HEK 293 cells were transfected with a vector expressing wild-type GST–FKHR. At 16 h post-transfection, the cells were serum starved for a further 20 h then stimulated for 10 min without (–) or with (+) IGF-1 (100 ng/ml) prior to lysis. Aliquots of the cell lysates (10 µg of protein) were then immunoblotted with antibodies that recognize FKHR phosphorylated at Ser325 (anti-pSer325), Thr24 (anti-pThr24), Ser319 (anti-pSer319) or with an antibody that recognizes phosphorylated and unphosphorylated FKHR equally well (anti-FKHR). (B) As (A), except that a vector expressing GST–FKHR mutated at Ser329 to alanine was also used. The results shown in this figure are typical of experiments performed in duplicate at least three times.
The observation that Ser325 phosphorylation was enhanced by stimulation with IGF-1 suggested that GSK3 was not the protein kinase responsible for its phosphorylation, because GSK3 is fully active in unstimulated cells and its activity is decreased in response to agonists that activate PI 3-kinase and PKB (Cross et al., 1995). Thus IGF-1 would be expected to suppress, rather than increase the phosphorylation of Ser325 if phosphorylation was mediated by GSK3. Similarly, the mutation of Ser329 to alanine should have abolished the GSK3-catalysed phosphorylation of Ser325, but the phosphorylation of this site was unaffected by the mutation of Ser329 to alanine (Figure 3B).
The phosphorylation of Ser319 is required for the phosphorylation of Ser325
We then noticed that residue 322 was serine in every mammalian FKHR homologue (Figure 1), suggesting another route to the phosphorylation of Ser325. The protein kinase CK1 is known to phosphorylate serine residues that lie in pSer–Xaa–Xaa–Ser sequences (Flotow et al., 1990). We therefore hypothesized that the phosphorylation of Ser319 by PKB might ‘prime’ the phosphorylation of Ser322 by CK1, which would then prime the phosphorylation of Ser325 by CK1. An analogous situation occurs in glycogen synthase, where the phosphorylation of Ser7 primes the subsequent phosphorylation of Ser10 by CK1 (Nakielny et al., 1991). This hypothesis was attractive, because it explained why the phosphorylation of Ser325 was stimulated by IGF-1, i.e. it would be dependent on the prior phosphorylation of another IGF-1-sensitive site, Ser319.
In order to test this idea, we raised an antibody that recognizes FKHR only if it is phosphorylated at Ser322. The specificity of this antibody was verified in a similar manner to that described earlier for the antibody that recognizes FKHR phosphorylated at Ser325 (Figure 4A). We found that if FKHR was not pre-phosphorylated by PKB, then CK1 did not phosphorylate Ser322 or Ser325. However, the CK1-catalysed phosphorylation of Ser322 and Ser325 took place readily if FKHR was first phosphorylated by PKB (Figure 4B). Phosphorylation of Ser322 and Ser325 approached 1 mol/mol after prolonged incubation with CK1 (data not shown).


Fig. 4. Phosphorylation of FKHR at Ser319 by PKB ‘primes’ the subsequent phosphorylation of Ser322 and Ser325 by CK1 in vitro. (A) Bacterially expressed GST–FKHR was left unphosphorylated (U) or maximally phosphorylated with PKB (PKB), CK1 (CK1) or PKB followed by CK1 (PKB/CK1), and aliquots spotted onto a nitrocellulose membrane. They were then immunoblotted with the phospho-specific Ser322 antibody (p322) and an antibody that recognizes phosphorylated and dephosphorylated FKHR equally well (FKHR) and in the absence (NONE) or presence of the peptides shown on the right (each at 10 µM). The sequences of these peptides are given in Materials and methods. The prefix ‘p’ denotes the phosphorylated form of the peptide. (B) Wild-type FKHR (2 µM) was either left unphosphorylated (–) or phosphorylated (+) for 30 min using unlabelled ATP and activated GST–PKBα. Buffer (–) or CK1 (+) were then added to give a final CK1 concentration of 1 U/ml and phosphorylation continued for a further 30 min after the addition of [γ-32P]ATP. Aliquots of each reaction were subjected to electrophoresis on SDS–polyacrylamide gels, and either autoradiographed to show 32P radioactivity (32P) incorporated into FKHR, or transferred to nitrocellulose membranes and immunoblotted with the anti-pSer322 phospho-specific antibody tested in (A), in addition to those used in Figure 3A. (C) As (B), except that FKHR mutated at Ser319 to alanine was used instead of the wild-type protein. (D) Bacterially expressed wild-type GST–FKHR and GST–FKHR[S319A] were either left unphosphorylated (U) or maximally phosphorylated with DYRK followed by GSK3 (DYRK/GSK3), and aliquots spotted onto a nitrocellulose membrane. They were then immunoblotted with the phospho-specific Ser325 antibody. The results shown in this figure are typical of experiments performed in duplicate at least three times.
In contrast, if Ser319 was mutated to alanine, CK1 did not phosphorylate Ser325, even after prior phosphorylation with PKB (Figure 4C). It could be argued that the lack of phosphorylation of Ser325 is an artefact explained by the mutation of Ser319 to alanine preventing recognition of the phospho-Ser325 epitope, but a control experiment showed that this was not the case. The phospho-specific Ser325 antibody recognized phospho-Ser325 equally well when either wild-type FKHR or FKHR[Ser319Ala] was phosphorylated by DYRK plus GSK3 (Figure 4D).
In contrast, the mutation of Ser319 to alanine disrupted the recognition of phospho-Ser322 because the Ser322 phospho-specific antibody recognized the synthetic peptide RTSSNApSTISGRL but not the peptide RTSANAp STISGRL (data not shown). An analogous experiment showed that the mutation of Ser322 to alanine prevented the recognition of phospho-Ser325 by the phospho-specific Ser325 antibody (data not shown).
It should be noted that phosphorylation by CK1 impaired the recognition of pSer319 by the anti-pSer319 antibody (Figure 4B), presumably because dephosphorylated Ser322, and perhaps Ser325, are part of the epitope recognized by the antibody. An analogous situation has been observed in eIF2B (Woods et al., 2001a). This may explain the difficulty that some investigators have had in detecting the phosphorylation of Ser319 in cells (Biggs et al., 1999; Brunet et al., 1999).
We next examined the phosphorylation of Ser325 in 293 cells transfected with the appropriate FKHR mutants (Figure 5). These experiments demonstrated that the mutation of Ser319 to alanine abolished the IGF-1-induced phosphorylation of Ser325. However, the mutation of Ser325 to alanine did not affect the phosphorylation of Ser319. These results are consistent with a hierarchical phosphorylation in which Ser319, Ser322 and Ser325 are phosphorylated sequentially.

Fig. 5. Phosphorylation of Ser319 is required for phosphorylation of Ser325 in cells. HEK 293 cells were transfected with vectors expressing wild-type GST–FKHR, GST–FKHR[S319A] or GST–FKHR[S325A]. Cells were stimulated with IGF-1, lysed and prepared for immunoblotting as in Figure 2. Immunoblotting was performed with antibodies that recognize FKHR phosphorylated at Ser319 (anti-pSer319), Ser325 (anti-pSer325), Ser329 (anti-pSer329) or Thr24 (anti-pThr24), or with an antibody that recognizes phosphorylated and unphosphorylated FKHR equally well (anti-FKHR). The results shown in this figure are typical of experiments performed with duplicate lysates at least three times.
Phosphorylation of the endogenous FKHR at Thr24, Ser256, Ser319, Ser322 and Ser325 is blocked by wortmannin in 293 cells, but not by PD 98059 or rapamycin
The IGF-1-induced phosphorylation of overexpressed FKHR at Thr24, Ser256 and Ser319 is inhibited by pre-incubation with wortmannin (Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Nakae et al., 1999; Rena et al., 1999), an inhibitor of PI 3-kinase, but is unaffected by rapamycin (Nakae et al., 1999; Rena et al., 1999) (an inhibitor of the protein kinase mTOR) or by PD 98059 (Rena et al., 1999) (an inhibitor of the classical mitogen-activated protein kinase cascade). In order to provide further evidence that Ser322 and Ser325 are phosphorylated in a hierarchy following Ser319 phosphorylation, we examined the signalling pathways that mediate the IGF-1-induced phosphorylation of the endogenous FKHR at Ser322 and Ser325, as well as at the three sites of PKB phosphorylation. This was achieved by immunoprecipitating relatively large amounts of endogenous FKHR from 293 cell lysates to facilitate subsequent detection of phosphorylation using appropriate phospho-specific antibodies (Woods et al., 2001b).
The IGF-1-induced phosphorylation of Thr24, Ser256, Ser319, Ser322 and Ser325 was found to be prevented by wortmannin, but not rapamycin or PD 98059 (Figure 6A). These experiments establish that the phosphorylation of Ser322 and Ser325 is dependent on the activation of PI 3-kinase. The phosphorylation of endogenous FKHR at Ser329 was not stimulated by IGF-1 or affected by any of the inhibitors, confirming previous results (Woods et al., 2001b). Similar results were obtained after stimulation with epidermal growth factor (EGF) instead of IGF-1 (data not shown). As reported previously (Woods et al., 2001b), the phospho-specific FKHR antibodies recognized two proteins in 293 cells. The lower band co-migrated with the anti-FKHR antibody, while the upper band was recognized particularly strongly by the anti-pThr24 antibody and the anti-pSer325 antibody. Since the anti-Thr24 antibody was raised against a synthetic phosphopeptide corresponding to the sequence of the FKHR-L1 isoform, it is likely that the upper band is FKHR-L1. The fact that the upper band becomes phosphorylated in response to IGF-1 and that phosphorylation is prevented by wortmannin is consistent with this interpretation.
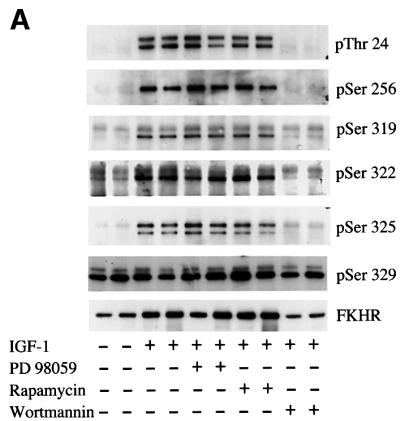
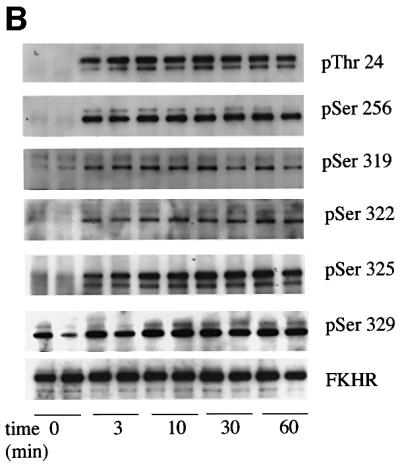
Fig. 6. Phosphorylation of the endogenous FKHR in 293 cells. (A) HEK 293 cells were serum starved for 4 h, then stimulated for 30 min without (–) or with (+) 100 ng/ml IGF-1. Aliquots of the cell lysates (2.5 mg of protein) were then immunoprecipitated with 10 µg of anti-FKHR antibody raised against the whole protein. After washing and denaturation in SDS, aliquots of the solubilized material were prepared for immunoblotting (as in Figure 2B) using the phospho-specific antibodies indicated. Where indicated, the cells were incubated for 10 min with 100 nM wortmannin or for 60 min with 100 nM rapamycin or 50 µM PD 98059, prior to stimulation with IGF-1. (B) As (A), except that cells were stimulated with 100 ng/ml IGF-1 for the times indicated. The results shown in this figure are typical of experiments performed with duplicate lysates at least three times.
We also examined the rate at which the different sites on FKHR were phosphorylated. The rates of phosphorylation of Ser319, Ser322 and Ser325 were similar in response to either IGF-1 (Figure 6B) or EGF (data not shown), suggesting that Ser322/325 kinase activity is not rate limiting for the phosphorylation of Ser322 and Ser325.
Site-specific phosphorylation of the endogenous FKHR in wild-type and PDK1–/– embryonic stem cells
The results presented in the preceding section were consistent with a requirement for the phosphorylation of Ser319 via a PI 3-kinase-dependent pathway to permit the subsequent phosphorylation of Ser322 and Ser325. If this were the case, then the phosphorylation of Ser322 and Ser325 should be dependent on the activity of PDK1, the immediate upstream activator of PKB and SGK, the protein kinases reported to phosphorylate Ser319 (see Introduction). We therefore examined the phosphorylation of the different sites in FKHR using wild-type embryonic stem (ES) cells and ES cells that carry a targeted disruption of the gene encoding PDK1. These experiments revealed that, as in 293 cells, IGF-1 induced phosphorylation of the endogenous FKHR at Ser322, Ser325 and the three sites phosphorylated by PKB/SGK in wild-type ES cells. Phosphorylation of each site was abolished if the cells were first pre-incubated with the PI 3-kinase inhibitor wortmannin (Figure 7). No phosphorylation of any of these sites was detected in PDK1–/– cells, consistent with the requirement for phosphorylation of Ser319 to prime the phosphorylation of Ser322 and Ser325 in vivo. In contrast, neither the expression of FKHR nor the phosphorylation of Ser329 were affected significantly in the PDK1–/– cells and, as expected (Woods et al., 2001b), the phosphorylation of Ser329 was unaffected by IGF-1 (Figure 7). Similar results were obtained when IGF-1 was replaced by EGF (data not shown).

Fig. 7. Phosphorylation of FKHR in wild-type ES cells and cells carrying a targeted disruption of the PDK1 gene. Wild-type murine ES cells (termed PDK1+/+ cells) and PDK1–/– ES cells were serum starved for 4 h, then stimulated for 30 min with or without 100 ng/ml IGF-1. Where indicated, the cells were incubated for 10 min with 100 nM wortmannin prior to stimulation with IGF-1. Immunoprecipitation, electrophoresis and immunoblotting with the indicated antibodies were performed as in Figure 6. Note that FKHR expression is much lower in ES cells than in 293 cells (unpublished observations), resulting in weaker signals. In addition, the pSer319 antibody signal is partly suppressed by phosphorylation of Ser322 and/or Ser325; see Figure 4B and text. The results shown in this figure are typical of experiments performed with duplicate lysates at least three times.
Multisite phosphorylation of the region containing Ser319, Ser322, Ser325 and Ser329 provides a signal for the nuclear exclusion of FKHR
We have reported that the mutation of Ser319 to alanine increases the proportion of FKHR in the nucleus of unstimulated cells and decreases the rate at which FKHR is excluded from the nucleus in response to IGF-1 (Rena et al., 2001). In order to assess the relative importance of each phosphorylation site to the subcellular localization of FKHR, we assessed the rate at which wild-type and mutated forms of FKHR were redistributed between the nucleus and cytoplasm in single living cells, by monitoring the loss of FKHR–green fluorescent protein (GFP) fluorescence from nuclei after stimulation with IGF-1.
We observe that the intensity of nuclear wild-type FKHR in a cell and the initial rate at which it redistributes to the cytoplasm depend on its nuclear concentration; the higher the concentration of nuclear FKHR, the faster the rate of redistribution. The mutation of Thr24, Ser256 or Ser329 to alanine does not affect this proportionality and the four populations of cells may be plotted together to produce a single line (Figure 8A). In contrast, we find that the mutation of either Ser319 or Ser322 to alanine greatly decreases the slope and position of the line, indicating that these mutations decrease the rate of nuclear exclusion (Figure 8A and B). Pre-incubation of cells with the nuclear export inhibitor leptomycin B blocked nuclear exclusion of wild-type FKHR (Figure 8B).

Fig. 8. The rate of nuclear exclusion of FKHR mutants from single live cells. HEK 293 cells were transfected with wild-type FKHR or the indicated FKHR–GFP mutants. At 24 h post-transfection, the cells were serum starved for 20 h then stimulated with IGF-1 and 10% fetal calf serum. Initial rates of nuclear exclusion were determined by time-lapse imaging of live cells using confocal microscopy on a heated stage. (A) Each point represents the mean nuclear exclusion rate from 25 cells, plotted versus the mean nuclear concentration of FKHR in the cells. Closed circles are a data set that includes wild-type FKHR, FKHR[S256A], FKHR[T24A] and FKHR[S329A]. Open circles are a data set that includes FKHR[S319A], FKHR[S322A] and FKHR[S319A, S322A, S325A, S329A]. (B) As (A), except that individual data sets for wild-type and each mutant are presented. In addition, data are presented from cells that were pre-incubated for 2 h with leptomycin B (5 ng/ml) before stimulation with IGF-1 and serum (closed square).
Retarded nuclear exclusion due to the mutation of Ser319 or Ser322 to alanine could be explained by one or more of the following mechanisms: (i) slower nuclear export; (ii) faster nuclear import; or (iii) nuclear retention. In order to distinguish between these possibilities, we deprived the cells of serum for only 2 h, which results in a higher proportion of cytosolic FKHR than occurs after overnight starvation. We then measured nuclear import alone by inhibiting nuclear export with leptomycin B. We found that the mutation of Ser319 or Ser322 to alanine causes, if anything, a small reduction of nuclear import compared with wild-type FKHR (Table I). This suggests that these mutations do not reduce nuclear exclusion by accelerating import; rather they must decrease the rate of nuclear export and/or increase nuclear retention.
Table I. Rates of nuclear import of wild-type and mutant FKHR.
| Mutation | Mean cytoplasmic concentration (arbitrary units) | Initial rate of import (U/min) |
|---|---|---|
| Wild type | 37 ± 3 | 0.20 ± 0.02 |
| Thr24Ala | 28 ± 4 | 0.22 ± 0.05 |
| Ser256Ala | 16 ± 1 | 0.24 ± 0.02 |
| Ser319Ala | 28 ± 2 | 0.17 ± 0.03 |
| Ser322Ala | 35 ± 4 | 0.12 ± 0.03 |
| Ser329Ala | 34 ± 3 | 0.23 ± 0.04 |
| Ser319Ala, Ser322Ala, Ser325Ala, Ser329Ala | 29 ± 4 | 0.17 ± 0.02 |
FKHR–GFP was transfected as described in the legend of Figure 8. After serum starvation for 2 h, and pre-incubation with leptomycin B (5 ng/ml) for 20 min, rates of nuclear import were determined in the presence of IGF-1 and 10% serum as described in Materials and methods.
Phosphorylation of the region containing Ser319, Ser322, Ser325 and Ser329 stimulates association with a protein complex that contains Ran
A protein complex that contains the GTPase Ran and chromosomal region maintenance protein-1 (CRM1) is known to control leptomycin B-sensitive nuclear exit of proteins (reviewed in Komeili and O’Shea, 2000). We therefore investigated whether FKHR was capable of binding to this complex and whether binding was affected by phosphorylation.
We found that overexpressed FKHR could be immunoprecipitated from cell extracts with an anti-Ran antibody and that the amount bound was increased modestly by stimulation for 10 min with IGF-1. The mutation of Ser319 to alanine has been reported to have no effect on the binding of AFX to the Ran–CRM1 complex (Brownawell et al., 2001), and we have confirmed this result for FKHR (Figure 9). This indicates that the phosphorylation of Ser319, Ser322 and Ser325 does not disrupt interaction with the FKHR–Ran complex. However, interestingly, the mutation of either Ser322 or Ser325 to alanine drastically reduced binding to Ran in either unstimulated or IGF-1-stimulated cells (Figure 9). The mutation of Ser322 or Ser325 to alanine also abolished the binding of FKHR to the GTPase-deficient mutant Ran[Q69L] in the presence of GTP (data not shown). These results suggest that Ser322 and Ser325 may be important for the interaction of FKHR with the Ran complex. The mutation of Ser322 or Ser325 does not disrupt the structure of FKHR, because these mutants are just as effective as wild-type FKHR in stimulating FKHR-dependent gene transcription in cell-based assays (L.Gan and T.Unterman, unpublished work).

Fig. 9. Co-immunoprecipitation of FKHR with Ran GTPase. HEK 293 cells were transfected with wild-type or the indicated FKHR mutants, then stimulated without (–) or with (+) IGF-1 for 10 min. Ran was then immunoprecipitated (IP) from the cell lysates (100 µg of protein) using 10 µg of an anti-Ran antibody. Samples were then immunoblotted as in Figure 2B using an FKHR antibody raised against the whole protein that recognizes the unphosphorylated and phosphorylated forms of the protein equally well (upper panel). The level of expression of FKHR in the lysates was assessed by immunoblotting with the same antibody (lower panel). Wort = wortmannin. The results shown in this figure are typical of experiments performed with duplicate lysates at least three times.
These findings raise the question of whether FKHR binds directly to Ran or to a Ran-interacting protein, such as CRM1. The ability of leptomycin B, a CRM1-mediated nuclear export inhibitor, to block the nuclear exclusion of FKHR provides strong evidence that CRM1 is required for the nuclear export of FKHR. We have observed that purified recombinant Ran binds to FKHR–GFP immunoprecipitates, even after extensive washing in 0.5 M NaCl, but have been unable to detect any interaction with purified recombinant CRM-1 under the same conditions (data not shown). These observations indicate that FKHR can bind to Ran, although a weaker interaction with CRM1 is not excluded, or may depend on other factors such as the Ran-binding protein RanBP3 (Englmeier et al., 2001). These investigators also noted the difficulty in detecting the binding of purified CRM1 to many ‘cargo’ proteins.
Hyperphosphorylation generates acidic patches that regulate nuclear export
Phosphorylation site repeats are found in many proteins that shuttle between the nucleus and cytoplasm, including p53 (Dumaz et al., 1999), NFAT2 (Beals et al., 1997), NFAT4 (Zhu et al., 1998), mPER (Vielhaber et al., 2000), Dictyostelium STAT (Ginger et al., 2000) and the yeast transcription factor Pho4 (Komeili and O’Shea, 1999), as well as FKHR. The CK1-catalysed phosphorylation of multiple sites is reported to promote nuclear exclusion of NFAT4 and mPER, while the phosphorylation of multiple sites by GSK3 is reported to promote the nuclear exclusion of NFAT2 and Dictyostelium STAT. The studies with yeast Pho4 and Dictyostelium STAT demonstrated that, in contrast to nuclear import which depends on basic residues on cargo, nuclear export can be promoted by acidic patches due to multisite phosphorylation. Our results are consistent with a similar mechanism operating in mammalian cells to regulate the subcellular redistribution of FKHR.
In summary, the phosphorylation of cargo proteins can promote nuclear exclusion in at least three main ways, all of which may operate in FKHR (Table II). First, phosphorylation may retard nuclear import by masking a basic nuclear localization signal. Secondly, 14-3-3s by binding to phosphoproteins may retain them in the cytosol. Thirdly, hyperphosphorylation of cargo proteins generates acidic patches that may accelerate nuclear export. The simultaneous regulation of nuclear import and export by these mechanisms is likely to facilitate the rapid redistribution of FKHR and other proteins between the cytosol and the nucleus.
Table II. Summary of functions proposed for different phosphorylation sites on FKHR.
| Phosphorylation site | Putative upstream kinase | Proposed function |
|---|---|---|
| Thr24 | PKB/SGK | 14-3-3 binding (Brunet et al., 1999; Rena et al., 2001), perhaps to block DNA binding (Cahill et al., 2001) and/or to promote cytoplasmic retention (Brunet et al., 1999) |
| Ser256 | PKB/SGK | Inhibition of transactivation (Guo et al., 1999), masking of adjacent nuclear localization signal (Brownawell et al., 2001; Rena et al., 2001) |
| Ser319 | PKB/SGK | Regulation of nuclear export (this study) |
| Ser322 and Ser325 | CK1 | Regulation of nuclear export (this study) |
| Ser329 | DYRK | Mutation of this constitutive site increases transactivation, at least in part by increasing basal nuclear localization (Woods et al., 2001b) |
Conclusions
We have identified two new phosphorylation sites in FKHR, Ser322 and Ser325, and shown that they are phosphorylated sequentially, probably by CK1, following the phosphorylation of Ser319 by PKB or SGK. We have also provided the first genetic evidence in mammalian cells that the phosphorylation of FKHR depends on PDK1 and hence on protein kinases, such as PKB and SGK, that lie downstream of PI 3-kinase. The cluster of four phosphorylation sites between residues 319 and 329 plays a key role in stimulating redistribution of FKHR between the nucleus and cytoplasm.
Materials and methods
Materials
Restriction enzymes were purchased from NEB (UK) Ltd (Hitchin) and MBI (Vilnius, Lithuania), and leptomycin B from Sigma (Poole, UK). An antibody raised against Ran (sc 1156) was from Santa Cruz, USA. An FKHRL1 antibody and a phospho-specific antibody that recognizes FKHR isoforms only when phosphorylated at sites equivalent to Thr24 of FKHR or Thr32 of FKHRL1 were from Upstate Biotechnology (Lake Placid, NY). Phospho-specific antibodies that recognize FKHR phosphorylated specifically at either Ser319, Ser322 or Ser325 were raised against the synthetic phosphopeptides TFRPRTSpSNASTIS, RTS SNApSTISGRL and NASTIpSGRLSPIMTEQ, corresponding to residues 312–326, 316–328 and 320–335, respectively. These peptides were synthesized by Dr Graham Bloomberg, University of Bristol, UK. The antibodies were affinity purified against the appropriate phosphopeptide bound to Sepharose. These antibodies were produced by Drs Jane Leitch and Chris Armstrong (Division of Signal Transduction Therapy, University of Dundee). The 4–12% polyacrylamide gels used were from InVitrogen. Sources of other materials have been given previously (Rena et al., 1999, 2001).
Cell culture, transient transfection and cell lysis
293 cells were cultured and transfected with FKHR as described previously (Rena et al., 1999). Wild-type ES cells and PDK1–/– ES cells that do not express PDK1 were cultured as described previously (Williams et al., 2000). Cells were serum starved prior to stimulation with IGF-1 (100 ng/ml). The cells were then lysed in 1 ml of ice-cold buffer A [50 mM Tris-acetate pH 7.5, 1 mM EGTA, 1% (w/v) Triton X-100, 1 mM EDTA, 50 mM NaF, 10 mM sodium β-glycerophosphate, 5 mM sodium pyrophosphate, 1 mM benzamidine, 0.2 mM phenylmethylsulfonyl fluoride and 0.1% (v/v) β-mercaptoethanol]. The lysates were centrifuged at 13 000 g and the supernatants removed, frozen immediately in liquid nitrogen and stored at –80°C until use.
Expression, immunoprecipitation and phosphorylation of wild-type and mutated GST–FKHR
FKHR mutants in which either Ser322 or Ser325 alone or Ser319, Ser322, Ser325 and Ser329 in combination were changed to alanine were made by PCR-based methods and subcloned into the TA cloning vector pCR2.1. Mutations were incorporated into the vector pEBG-2T by replacement of the region of the 3′ end of wild-type FKHR digested by PflMI and KpnI. The final construct encoded GST immediately followed by haemagglutinin (HA)-FKHR (herein termed GST–FKHR) and was used for expression in mammalian cells. Wild-type FKHR, a Ser322 to alanine single mutant and a Ser319, Ser322, Ser325 and Ser329 to alanine quadruple mutant were also subcloned into the vector pEGFP-N1 by replacement of an XmnI and HindIII fragment in a Ser319 to alanine mutant. Each construct encoded FKHR, with the stop codon removed, immediately followed by GFP, hereafter termed FKHR–GFP. Each construct was sequenced to establish the accuracy of the DNA construct. For immunoprecipitation experiments, 10 µg of Ran antibody or FKHR antibody was used. Ran immunoprecipitations were performed with 100 µg of lysate, while FKHR immunoprecipitations were performed with 2.5 mg of lysate. Immunoprecipitates were washed twice with 1 ml of buffer A plus 500 mM NaCl (FKHR) or 100 mM NaCl (Ran), followed by two washes in buffer A alone. After denaturation in SDS, 10% of the solubilized material was run in each gel lane.
Confocal microscopy and determination of the rate of nuclear exclusion and import
Experiments with FKHR–GFP constructs were carried out without fixation in transfected living cells in a heated chamber at 37°C in 10 mM HEPES-buffered Dulbecco’s modified Eagle’s medium pH 7.5 and imaged using a Zeiss laser scanning confocal microscope 410 using a ×63 plan-apo(NA1-4) objective (Rena et al., 2001). In order to pool standardized results from different cells across different experiments, the initial nuclear concentration of FKHR in each cell was determined, using the NIH Image 1.61 software package. The concentration of FKHR is measured in arbitrary units. Zero is the mean intensity of nuclei of cells transfected with the cytosolic mutant RRR/SAS (Rena et al., 2001). To collect data from which the initial rate of exclusion could be calculated, the nuclear concentration of FKHR was also determined for each cell at 5 min time points in a 90 min time course. Using Cricket Graph software, the data were plotted as the concentration of nuclear FKHR versus time. The curve fitting tool in the software package allowed initial exclusion rates to be determined. Nuclei were only measured if they remained in the plane of focus for the duration of the experiment. In order to eliminate dead and dying cells, nuclei with a mean intensity of >60 U of FKHR at t = 0 were not analysed, and the slowest 20% of the analysed cells were eliminated in each experiment. The initial rate of nuclear import was measured similarly, where import is measured as nuclear accumulation of FKHR. The main difference is that import experiments were standardized by measurement of the initial cytoplasmic concentration of FKHR in each cell. Data points are presented as means ± SE.
Acknowledgments
Acknowledgements
We are grateful to Dr David Meek (University of Dundee Medical School, Ninewells Hospital) for bacteria expressing the GST fusion protein of rat CK1δ[1–294], Dr Walter Becker (Institute for Pharmacology and Toxicology, Aachen, Germany) for DYRK1A, Dr Paul Clarke (University of Dundee Medical School, Ninewells Hospital) for generously providing Ran Q69L and CRM1 protein, Dr Dario Alessi (MRC Protein Phosphorylation Unit, Dundee) for the PDK1–/– cells, Agnieszka Kieloch (Division of Signal Transduction Therapy, Dundee) for cell culture, and Professor Angus Lamond (Dundee) for use of the confocal microscope. We thank the UK Medical Research Council for a studentship (to Y.L.W.) and the MRC, The Royal Society, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, NovoNordisk and Pfizer for financial support.
References
- Alessi D.R. and Cohen,P. (1998) Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev., 8, 55–62. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Caudwell,F.B. Andjelkovic,M., Hemmings,B.A. and Cohen,P. (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett., 399, 333–338. [DOI] [PubMed] [Google Scholar]
- Beals C.R., Sheridan,C.M., Turck,C.W., Gardner,P. and Crabtree,G.R. (1997) Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science, 275, 1930–1933. [DOI] [PubMed] [Google Scholar]
- Biggs W.H., Meisenhelder,J., Hunter,T., Cavenee,W.K. and Arden,K.C. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl Acad. Sci. USA, 96, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownawell A.M., Kops,G.J., Macara,I.G. and Burgering,B.M. (2001) Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol., 21, 3534–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A. et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Brunet A., Park,J., Tran,H., Hu,L.S., Hemmings,B.A. and Greenberg,M.E. (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol., 21, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D.A.E., Alessi,D.R., Cohen,P., Andjelkovich,M. and Hemmings,B.A. (1995) Inhibition of glycogen-synthase kinase-3 by insulin mediated by protein-kinase-B. Nature, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet,A. and Greenberg,M.E. (1999) Cellular survival: a play in three Akts. Genes Dev., 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- del Peso L., Gonzalez,V.M., Hernandez,R., Barr,F.G. and Nunez,G. (1999) Regulation of the forkhead transcription factor FKHR, but not the PAX3–FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene, 18, 7328–7333. [DOI] [PubMed] [Google Scholar]
- de Ruiter N.D., Burgering,B.M. and Bos,J.L. (2001) Regulation by the forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol. Cell. Biol., 21, 8225–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N., Milne,D.M. and Meek,D.W. (1999) Protein kinase CK1 is a p53-threonine 18 kinase which requires prior phosphorylation of serine 15. FEBS Lett., 463, 312–316. [DOI] [PubMed] [Google Scholar]
- Englmeier L., Fornerod,M., Bischoff,F.R., Petosa,C., Mattaj,I.W. and Kutay,U. (2001) RanBP3 influences interactions between CRM1 and its nuclear protein export complexes. EMBO Rep., 2, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H., Graves,P.R., Wang,A.Q., Fiol,C.J., Roeske,R.W. and Roach,P.J. (1990) Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem., 265, 14264–14269. [PubMed] [Google Scholar]
- Ginger R.S., Dalton,E.C., Ryves,W.J., Fukuzawa,M., Williams,J.G. and Harwood,A.J. (2000) Glycogen synthase kinase-3 enhances nuclear export of a Dictyostelium STAT protein. EMBO J., 19, 5483–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.D., Rena,G., Cichy,S., He,X.W., Cohen,P. and Unterman,T.G. (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem., 274, 17184–17192. [DOI] [PubMed] [Google Scholar]
- Komeili A. and O’Shea,E.K. (1999) Roles of phosphorylation sites in regulating activity of the transcription factor Pho4. Science, 284, 977–980. [DOI] [PubMed] [Google Scholar]
- Komeili A. and O’Shea,E.K. (2000) Nuclear transport and transcription. Curr. Opin. Cell Biol., 12, 355–360. [DOI] [PubMed] [Google Scholar]
- Kops G.J.P.L., de Ruiter,N.D., De Vriets-Smits,A.M.M., Powell,D.R., Bos,J.L. and Burgering,B.M.T. (1999) Direct control of the forkhead transcription factor AFX by protein kinase B. Nature, 398, 630–634. [DOI] [PubMed] [Google Scholar]
- Nakae J., Park,B.C. and Accili,D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a wortmannin-sensitive pathway. J. Biol. Chem., 274, 15982–15985. [DOI] [PubMed] [Google Scholar]
- Nakae J., Barr,V. and Accili,D. (2000) Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J., 19, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S., Campbell,D.G. and Cohen,P. (1991) The molecular mechanism by which adrenaline inhibits glycogen synthesis. Eur. J. Biochem., 199, 713–722. [DOI] [PubMed] [Google Scholar]
- Paraskeva E., Izaurralde,E., Bischoff,F.R., Huber,J., Kutay,U., Hartmann,E., Luhrmann,R. and Gorlich,D. (1999) CRM1-mediated recycling of snurportin 1 to the cytoplasm. J. Cell Biol., 145, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G., Guo,S.D., Cichy,S.C., Unterman,T.G. and Cohen,P. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem., 274, 17179–17183. [DOI] [PubMed] [Google Scholar]
- Rena G., Prescott,A.R., Guo,S.D., Cohen,P. and Unterman,T.G. (2001) Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem. J., 354, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi H. et al. (1999) Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc. Natl Acad. Sci. USA, 96, 11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang E.D., Nunez,G., Barr,F.G. and Guan,K.L. (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem., 274, 16741–16746. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B. and Alessi,D.R. (2000) The PI3K–PDK1 connection: more than just a road to PKB. Biochem. J., 346, 561–576. [PMC free article] [PubMed] [Google Scholar]
- van Weeren P.C., de Bruyn,K.M.T., de Vries-Smits,A.M.M., van Lint,J. and Burgering,B.M.T. (1998) Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation—characterization of dominant-negative mutant of PKB. J. Biol. Chem., 273, 13150–13156. [DOI] [PubMed] [Google Scholar]
- Vielhaber E., Eide,E., Rivers,A., Gao,Z.H. and Virshup,D.M. (2000) Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase Iε. Mol. Cell. Biol., 20, 4888–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.R., Arthur,J.S.C., Balendran,A., van der Kaay,J., Poli,V., Cohen,P. and Alessi,D.R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol., 10, 439–448. [DOI] [PubMed] [Google Scholar]
- Woods Y.L., Cohen,P., Becker,W., Jakes,R., Goedert,M., Wang,X. and Proud,C.G. (2001a) The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2B at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J., 355, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods Y.L., Rena,G., Morrice,N., Barthel,A., Becker,W., Guo,S., Unterman,T.G. and Cohen,P. (2001b) The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J., 355, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.Y., Shibasaki,F., Price,R., Guillemot,J.C., Yano,T., Dotsch,V., Wagner,G., Ferrara,P. and McKeon,F. (1998) Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell, 93, 851–861. [DOI] [PubMed] [Google Scholar]